

Abstract
Biomimetic, strain‐stiffening materials are reported, made through self‐assembly and covalent fixation of small building blocks to form fibrous hydrogels that are able to stiffen by an order of magnitude in response to applied stress. The gels consist of semi‐flexible rodlike micelles of bisurea bolaamphiphiles with oligo(ethylene oxide) (EO) outer blocks and a polydiacetylene (PDA) backbone. The micelles are fibers, composed of 9–10 ribbons. A gelation method based on Cu‐catalyzed azide–alkyne cycloaddition (CuAAC), was developed and shown to lead to strain‐stiffening hydrogels with unusual, yet universal, linear and nonlinear stress–strain response. Upon gelation, the X‐ray scattering profile is unchanged, suggesting that crosslinks are formed at random positions along the fiber contour without fiber bundling. The work expands current knowledge about the design principles and chemistries needed to achieve fully synthetic, biomimetic soft matter with on‐demand, targeted mechanical properties.
Keywords: bisurea, covalent fixation, polydiacetylene, self-assembly, strain-stiffening
Many natural soft tissues respond to small strains with a large change in mechanical properties. A particularly advantageous response of natural materials is to stiffen when exposed to small strains. This behavior can counteract large deformations, which otherwise might compromise their integrity. Such complex, non‐linear mechanical behavior is shared by a number of proteins arranged into network architectures, including actin,1, 2 collagen,3, 4 fibrin,5, 6 and all types of intermediate filaments.7 However, this type of adaptivity is unmatched in the vast majority of synthetic materials, including many artificial extracellular matrices.
Considerable effort has gone into the creation of synthetic materials that are mechanically indistinguishable from natural systems, for potential application in tissue engineering or regenerative medicine. In this context, a variety of theoretical models has been proposed to establish the fundamental design principles of synthetic biogels.8, 9, 10 Early theoretical work8 suggests that strain‐stiffening is inherent to any connected mesh of semi‐flexible filaments. In later work,10 it was shown that even for stiff polymers, stiffening is universally expected for generic, geometric reasons. For all its ubiquity in natural materials, the general absence of strong strain‐stiffening in synthetic gels is perhaps all the more remarkable. The reason for this, largely, has been the difficulty to obtain semi‐flexible or even stiff polymers (synthetic polymers are generally very flexible) with strong crosslinks or long‐lived entanglements which can, moreover, remain intact at sufficiently large stresses to permit the polymers to enter their nonlinear extensional regimes.
Recent work of Kouwer et al.11 on entangled networks of bundled polyisocyanopeptides (PICs), presented the first synthetic system mimicking the strain‐stiffening mechanisms of biopolymer networks and showed considerable potential to further exploit the high degree of control over design parameters in synthetic molecules. In the current work, we introduce a novel approach, complementary to the PIC strategy, to create strain stiffening gels. In our system, actual crosslinking is incorporated rather than relying on physical entanglements for connectivity. This provides with control over the mesh size, one of the key structural parameters of network gels and crucially implicated in their mechanical response. We use a combination of self‐assembly, covalent fixation and click chemistry to develop a novel class of materials that responds to very low stresses. The strategy towards these gels is shown in Scheme 1 a and entails the use of a set of three oligo(EO)‐grafted bisurea bolaamphiphiles (DA, DA‐Ac and DA‐N3) containing a topochemically polymerizable diacetylene unit in between ureas.12 In water, these molecules aggregate into long, semi‐flexible rodlike micelles through urea hydrogen‐bonding and hydrophobic interactions. In previous work we demonstrated that when the fibers are covalently fixated by topochemical polymerization of the diacetylene groups to gain mechanical strength, no gelation takes place.12
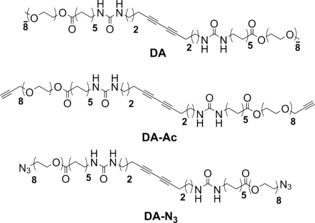
Scheme 1.
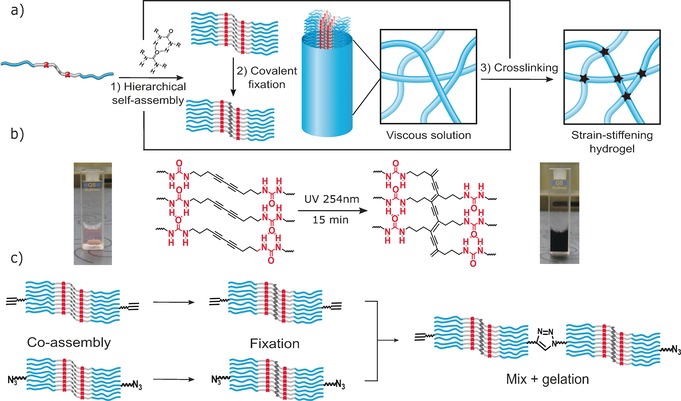
a) Hierarchical self‐assembly through intermolecular H‐bonding and hydrophobic interactions of diacetylene bisurea bolaamphiphiles followed by topochemical polymerization of the stacked diacetylenes. Aggregation of 9–10 ribbons into semi‐flexible fibres followed by covalent crosslinking into strain‐stiffening networks. b) Photo‐polymerization of the assembled diacetylene groups (i.e. covalent fixation) and the resulting PDAs. c) Gel preparation method involving separate co‐assembly and covalent fixation of DA‐Ac and DA‐N3 analogues with DA followed by mixing and gelation through CuAAC cycloaddtion reaction.
In this work, gels are obtained by a Cu‐catalyzed click reaction of azide and acetylene groups that crosslinks fibers without affecting their size (Scheme 1). The solubility of DA bisurea bolaamphiphiles in water combined with precise control over the crosslink density allows us to finely tune the network stiffness to access a broad range of storage modulus G′ values.
Polydiacetylene PDA was selected due to its previously reported high DP and its tendency to form long semi‐flexible rods in water both before and after topochemical polymerization.12 In order to determine the average values of the contour length L c and the persistence length l p in their native environment, a series of cryo‐TEM images of PDA fibers (Figure 1 and Figure S5 of the Supporting Information) were subjected to statistical analysis. From an image with 84 rods, L c and l p were determined and compared to those of neurofilaments (NFs) (Table 1). We found that the persistence length of PDA fibers is on the order of 280 nm, comparable to typical NFs, although NFs vary in length across several microns, while the PDA fibers have an average length of 0.16 μm.
Figure 1.
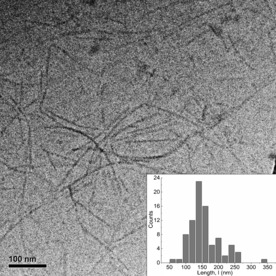
Cryo‐TEM image of PDA in aqueous solution (1 mm). Inset: Histogram of contour length distribution calculated for a sample of N=84 fibers from a set of cryo‐TEM images.
Table 1.
Comparison of PDA gels with neurofilaments.
| Characteristic network parameter | DA | NFs7, 13, 14 |
|---|---|---|
| Crosslink distance | 50–80[a] | 300 |
| Contour length, L c [nm] | 157[b] | 2000–10 000 |
| Persistence length, l p [nm] | 280[b] | 200 |
| Concentration [mg mL−1] | 10–30 | 0.2–5 |
| G 0 [Pa] | 4–4000 | 0.5–30 |
| Critical stress (σ c) [Pa] | 1–86 | 0.1–4 |
| High stress regime | K′∝σ 1 | K′∝σ 1 |
| Fibre diameter [nm] | 6.6[b] | 10 |
[a] Determined from modeling detailed in the Supporting Information. [b] Calculated from cryo‐TEM graphical analysis.
The structure of DA and PDA rods was further investigated with small‐angle X‐ray scattering (SAXS). The scattering profiles of both objects displayed in Figure 2 a overlap over the whole q‐range indicating no structural changes upon topochemical polymerization. Furthermore, in the low q‐regime the slope is close to unity, indicative of high‐aspect‐ratio scatterers with lengths beyond the resolution of the experiment (given by Q min −1≈30 nm). These results are consistent with those derived from cryo‐TEM,12 showing rodlike micelles whose morphology is retained when undergoing photochemical polymerization of the assembled diacetylene groups. By fitting the scattering data (Figure S8), a fiber radius of 3.1 nm and a cross‐sectional mass per unit length (M L) of 4.70×1020 g nm−1 was calculated, indicating that each fiber is composed of 9–10 ribbons, on average. Similar aggregation numbers have been reported for other bolaamphiphile wormlike micelles and have been ascribed to the hydrophobic segregation of the micellar cores.15, 16 As previously reported, rodlike micelles of bisurea bolaamphiphiles do not form elastic gels on their own.17 In order to induce gelation by covalent crosslinking, DA‐Ac and DA‐N3, (analogues of DA with matching hydrophobic domains and octaethylene glycol segments functionalized with acetylene and azide end‐groups) were incorporated into the rods. These groups undergo a highly efficient cycloaddition reaction at low substrate concentrations using Cu catalysts with accelerating ligands.18, 19 In a protocol that was used to suppress reactions within fibers (Scheme 1 c), DA‐N3 and DA‐Ac (5 mol %) were separately blended with DA in chloroform. After allowing the solvent to evaporate overnight, DA/DA‐N3 and DA/DA‐Ac mixtures were dissolved in water and exposed to UV‐light (254 nm)12 with rapid evolution of a dark purple color (Scheme 1 b) due to extensive π‐electron delocalization along the PDA backbone. After covalent fixation, the solutions were mixed ensuring equimolar azide/acetylene ratios while preventing monomer exchange between micelles. Crosslinking of the rods was performed using a slightly modified standard protocol (see the Supporting Information),18 and after 2–3 h resulted in the formation of elastic gels able to support their own weight upon tilting the vials (Figure 3 a). Gel formation was further monitored with oscillatory rheology in which storage (G′) and loss (G′′) moduli were followed over time (Figure 3 b), requiring as much as 19 h before G′ and G′′ reached a plateau. To test the assumption that reduction of CuII to CuI is essential for the gelation process, we performed a crosslinking experiment of an aqueous solution of 2 wt % PDA co‐assembled with DA‐N3/DA‐Ac (5 mol %) in the absence of Na‐ascorbate. Figure S10 shows that without generation of CuI ions no measurable changes in moduli are observed.
Figure 2.
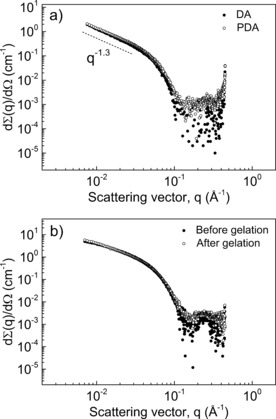
a) Small‐angle X‐ray scattering profiles of DA aqueous solutions (1 mg mL−1) before and after photo‐polymerization of the diacetylene unit. b) PDA (2.5 wt %) containing DA‐Ac and DA‐N3 (5 mol %) recorded before (solution) and 24 h after addition of Na‐ascorbate (gel).
Figure 3.
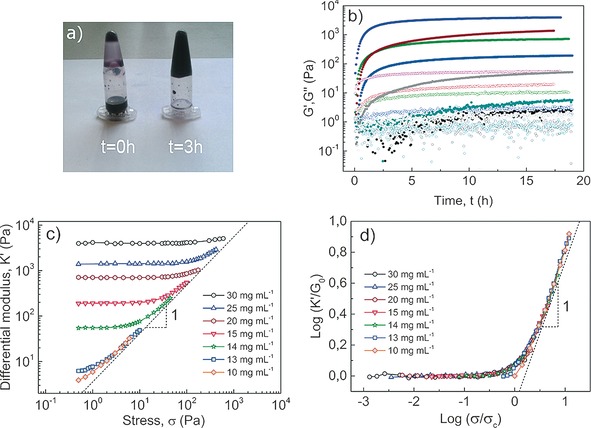
a) Aqueous PDA mixtures before and 3 h after addition of catalyst illustrate gelation. b) Time course of moduli during Cu‐catalyzed crosslinking of DA mixtures containing 5 mol % DA‐N3 and Da‐Ac. G′ (solid dots) and G′′ (open dots) as a function of reaction time at constant strain amplitude (0.1 %) and angular frequency (6.28 rad s−1) for different concentrations (in mg mL−1) of 30 (violet), 25 (wine), 20 (green), 15 (blue), 14 (gray), 13 (cyan) and 10 (black). c) Differential modulus against stress of PDA gels. d) Scaling of K′ with G 0 and σ with σ c with collapse into a master curve showing K′∝σ 1 dependence at high σ and low c.
To learn more about the mechanical properties of the so‐formed gels, a well‐stablished pre‐stress protocol was applied,20 which records the differential modulus K′ (defined as the derivative of stress σ with respect to shear strain γ, K′=dσ/dγ) as a function of the applied stress. The typical elastic response observed in crosslinked biopolymer networks, as well as synthetic PIC gels, features two distinct regimes: A linear, low stress regime where K′ is defined by the plateau storage modulus (K′=G′ 0) and a stress‐stiffening regime at higher stresses where the magnitude of K′ increases as a power law K′∝σ m. The power m is known as the stiffening index and is a direct measure of the degree of mechanical responsiveness of the material. In addition, we define σ c to be the critical stress at which the modulus begins to depart from its linear behavior. For biopolymer networks, σ c typically lies between 0.5 Pa≤σ c≤10 Pa.
In order to probe the mechanical properties of the PDA hydrogels, a concentration series of PDA was prepared while keeping the relative content of functionalized (DA‐N3 and DA‐Ac) to unfunctionalized DA constant (i.e. 5 mol %). Figure 3 c shows a strong concentration dependence of the linear response G 0. The value of K′ in the low‐stress regime ranges over nearly 3 decades. Figure 3 c shows, moreover, a universal high‐stress asymptote characterized by a stiffening exponent m=1. Similar indices have been reported by Rammensee et al.13 for in vitro NFs and, most notably, for reconstituted collagen type I networks which are known to exhibit highly unusual nonlinear mechanics.21 In the PIC gels, m=1 is typically seen close to the critical stress; interestingly the PDA gels seem not to cross over to the characteristic m=3/2 associated with the terminal scaling of semi‐flexible networks. We speculate that the origin of the absence (or strongly delayed onset) of this regime is the relatively high density of the networks; previous work on aggrecan gels,22 suggests that at increased concentrations m may be decreased to values as low as 0.6. In biological m=1 networks, the stress response is argued to be dominated by the release of mesoscopic bending of the fibrils in contrast to F‐Actin or intermediate filaments where the upper limit of m=3/2, associated to the stretching out of thermal undulations at short length scales, is typically accessible at high stress.2, 7 The PDA networks all have m=1, but the extent of this responsive regime is far larger at lower concentrations, where the largest range of stiffening is observed. If, as we hypothesize, this stiffening originates from the exhaustion of very soft bending modes (which may involve clusters of fibers, rather than single fibers) we can understand that increasing the concentration strongly increases the modulus, but at the same time the increased confinement suppresses those soft modes and the stiffening range decreases.
Figures 3 c and d summarize the findings: PDA networks exhibit a universal m=1 stiffening at large strains. Upon rescaling the stresses and moduli, a universal stiffening curve is obtained highlighting the common origins of linear and nonlinear elasticity in these materials. In the supplemental information, we present further analysis of the scaling behavior of the critical stress and the modulus.
SAXS scattering profiles of PDA were measured before and after gelation. To this end, 2.5 wt % aqueous solutions of PDA containing 5 mol % DA‐N3 and DA‐Ac were recorded before, and 24 h after, the addition of Na‐ascorbate (Figure 2 b). Gelation through bundle formation is a common feature of PIC gels11, 23, 24 as well as certain biopolymers, such as fibrin.6 In contrast, PDA gels follow a rather different gelation mechanism: there, the formation of covalent links occurs at random positions along the fiber contour (Scheme 1 a). In that case, there is no net increase of the bundle diameter as inferred from SAXS data that show similar forward scattering intensities while completely retaining the shape of the scattering profiles as the system undergoes gelation.
In summary, we present a novel approach towards biomimetic strain‐stiffening gels. Implementing requisite features of natural materials—semi‐flexible polymers and strong links—the bisurea bolaamphihiles covalently fixated into rodlike micelles with a conjugated PDA backbone represent the first synthetic system that just like natural protein materials relies on self‐assembly for the crucial step of fiber formation. The self‐assembled rodlike micelles exist in water as bundled structures containing multiple ribbons, which we argue is essential to endow fibers with the necessary high persistence lengths. In future work, control over bundling may be leveraged to gain control over the persistence length. The supramolecular nature of the rods, moreover, allows for the controlled incorporation of a variety of functional analogues which can be subsequently immobilized via covalent fixation. Here, this advantage is exploited to suppress the formation of inactive loops along the same rod. The hydrogelation process, as inferred from SAXS analysis, occurs without increase in fiber dimensions. In other words, the structure of the constituent rods remains intact as the system undergoes gelation, with superior control over the network architecture.
The ability to control both the stiffness and the degree of connectivity between networks of semi‐flexible rods breaks ground for truly biomimetic materials; indeed we show that in the first incarnation presented here the network shares crucial dimensional and elastic metrics with gels composed of neurofilament fibers. However, where nature is limited by properties of the fibrillar proteins at its disposal, the current synthetic route in principle allows for continuous variation of properties, opening up a class of transnatural, bio‐inspired materials that not only mimic, but conceivably outperform biological matter.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. K. Pieterse (ICMS Animation Studio) for help with the graphics and Dr. A. W. Bosman of SupraPolix for support and fruitful discussions. M.F.‐C.R. is financially supported by the Marie Curie FP7 SASSYPOL ITN programme (No. 607602). This work was supported by the Dutch Ministry of Education, Culture, and Science (Gravity program 024.001.035).
M. Fernandez-Castano Romera, R. P. M. Lafleur, C. Guibert, I. K. Voets, C. Storm, R. P. Sijbesma, Angew. Chem. Int. Ed. 2017, 56, 8771.
References
- 1. Gardel M. L., Shin J. H., MacKintosh F. C., Mahadevan L., Matsudaira P. A., Weitz D. A., Phys. Rev. Lett. 2004, 93, 188102. [DOI] [PubMed] [Google Scholar]
- 2. Gardel M. L., Shin J. H., MacKintosh F. C., Mahadevan L., Matsudaira P., Weitz D. A., Science 2004, 304, 1301–1305. [DOI] [PubMed] [Google Scholar]
- 3. Kurniawan N. A., Wong L. H., Rajagopalan R., Biomacromolecules 2012, 13, 691–698. [DOI] [PubMed] [Google Scholar]
- 4. Motte S., Kaufman L. J., Biopolymers 2013, 99, 35–46. [DOI] [PubMed] [Google Scholar]
- 5. Janmey P. A., Winer J. P., Weisel J. W., J. R. Soc. Interface 2009, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Piechocka I. K., Jansen K. A., Broedersz C. P., Kurniawan N. A., MacKintosh F. C., Koenderink G. H., Soft Matter 2016, 12, 2145–2156. [DOI] [PubMed] [Google Scholar]
- 7. Lin Y.-C., Yao N. Y., Broedersz C. P., Herrmann H., MacKintosh F. C., Weitz D. A., Phys. Rev. Lett. 2010, 104, 058101. [DOI] [PubMed] [Google Scholar]
- 8. Storm C., Pastore J. J., MacKintosh F. C., Lubensky T. C., Janmey P. A., Nature 2005, 435, 191–194. [DOI] [PubMed] [Google Scholar]
- 9. Broedersz C. P., Storm C., MacKintosh F. C., Phys. Rev. Lett. 2008, 101, 118103. [DOI] [PubMed] [Google Scholar]
- 10. Onck P. R., Koeman T., van Dillen T., van der Giessen E., Phys. Rev. Lett. 2005, 95, 178102. [DOI] [PubMed] [Google Scholar]
- 11. Kouwer P. H. J., Koepf M., Le Sage V. A. A., Jaspers M., van Buul A. M., Eksteen-Akeroyd Z. H., Woltinge T., Schwartz E., Kitto H. J., Hoogenboom R., et al., Nature 2013, 493, 651–655. [DOI] [PubMed] [Google Scholar]
- 12. Pal A., Voudouris P., Koenigs M. M. E., Besenius P., Wyss H. M., Degirmenci V., Sijbesma R. P., Soft Matter 2014, 10, 952–956. [DOI] [PubMed] [Google Scholar]
- 13. Rammensee S., Janmey P. A., Bausch A. R., Eur. Biophys. J. 2007, 36, 661–668. [DOI] [PubMed] [Google Scholar]
- 14. Dogic Z., Zhang J., Lau A. W. C., Aranda-Espinoza H., Dalhaimer P., Discher D. E., Janmey P. A., Kamien R. D., Lubensky T. C., Yodh A. G., Phys. Rev. Lett. 2004, 92, 125503. [DOI] [PubMed] [Google Scholar]
- 15. Obert E., Bellot M., Bouteiller L., Andrioletti F., Lehen-Ferrenbach C., Boué F., J. Am. Chem. Soc. 2007, 129, 15601–15605. [DOI] [PubMed] [Google Scholar]
- 16. Saez Talens V., Englebienne P., Trinh T. T., Noteborn W. E. M., Voets I. K., Kieltyka R. E., Angew. Chem. Int. Ed. 2015, 54, 10502–10506; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10648–10652. [Google Scholar]
- 17. Koenigs M. M. E., Pal A., Mortazavi H., Pawar G. M., Storm C., Sijbesma R. P., Macromolecules 2014, 47, 2712–2717. [Google Scholar]
- 18. Hong V., Presolski S. I., Ma C., Finn M. G., Angew. Chem. Int. Ed. 2009, 48, 9879–9883; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 10063–10067. [Google Scholar]
- 19. Presolski S. I., Hong V., Cho S.-H., Finn M. G., J. Am. Chem. Soc. 2010, 132, 14570–14576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Broedersz C. P., Kasza K. E., Jawerth L. M., Münster S., Weitz D. A., MacKintosh F. C., Soft Matter 2010, 6, 4120–4127. [Google Scholar]
- 21. Licup A. J., Münster S., Sharma A., Sheinman M., Jawerth L. M., Fabry B., Weitz D. A., MacKintosh F. C., Proc. Natl. Acad. Sci. USA 2015, 112, 9573–9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meechai N., Jamieson A. M., Blackwell J., Carrino D. A., Bansal R., J. Rheol. 2002, 46, 685–707. [Google Scholar]
- 23. Jaspers M., Pape A. C. H., Voets I. K., Rowan A. E., Portale G., Kouwer P. H. J., Biomacromolecules 2016, 17, 2642–2649. [DOI] [PubMed] [Google Scholar]
- 24. Jaspers M., Dennison M., Mabesoone M. F. J., MacKintosh F. C., Rowan A. E., Kouwer P. H. J., Nat. Commun. 2014, 5, 5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary