

Abstract
Enoate reductases from the family of old yellow enzymes (OYEs) can catalyze stereoselective trans‐hydrogenation of activated C=C bonds. Their application is limited by the necessity for a continuous supply of redox equivalents such as nicotinamide cofactors [NAD(P)H]. Visible light‐driven activation of OYEs through NAD(P)H‐free, direct transfer of photoexcited electrons from xanthene dyes to the prosthetic flavin moiety is reported. Spectroscopic and electrochemical analyses verified spontaneous association of rose bengal and its derivatives with OYEs. Illumination of a white light‐emitting‐diode triggered photoreduction of OYEs by xanthene dyes, which facilitated the enantioselective reduction of C=C bonds in the absence of NADH. The photoenzymatic conversion of 2‐methylcyclohexenone resulted in enantiopure (ee>99 %) (R)‐2‐methylcyclohexanone with conversion yields as high as 80–90 %. The turnover frequency was significantly affected by the substitution of halogen atoms in xanthene dyes.
Keywords: asymmetric reduction, enoate reductases, green chemistry, photocatalysis, redox enzymes
Enzyme‐catalyzed transformations provide unmatched high efficiency, regio‐, stereo‐, and enantioselectivities under mild conditions.1 Enoate reductases from the family of old yellow enzymes (OYEs) are a class of flavin‐containing enzymes that catalyze asymmetric reduction of activated (or conjugated) C=C bonds, producing industrially useful chiral alkanes.2 The redox enzymes contain a prosthetic group of flavin mononucleotide (FMN), which, in its reduced form (FMNH−), transfers a hydride by a Michael‐type reaction to the conjugated C=C double bond. The regeneration of the reduced flavin occurs by the transfer of two electrons and two protons,3 which is achieved in vivo through the mediation of reduced nicotinamide cofactors [NAD(P)H]. For the development of biocatalytic processes, however, NAD(P)H is too expensive to be supplied in a stoichiometric amount; thus, the majority of OYE applications have been made by mimicking the biological scheme through in situ regeneration of reduced nicotinamide cofactors using enzymatic,4 chemical,5 electrochemical,6 and photochemical7 approaches.
Herein, we report NAD(P)H‐free activation of flavin‐containing OYEs by molecular photosensitizers that allow for direct transfer of photoinduced electrons to the prosthetic flavin moiety. Rose bengal (4,5,6,7‐tetrachloro‐2′,4′,5′,7′‐tetraiodofluorescein, RB) and its xanthene derivatives are explored as photosensitizers to drive OYE‐catalyzed redox reactions under illumination with light‐emitting‐diodes (LEDs). Xanthene dyes are low‐cost photosensitizers, capable of capturing visible light energy with high quantum efficiency, which have been applied to reductive chemical reactions such as solar hydrogen evolution as well as photochemical NADH regeneration8 and for the reduction of metalloenzymes (for example, heme‐containing cytochrome P450s, metal cluster‐containing hydrogenases).9 Scheme 1 illustrates direct transfer of photoexcited electrons from RB to a flavin‐containing OYE homologue from Thermus scotoductus (TsOYE)10 upon visible light irradiation with triethanolamine (TEOA) as an electron donor. Successive transfer of electrons reduces the enzyme‐bound FMN moiety and drives the enantioselective conversion of 2‐methylcyclohexenone into (R)‐2‐methylcyclohexanone.
Scheme 1.
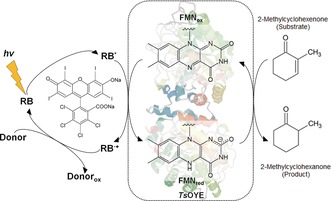
Illustration of the light‐driven activation of flavin‐containing TsOYE using rose bengal (RB) as a photosensitizer. Photoexcitation of the molecular photosensitizer reduces the active species of TsOYE to catalyze an enantioselective reduction of 2‐methylcyclohexenone to 2‐methylcyclohexanone.
Typically, OYEs are reduced by NAD(P)H that donate two electrons and one proton directly to the enzyme‐bound FMN moiety (Figure 1 A). A stepwise reduction by successive transfer of electron yields a single‐electron reduced semiquinone state of FMN, which can be fully reduced by taking an additional electron.11 We investigated the possibility of photosensitized reduction of TsOYE by RB using UV/Vis spectroscopy. TsOYE shows an absorbance peak at 464 nm due to a strong π–π* transition associated with the oxidized redox state of the enzyme‐bound FMN. Any observed decrease of the intensity can be attributed to the reduction of FMN to FMNH− through the stepwise transfer of electrons and protonation.12 According to our result (Figure 1 B), the absorbance peak at 464 nm decreased gradually during light irradiation in the presence of both RB and TEOA, which we attribute to the reduction of the prosthetic FMN by photoinduced electron transfer from RB to TsOYE with TEOA as an electron donor. Note that there was negligible change in the absorbance of TsOYE without either RB or TEOA even under illumination (Figure 1 C). The photoreduction of OYE‐bound FMN by RB was further confirmed by using another OYE homologue from Bacillus subtilis (YqjM)13 (Supporting Information, Figure S1). According to our result, three isosbestic points, at 318 nm, 382 nm, and 407 nm, were clearly observed during the photoreduction of prosthetic FMN by RB, which indicates that the semiquinone was built up as an intermediate during the stepwise reduction process. In addition, we observed spontaneous association of RB with TsOYE by measuring the changes in absorbance and photoluminescence spectra of RB upon addition of TsOYE. The absorbance of RB at 546 nm gradually red‐shifted with the increasing concentration of TsOYE (Figure 2 A), which indicates a change of the ionization state of RB upon its binding to the enzyme.14 The change in the absorbance at around 464 nm was caused by the increasing concentration of TsOYE. Furthermore, we observed that the addition of TsOYE induces quenching and red‐shift of the photoluminescence spectrum of RB (Supporting Information, Figure S2), which is attributed to fluorescence energy transfer from RB to TsOYE.
Figure 1.

A) Possible steps of enzyme‐bound FMN reduction. B) UV/Vis absorbance spectra of TsOYE‐RB mixture under visible light irradiation. C) The change in the absorbance of TsOYE at 464 nm with or without RB or TEOA. An aqueous solution containing 60 μm TsOYE, 2 μm RB, 5 mm CaCl2, and 200 mm TEOA was irradiated by a 450 W Xenon lamp equipped with 420 nm cut‐off filter.
Figure 2.
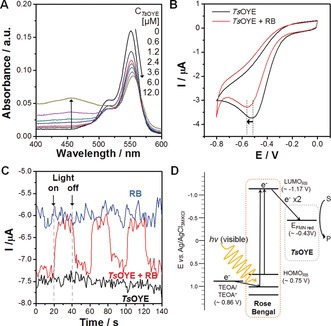
A) Spectrophotometric changes in the absorbance of RB upon addition of TsOYE. B) Cyclic voltammograms of TsOYE modified electrode in the presence or absence of RB. C) Photocurrent response of TsOYE, RB, and TsOYE with RB at an applied potential of −0.6 V (vs. Ag/AgCl). D) Proposed mechanism of electron transfer from RB to TsOYE upon visible‐light irradiation in the presence of sacrificial electron donor (TEOA).
We further investigated electrochemical relationship between RB and TsOYE using protein film voltammetry. For the analysis of electrochemical behavior of TsOYE, we prepared a TsOYE‐coated glassy carbon disk electrode by using didodecyldimethylammonium bromide as a support for protein adsorption.15 We observed TsOYE reduction at around −0.5 V (vs. Ag/AgCl) (Figure 2 B), which corresponds to the reduction of enzyme‐bound flavin cofactor.16 In the presence of RB, we detected a negative shift of approximately 60 mV in the reduction potential of TsOYE. Considering that reduction potential of RB exists at a more negative potential (ca. −0.92 V),8b this result suggests that TsOYE and RB form an electrochemically hybridized species that exhibits a co‐reducing property due to the binding affinity between the two components. Upon the addition of RB, TsOYE showed apparent increases in the reduction peak current in the presence of 2‐methylcyclohexenone, a substrate of TsOYE, indicating that the association of TsOYE with RB does not alter its catalytic activity (Supporting Information, Figure S3). We also observed a distinguishable feature of photochemical activity resulted by the co‐existence of TsOYE and RB (Figure 2 C); at an applied potential of −0.6 V, an anodic photocurrent (ca. 1.2 μA) was generated in the presence of both TsOYE and RB upon light illumination, which is attributed to the decrease in the number of electrons from the electrode to the enzyme due to the photosensitization of RB. In contrast, there was no photoresponse with either TsOYE or RB alone. Taken together, the spectroscopic, electrochemical, and photochemical results suggest a spontaneous association between TsOYE and RB through a direct energy transfer relationship, facilitating RB‐sensitized reduction of TsOYE.
Next, we investigated the asymmetric reduction of conjugated C=C double bonds via light‐driven activation of TsOYE. As shown in the Supporting Information, Figure S4, we observed a gradual conversion of 2‐methylcyclohexenone to enantiopure (R)‐2‐methycyclohexanone (ee>99 %) with 76 % yield after illumination of white LED (4.74 mW cm−2) for 150 min. A series of control experiments in the absence of each reaction component (that is, light, electron donor, photosensitizer, and enzyme) revealed that only trace amounts (ca. 0.2 mm at maximum) of enantiopure product was detected without light, TEOA, or RB (Supporting Information, Figure S5A), and no product was formed in the absence of TsOYE. This result indicates that photosensitization is essential for generating high energy electrons to initiate photoinduced electron transfer for OYE catalysis and, to sustain light‐driven enzymatic turnover, both light‐harvesting photosensitizer (that is, RB) and electron‐supplying agent (TEOA) are required. We also observed a clear dependency of initial reaction rate and product yield on the intensity of white LED light (Supporting Information, Figure S5B). To elucidate the rate‐determining factor in the photobiocatalytic reaction, we measured changes in the initial rate and product yield according to the concentrations of TsOYE and RB, respectively. As shown in the Supporting Information, Figure S6A, proportional enhancements of reaction rate and product yield were observed with the increasing concentration of RB (from 0 to 100 μm) at a fixed TsOYE concentration (18 μm). The initial turnover frequency (TOF) of TsOYE was calculated to be 256.1±4.9 h−1, which was saturated with a RB concentration at over 100 μm. The TOF value is comparable to those of previously reported methyl viologen‐mediated, Ru‐polypyridyl‐sensitized OYE reactions.17 The apparent saturation kinetics, which levels off at higher RB concentration, indicate a Michaelis–Menten‐type behavior of TsOYE with RB as a co‐substrate, instead of NADH. In a control experiment conducted for comparison using NADH as a cosubstrate (Supporting Information, Figure S6B), we found that the reaction rate saturated at around 2 mm NADH and TOF was estimated to be 734±18 h−1 at maximum. The stoichiometric ratio of TsOYE and RB (1:5) at the saturation was two‐order lower than that of TsOYE and NADH (1:333), indicating high efficiency of RB for the activation TsOYE. We found that the reaction rate reaches a saturation at around the RB/TsOYE ratio of approximately five, regardless of the TsOYE concentration, indicating that the formation of RB/TsOYE complex is critical for driving the photobiocatalytic reaction with a threshold ratio of approximately five (Supporting Information, Figure S7). When we varied TsOYE concentration at a fixed amount of RB (60 μm; Supporting Information, Figure S8A), the reaction showed a maximum rate of about 2.92 mm h−1 with 12 μm TsOYE. A decrease in the reaction rate was observed at the TsOYE concentration higher than 12 μm, which we attribute to the drastic decrease in the RB/TsOYE ratio from 5 to 1.6 (Supporting Information, Figure S8B). A reduced association of RB per TsOYE is expected because the molar ratio of RB to TsOYE decreases with the increasing TsOYE concentration at a fixed RB concentration. Figure 2 D illustrates an energy diagram for photoinduced electron transfer from RB to TsOYE. Upon visible‐light absorption, RB promotes an internal conversion of its electrons from a ground state (HOMORB≈0.75 V vs. Ag/AgCl) to the excited state (LUMORB≈−1.17 V).18 The excited electrons in RB possess enough potential energy to drive the reduction of TsOYE‐bound FMN (E reduction≈−0.43 V), and the oxidation of TEOA (E oxidation≈0.86 V) stabilizes RB to sustain its photocatalytic turnover. The repeated transfer of photoexcited electrons from RB to TsOYE fully reduces enzyme‐bound FMN to FMNH−, driving the catalytic cycle for asymmetric C=C reduction.
We further investigated different xanthene molecules and natural flavin mononucleotide (FMN) for direct photoactivation of TsOYE. According to our result (Table 1), halogenated derivatives of xanthenes, such as eosin Y (EY), erythrosine B (ErB), phloxine B (PhB), and RB showed high activity in TsOYE‐catalyzed reduction of 2‐methylcyclohexenone; EY exhibited best performance in terms of reaction rate with a TOF of 118 and ErB resulted in the highest conversion yield (73.9 %). Our result indicates that the substitution of a heavier halogen atom in xanthenes resulted in slower reaction rate (that is, TOFs in the order of EY>ErB>PhB>RB). We ascribe the result to an increased non‐specific association of halogenated xanthene dyes to TsOYE, considering that hydrophobic interaction of xanthenes with proteins is known to increase with their halogenations in the order of EY<ErB<RB.19 On the other hand, non‐halogenated derivatives of xanthene dyes, such as fluorescein, rhodamine B, and rhodamine 6G, showed significantly lower (by more than one order of magnitude) performance compared to halogenated xanthenes. For example, fluorescein showed an initial TOF of 4.6 h−1 and a product yield of 9.7 %. We attribute the low photocatalytic activities of non‐halogenated derivatives (fluorescein, rhodamines) to their higher fluorescence quantum yields (Φ fluorescein, rhodamine B≈0.95) than those of other halogenated species (Φ EY=0.57),20 which causes a shorter life time of excited triplet state due to the high fluorescence. Halogen substitution enhances the singlet–triplet transition state of aromatic molecules by increasing spin–orbit mixing, and the substitution of heavier halogen atom makes the molecule easier to associate with other species owing to the decrease in the electronegativity.8b, 21 In the case of FMN, we observed a very low TOF and product yield, comparable to that of fluorescein, which should be caused by high reactivity of photoexcited flavins with dissolved oxygen, producing reactive radical species that denature enzymes; thus, the use of free FMN as a photocatalyst requires deaeration of the reaction medium to remove dissolved oxygen and drive substrate conversion.22 In contrast, the photoreaction carried out by RB was not affected under O2‐rich conditions; our experiments conducted with three different ratios (2, 5, 10) of RB/TsOYE under O2‐purged or N2‐purged conditions also confirms that excessive RB over TsOYE under O2‐rich conditions had negligible effect on RB‐mediated reductions (Supporting Information, Figure S9). A nonspecific binding of RB with protein induces static quenching of the excited state of RB that prevents photosensitized production of singlet oxygen.23 The spontaneous association of RB with TsOYE should facilitate the reduction of enzyme‐bound FMN, instead of singlet oxygen production. Furthermore, the fast reduction of RB in the presence of TEOA would have prevented the generation of singlet oxygen by transforming the singlet‐excited RB to its reduced state.
Table 1.
Reduction of 2‐methylcyclohexenone (R)‐2‐methycyclohexanone by TsOYE in NAD(P)H‐free, light‐driven biocatalytic platform employing different molecular photosensitizers.
| Photosensitizer (PS) | TOF[a] [h−1] | Yield[b] [%] | TTNPS [b] | TTNOYE [b] | ee [b] [%] |
|---|---|---|---|---|---|
| Eosin Y | 118 | 66.5 | 106 | 295 | >99 (R) |
| Erythrosine B | 100 | 73.9 | 118 | 328 | >99 (R) |
| Phloxine B | 78 | 45.5 | 73 | 202 | >99 (R) |
| Rose Bengal | 52 | 53.1 | 85 | 235 | >99 (R) |
| Fluorescein | 4.6 | 9.7 | 15 | 43 | >99 (R) |
| Rhodamine B | 0 | 1.1 | 1.8 | 5 | >99 (R) |
| Rhodamine 6G | 2.4 | 1.1 | 1.7 | 4.9 | >99 (R) |
| FMN | 4.8 | 4.1 | 6.5 | 18 | >99 (R) |
[a] Turnover frequency of photosensitizers determined after 30 min. [b] Determined by GC analysis after 180 min reaction. ee: enantiomeric excess; reaction conditions: 50 μm [PS], 18 μm [TsOYE], 10 mm CaCl2, 8 mm 2‐methylcyclohexenone in a 200 mm TEOA buffer, pH 7.5.
We confirmed a broad applicability of the NADH‐free, xanthene‐sensitized photobiocatalytic platform by extending it to YqjM (Table 2). The catalytic reaction by YqjM showed a product yield of 84 % with a TTN of 282, which is comparable to the result with TsOYE (yield 76 %, TTN 254). The catalytic performances of both enzymes were also dependent on the concentration of RB. Furthermore, we performed RB‐sensitized photobiocatalytic reduction of α,β‐unsaturated aldehyde (cinnamaldehyde) by TsOYE and YqjM. Consensus to the reaction with 2‐methylcyclohexenone, both enzymes showed apparent activity towards the reduction of the unsaturated aldehyde with a product yield around 30 %. The lower catalytic activity compared to 2‐methylcyclohexenone is attributed to the inhibitory effect of the product on OYE and poor solubility of the substrate in the aqueous medium,17 which should be improved by applying a biphasic reaction system in future studies. Overall, the results show that the xanthene‐sensitized photoactivation can be applied to broader types of OYEs for different substrates.
Table 2.
Photoenzymatic reduction of 2‐methylcyclohexenone and cinnamaldehyde by different OYEs (TsOYE and YqjM).
| Substrate | OYE | [RB] [μm] | TTNOYE [a] | Yield[a] [%] | ee [a] |
|---|---|---|---|---|---|

|
TsOYE | 80 | 169 | 51 | >99 (R) |
| TsOYE | 160 | 254 | 76 | >99 (R) | |
| YqjM | 80 | 149 | 44 | >99 (R) | |
| YqjM | 160 | 282 | 84 | >99 (R) | |

|
TsOYE | 80 | 120 | 36 | – |
| TsOYE | 160 | 115 | 34 | – | |
| YqjM | 80 | 77 | 23 | – | |
| YqjM | 160 | 106 | 32 | – |
[a] Determined by GC analysis after 180 min reaction. Reaction conditions: 24 μm [OYE], 10 mm CaCl2, 8 mm substrate in a 200 mm TEOA buffer pH 7.5.
Photosensitized activation of redox enzymes is an attractive approach to promote electron transfer by utilizing clean light energy towards realizing green and sustainable chemistry.7, 24 Herein, we have demonstrated a mediator‐free, visible‐light driven activation of OYEs through direct transfer of photoinduced electrons from molecular dyes to the prosthetic flavin moiety. Previous studies for light‐driven activation of OYEs often involved Ru‐based organometallic photosensitizers or CdSe quantum dots that necessitate a redox mediator, such as toxic methyl viologen, to shuttle the electrons between photosensitizer and the enzyme.17, 25 Overcoming this limitation, we established direct photoinduced electron transfer from RB and its derivatives to OYEs by eliminating the necessity for the supply of expensive nicotinamide cofactors and its regeneration, as well as harmful electron mediators. Our spectroscopic assays revealed a spontaneous association between TsOYE and RB and protein film voltammetric analysis showed their co‐reducing behavior with strong photoelectrochemical responses. The RB‐sensitized photoenzymatic conversion of 2‐methylcyclohexenone resulted in a maximum 90 % yield of enantiopure (R)‐2‐methycyclohexanone (ee >99 %) with a TOF of 256 h−1. A decreasing order of photoenzymatic activity was observed with the increasing weight of substituted halogen atoms in xanthenes. A general applicability of the NADH‐free, RB‐sensitized photobiocatalytic system was demonstrated by using different OYE homologues (for example, TsOYE, YqjM) and substrates (for example, 2‐methylcyclohexenone, cinnamaldehyde). We anticipate our strategy would provide a simple and cost‐effective platform for developing scalable and sustainable biocatalytic processes utilizing light energy.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the National Research Foundation (NRF) via the Creative Research Initiative Center (Grant number: NRF‐2015 R1A3A2066191), Republic of Korea, and the Netherlands Organisation for Scientific Research by a VICI grant (Grant number: 724.014.003).
S. H. Lee, D. S. Choi, M. Pesic, Y. W. Lee, C. E. Paul, F. Hollmann, C. B. Park, Angew. Chem. Int. Ed. 2017, 56, 8681.
Contributor Information
Prof. Dr. Frank Hollmann, Email: f.hollmann@tudelft.nl
Prof. Dr. Chan Beum Park, Email: parkcb@kaist.ac.kr.
References
- 1.
- 1a. Aldridge S., Nat. Biotechnol. 2013, 31, 95–96; [DOI] [PubMed] [Google Scholar]
- 1b. Hollmann F., Arends I. W. C. E., Holtmann D., Green Chem. 2011, 13, 2285–2314; [Google Scholar]
- 1c. Nestl B. M., Hammer S. C., Nebel B. A., Hauer B., Angew. Chem. Int. Ed. 2014, 53, 3070–3095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3132–3158. [Google Scholar]
- 2.
- 2a. Toogood H. S., Gardiner J. M., Scrutton N. S., ChemCatChem 2010, 2, 892–914; [Google Scholar]
- 2b. Toogood H. S., Scrutton N. S., Curr. Opin. Chem. Biol. 2014, 19, 107–115; [DOI] [PubMed] [Google Scholar]
- 2c. Kitzing K., Fitzpatrick T. B., Wilken C., Sawa J., Bourenkov G. P., Macheroux P., Clausen T., J. Biol. Chem. 2005, 280, 27904–27913. [DOI] [PubMed] [Google Scholar]
- 3. Lonsdale R., Reetz M. T., J. Am. Chem. Soc. 2015, 137, 14733–14742. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Hall M., Stueckler C., Ehammer H., Pointner E., Oberdorfer G., Gruber K., Hauer B., Stuermer R., Kroutil W., Macheroux P., Faber K., Adv. Synth. Catal. 2008, 350, 411–418; [Google Scholar]
- 4b. Köninger K., Gómez Baraibar Á., Mügge C., Paul C. E., Hollmann F., Nowaczyk M. M., Kourist R., Angew. Chem. Int. Ed. 2016, 55, 5582–5585; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5672–5675; [Google Scholar]
- 4c. Hall M., Stueckler C., Kroutil W., Macheroux P., Faber K., Angew. Chem. Int. Ed. 2007, 46, 3934–3937; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 4008–4011. [Google Scholar]
- 5. Okamoto Y., Köhler V., Paul C. E., Hollmann F., Ward T. R., ACS Catal. 2016, 6, 3553–3557. [Google Scholar]
- 6. Kohlmann C., Märkle W., Lütz S., J. Mol. Catal. B 2008, 51, 57–72. [Google Scholar]
- 7.
- 7a. Lee S. H., Kim J. H., Park C. B., Chem. Eur. J. 2013, 19, 4392–4406; [DOI] [PubMed] [Google Scholar]
- 7b. Kim J. H., Nam D. H., Park C. B., Curr. Opin. Biotechnol. 2014, 28, 1–9. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Lazarides T., McCormick T., Du P., Luo G., Lindley B., Eisenberg R., J. Am. Chem. Soc. 2009, 131, 9192–9194; [DOI] [PubMed] [Google Scholar]
- 8b. Lee S. H., Nam D. H., Park C. B., Adv. Synth. Catal. 2009, 351, 2589–2594; [Google Scholar]
- 8c. Lee S. H., Nam D. H., Kim J. H., Baeg J.-O., Park C. B., ChemBioChem 2009, 10, 1621–1624. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Park J. H., Lee S. H., Cha G. S., Choi D. S., Nam D. H., Lee J. H., Lee J. K., Yun C. H., Jeong K. J., Park C. B., Angew. Chem. Int. Ed. 2015, 54, 969–973; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 983–987; [Google Scholar]
- 9b. Sakai T., Mersch D., Reisner E., Angew. Chem. Int. Ed. 2013, 52, 12313–12316; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12539–12542; [Google Scholar]
- 9c. Lee J. H., Nam D. H., Lee S. H., Park J. H., Park S. J., Lee S. H., Park C. B., Jeong K. J., J. Ind. Eng. Chem. 2016, 33, 28–32. [Google Scholar]
- 10.
- 10a. Opperman D. J., Piater L. A., van Heerden E., J. Bacteriol. 2008, 190, 3076–3082; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Opperman D. J., Sewell B. T., Litthauer D., Isupov M. N., Littlechild J. A., van Heerden E., Biochem. Biophys. Res. Commun. 2010, 393, 426–431. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Stewart R. C., Massey V., J. Biol. Chem. 1985, 260, 13639–13647; [PubMed] [Google Scholar]
- 11b. Crozier-Reabe K., Moran G., Int. J. Mol. Sci. 2012, 13, 15601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Kohli R. M., Massey V., J. Biol. Chem. 1998, 273, 32763–32770; [DOI] [PubMed] [Google Scholar]
- 12b. Hopkins N., Stanley R. J., Biochemistry 2003, 42, 991–999. [DOI] [PubMed] [Google Scholar]
- 13. Classen T., Korpak M., Schölzel M., Pietruszka J., ACS Catal. 2014, 4, 1321–1331. [Google Scholar]
- 14. Waheed A., Rao K. S., Gupta P., Anal. Biochem. 2000, 287, 73–79. [DOI] [PubMed] [Google Scholar]
- 15. Udit A. K., Hagen K. D., Goldman P. J., Star A., Gillan J. M., Gray H. B., Hill M. G., J. Am. Chem. Soc. 2006, 128, 10320–10325. [DOI] [PubMed] [Google Scholar]
- 16. Fleming B. D., Tian Y., Bell S. G., Wong L. L., Urlacher V., Hill H. A. O., FEBS J. 2003, 270, 4082–4088. [DOI] [PubMed] [Google Scholar]
- 17. Peers M. K., Toogood H. S., Heyes D. J., Mansell D., Coe B. J., Scrutton N. S., Catal. Sci. Technol. 2016, 6, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sayyed S. A., Beedri N. I., Kadam V. S., Pathan H. M., B. Mater. Sci. 2016, 39, 1381–1387. [Google Scholar]
- 19. Egbaria K., Friedman M., Pharm. Res. 1992, 9, 629–635. [DOI] [PubMed] [Google Scholar]
- 20. Xu J., Shanmugam S., Duong H. T., Boyer C., Polym. Chem. 2015, 6, 5615–5624. [Google Scholar]
- 21. Jhonsi M. A., Srinivasan V., Kathiravan A., Synth. Met. 2014, 196, 131–138. [Google Scholar]
- 22. Grau M. M., van der Toorn J. C., Otten L. G., Macheroux P., Taglieber A., Zilly F. E., Arends I. W., Hollmann F., Adv. Synth. Catal. 2009, 351, 3279–3286. [Google Scholar]
- 23. Turbay M. B. E., Rey V., Argañaraz N. M., Vieyra F. E. M., Aspée A., Lissi E. A., Borsarelli C. D., J. Photochem. Photobiol. B 2014, 141, 275–282. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Bachmeier A., Armstrong F., Curr. Opin. Chem. Biol. 2015, 25, 141–151; [DOI] [PubMed] [Google Scholar]
- 24b. Maciá‐Agulló J. A., Corma A., Garcia H., Chem. Eur. J. 2015, 21, 10940–10959. [DOI] [PubMed] [Google Scholar]
- 25. Burai T. N., Panay A. J., Zhu H., Lian T., Lutz S., ACS Catal. 2012, 2, 667–670. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary