

Abstract

Coupling reactions between benzylamines and boronic esters have been investigated. ortho-Lithiated benzylamines react with boronic esters and a N-activator to afford ortho-substituted benzylic boronic esters with formal 1,1′-benzylidene insertion into the C–B bond. The reaction occurs by a SN2′ elimination and 1,2-metalate rearrangement of the N-activated boronate complex to afford a dearomatized intermediate, which undergoes a Lewis-acid catalyzed 1,3-borotropic shift to afford the boronic ester products in high yield and with excellent enantiospecificity. The use of enantioenriched α-substituted benzylamines gave the corresponding secondary boronic esters with high ee.
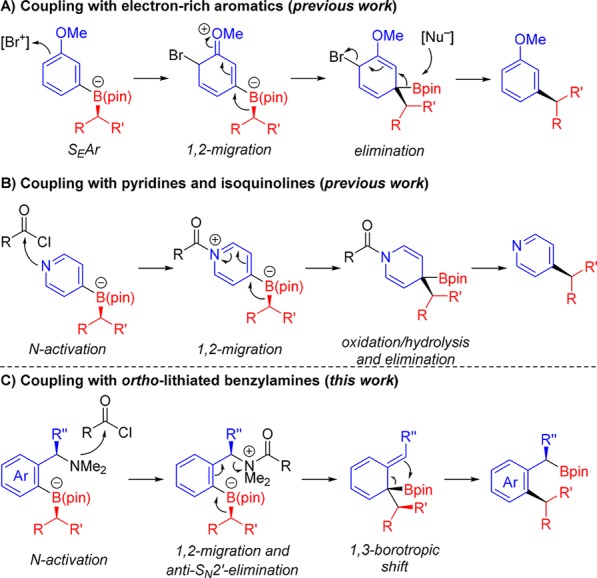
The enantiospecific coupling of secondary and tertiary boronic esters to aromatics through transition-metal catalyzed processes is challenging.1 Although some progress has been made enabling certain secondary boronic esters to be employed, the reactions are not generally applicable.2,3 We recently reported an alternative transition metal-free and stereospecific sp2–sp3 coupling of chiral secondary and tertiary boronic esters with electron-rich aryl lithium reagents that showed considerable scope (Scheme 1a).4 In related work, we5 (and Ready6) showed that pyridines could also be coupled with complete stereospecificity to a similar range of boronic esters (Scheme 1b). These processes involve four basic steps: (i) boronate formation, (ii) activation, (iii) 1,2-migration, and (iv) elimination/rearomatization. We reasoned that this methodology would be significantly enhanced if the boron moiety could be retained in the product because of its broad versatility.7 Inspired by the coupling reactions with pyridines, we considered a related transformation with ortho-lithiated benzylamines (Scheme 1c).8 We considered that N-activation of the corresponding boronate complex would trigger the 1,2-migration/anti-SN2′ elimination of the carbamate to give a dearomatized intermediate, which should undergo a suprafacial 1,3-borotropic shift.9 This would lead to the desired aromatic product in which the boron moiety is retained. Although the 1,3-borotropic shift of allylic boronic acid pinacol esters is exceedingly slow,10 we reasoned that the driving force of aromatization would facilitate this rearrangement. In this communication, we describe the realization of this transformation to give a variety of ortho-substituted benzylic boronic esters in high yield and with excellent enantiospecificity.
Scheme 1. Enantiospecific sp2–sp3 Couplings of Boronic Esters with Electron-Rich Aromatic (A) or Pyridines (B) and Proposed Work.
We began our study by reacting (2-((dimethylamino)methyl)phenyl)lithium (Li−1a) with CyBpin (2a) to give the corresponding boronate complex and screened a broad range of N-activators (see SI for details). Among the electrophiles tested, ClCO2CMe2CCl3 (Me2Troc–Cl) was highly selective giving the dearomatized intermediate over borinic ester side products, rapidly, even at low temperature. However, despite having the driving force of rearomatization, the subsequent 1,3-borotropic shift was very slow even at elevated temperature, and considerable protodeboronation occurred. DFT calculations showed that rearomatization lowered the barrier for the 1,3-borotropic shift from 37 to 25 kcal/mol (see SI for details). We therefore investigated the use of Lewis acids to promote the 1,3-borotropic shift by coordinating to oxygen, hence reducing the pO–pB π-donation.11 The dramatic effect π-donation of the ligand to boron has on the rate of the 1,3-borotropic shift is shown by the fact that allyl boranes12 undergo 1,3-borotropic shifts much faster than pinacol boronic esters (−78 °C vs >120 °C),10 whereas diamino boranes13 require very high temperatures (>200 °C).12 Unfortunately, most of the Lewis acids tested led to decomposition of the dearomatized intermediate (see SI for details). Interestingly, the addition of 12-crown-4 prevented the 1,3-borotropic shift, indicating that the LiCl generated during the reaction was catalyzing the rearrangement. To increase its potency, we performed an in situ salt metathesis with NaBPh4 and a solvent exchange to noncoordinating chloroform to give “naked” LiBPh4,14 which then efficiently catalyzed the reaction affording the benzylic boronate product 3aa in 85% NMR and 67% isolated yield.
Following this protocol, a series of ortho-lithiated benzylamines Li–1 (generated either by Br/Li exchange or directed lithiation, see SI for details) was tested with CyBpin (2a) to assess the scope of the aromatic component that can be employed in this transformation (Scheme 2). A broad range of electron-rich (3ab–3af) and electron-poor (3ag–3ai) aromatics can be used in this coupling reaction, as well as heteroaromatic compounds exemplified by 3aj and 4ak. Finally, an allylic amine was also tested to afford allylic boronic ester 3al (l:b = 1:1.2). Products 3aa–3al were obtained in 52–92% NMR yield, with slightly lower isolated yields due to the moderate stability of primary benzylic boronic esters on silica gel. Furthermore, the reaction was readily scalable, and 3aa was obtained in 64% isolated yield on a 10 mmol scale employing a catalytic amount of NaBPh4 (10 mol %).
Scheme 2. Scope of Lithiated Benzylamines.

Reactions were carried out with 0.50 mmol of boronic ester, 1.05 equiv of Ar–Li, 1.10 equiv of Me2Troc–Cl and 1.00 equiv of NaBPh4. Yields recorded are those of isolated material (NMR yields in brackets).
1,3-Borotropic shift conducted at 65 °C.
Product isolated after oxidation with sodium perborate (see SI for details).
The scope of the boronic ester was also explored (Scheme 3) and included primary, secondary, aryl, and alkenyl boronic esters (3aa–3ga). Furthermore, the tertiary boronic ester AdBpin (2e) and even B2pin2 (2h) afforded the corresponding products in 64% and 44% isolated yield, respectively. Essentially perfect enantiospecificity was observed for enantio- and diastereo-enriched boronic esters (2i–2v) to afford the corresponding products 3ia–3va in high enantiomeric purity (er = 95:5 to 99:1, dr >20:1). Importantly, a broad range of functional groups were well tolerated, highlighting synthetic utility of this dearomatizing 1,2-metalate rearrangement/rearomatizing 1,3-borotropic shift process.
Scheme 3. Stereospecific Coupling of ortho-Lithiated N,N-Dialkyl Benzylamine with Boronic Esters.

Reactions were carried out with 0.50 mmol of boronic ester, 1.05 equiv of Ar–Li, 1.10 equiv of Me2Troc–Cl, and 1.00 equiv of NaBPh4. Yields recorded are those of isolated material; er determined by HPLC analysis.
Product isolated after oxidation (see SI for details).
1,3-Borotropic shift conducted at rt.
Boronate complex formed at −100 °C.
We also investigated the use of readily available enantioenriched α-substituted benzylamines (Scheme 4). Following the standard procedure, reaction of 2a with ortho-lithiated benzylamine Li–1m yielded the corresponding secondary boronic ester 3am with 94:6 er and formal retention of configuration, indicating a high preference for the anti-SN2′ elimination pathway. Similarly, the reaction with 2d afforded 3dm with 96:4 er, whereas primary boronic esters 2b and 2c gave slightly reduced enantiospecificity (89:11 and 91:9 er, respectively). Importantly, the combination of enantioenriched boronic esters with either enantiomer of the ortho-lithiated α-methylbenzylamine afforded the corresponding, diastereomeric products 3im–3tn with excellent diastereomeric ratios, indicating that the reaction does not suffer from significant matched/mismatched effects.
Scheme 4. ortho-Lithiated α-Methylbenzylamines.

Reactions were carried out with 0.50 mmol of boronic ester, 1.05 equiv of Ar–Li, 1.10 equiv of Me2Troc–Cl, and 1.00 equiv of NaBPh4. Yields recorded are those of isolated material; es determined by HPLC analysis; dr determined by 1H NMR analysis of purified product.
Enantiomeric (S)-(2-(1-(dimethylamino)ethyl)phenyl)lithium Li–1n was used instead.
To determine the origin of both the sense and the level of stereospecificity, we performed further experimental and computational investigation. For the transformation of secondary boronic ester 2i (95:5 er) with the R enantiomer of the ortho-lithiated benzyl amine Li–1m (99:1 er) to give boronic ester product 3im, NMR analysis of the dearomatized intermediate showed a dr of 92:8, suggesting that the acylation-triggered 1,2-metalate rearrangement/fragmentation proceeded with 94% stereospecificity (Scheme 5), similar to that of secondary boronic esters 2a and 2d. Upon 1,3-borotropic shift, which was initiated through salt metathesis/solvent exchange, the benzylic boronic ester product was isolated in similar levels of diastereoselectivity,15 pointing toward a highly stereospecific 1,3-borotropic shift, as expected.
Scheme 5. Mechanistic Analysis with Boronic Ester 2i.

DFT calculations (B3LYP/6-31G*) revealed that the less-than-perfect level of stereospecificity of the acylation-triggered 1,2-metalate rearrangement/fragmentation step was locked in at the acylation event (see SI for details). Specifically, both the syn and anti conformers of a zwitterionic N-acylated amino boronate of the type under investigation undergo C–N bond cleavage with a very low-barrier (<2 kcal/mol), significantly more facile than interconversion of the conformers through bond rotation (>17 kcal/mol; Figure 1). As expected, for all low-energy conformers, the benzylic hydrogen atom lies approximately in the plane of the aromatic ring pointing toward the boronate moiety. The resulting zwitterionic boronate carbenium species then undergoes a similarly facile 1,2-metalate rearrangement (∼1.4 kcal/mol). A low-energy transition state for a more concerted 1,2-metalate rearrangement/fragmentation for either the syn or the anti conformer could not be identified, presumably owing to steric hindrance. These results show that both syn and anti conformers of the putative intermediate cannot interconvert and lead to stereoisomeric products with equal facility. Therefore, the higher levels of enantiospecificity for the transformation of secondary boronic esters (>90%) relative to that of primary boronic esters (∼80%; see Scheme 4) has its origins in the former engendering a more selective N-acylation of the anti conformer of the amino boronate intermediate relative to the corresponding syn conformer. The surprisingly precarious origin of stereospecificity, as suggested by computation, was borne out experimentally where simply replacing the dimethyl amino group with a diethyl amino group for the transformation of CyBpin led to a switch in the sense of stereospecificity (94:6 versus 25:75 er; see SI for details).
Figure 1.

Reaction profile of the acylation-triggered 1,2-metalate rearrangement/fragmentation step.
However, not all of the boronic esters tested worked. Notable exceptions included benzylic and tertiary boronic esters (with the exception of AdBpin (2e, vide supra)). In both cases, the 1,2-metalate rearrangement occurred, but the dearomatized intermediate did not undergo the 1,3-borotropic shift. In the former case, the intermediate underwent a Cope rearrangement to yield arylboronic ester 3wa′ instead (Scheme 6a). In the latter case, a retro-ene reaction intervened affording the ortho-tolylboronic ester 3xa′ (Scheme 6b). This reaction does not occur for AdBpin (2ea, Scheme 3), as an anti-Bredt product would arise. Finally, dearomatized intermediates generated from para-lithiated benzylamines did not undergo a double 1,3-borotropic shift sequence (Scheme 6c). Instead, a nonstereoselective von Auwers-type reaction16 afforded arylboronic ester 3uo′ with formal inverse 1,4′-benzylidene insertion.
Scheme 6. Unsuccessful Substrates.

The synthetic utility of the dearomatized intermediate is not restricted to the 1,3-borotropic-shift process (Scheme 7). For example, allylboration of benzaldehyde or ethyl glyoxalate afforded the corresponding alcohols 5 and 6 with excellent diastereo- and enantiocontrol (dr > 20:1, er = 95:5 and 91:9, respectively).17 Similarly to benzylic boronic esters (Scheme 6a), the intermediate prepared from allylBpin underwent an enantiospecific Cope rearrangement to give 7 with 97:3 er. That the Cope rearrangement is significantly more facile than the 1,3 borotropic shift was confirmed through computation (see SI for details). Finally, treatment of the intermediate with TBAF trihydrate18 selectively yielded protodeboronation product 8, an example in which the benzylic amine acted as a traceless directing group.
Scheme 7. Synthetic Utility of Intermediate.

In conclusion, a new strategy for the stereospecific synthesis of ortho-substituted benzylic boronic esters has been developed. The method relies on a 1,2-metalate rearrangement/anti-SN2′ reaction followed by a suprafacial 1,3-borotropic shift giving rise to sp2–sp3 cross-coupled products in high enantiopurity in which the boronic ester moiety is retained for further transformations.
Acknowledgments
We thank the ERC (670668) and the EPSRC (EP/I038071/1) for financial support. S.A. thanks the Austrian Science Fund (FWF) for an Erwin Schrödinger fellowship (J3919-N28). R.B. thanks the Swiss National Science Foundation fellowship program (P2EZP2_165268).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05880.
Detailed experimental procedures and characterization of all products (PDF)
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- For recent review articles, see:; a Leonori D.; Aggarwal V. K. Angew. Chem., Int. Ed. 2015, 54, 1082. 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]; b Wang C.-Y.; Derosa J.; Biscoe M. R. Chem. Sci. 2015, 6, 5105. 10.1039/C5SC01710F. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cherney A. H.; Kadunce N. T.; Reisman S. E. Chem. Rev. 2015, 115, 9587. 10.1021/acs.chemrev.5b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhou S.-M.; Deng M.-Z.; Xia L.-J.; Tang M.-H. Angew. Chem., Int. Ed. 1998, 37, 2845. 10.1002/(SICI)1521-3773(19981102)37:20<2845::AID-ANIE2845>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; b Rubina M.; Rubin M.; Gevorgyan V. J. Am. Chem. Soc. 2003, 125, 7198. 10.1021/ja034210y. [DOI] [PubMed] [Google Scholar]; c Ohmura T.; Awano T.; Suginome M. J. Am. Chem. Soc. 2010, 132, 13191. 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]; d Awano T.; Ohmura T.; Suginome M. J. Am. Chem. Soc. 2011, 133, 20738. 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]; e Glasspoole B. W.; Oderinde M. S.; Moore B. D.; Antoft-Finch A.; Crudden C. M. Synthesis 2013, 45, 1759. 10.1055/s-0033-1338875. [DOI] [Google Scholar]; f Imao D.; Glasspoole B. W.; Laberge V. S.; Crudden C. M. J. Am. Chem. Soc. 2009, 131, 5024. 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]; g Matthew S. C.; Glasspoole B. W.; Eisenberger P.; Crudden C. M. J. Am. Chem. Soc. 2014, 136, 5828. 10.1021/ja412159g. [DOI] [PubMed] [Google Scholar]
- For related trifluoroborates, see:; a Fang G.-H.; Yan Z.-J.; Deng M.-Z. Org. Lett. 2004, 6, 357. 10.1021/ol036184e. [DOI] [PubMed] [Google Scholar]; b Sandrock D. L.; Jean-Gérard L.; Chen C.; Dreher S. D.; Molander G. A. J. Am. Chem. Soc. 2010, 132, 17108. 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee J. C. H.; McDonald R.; Hall D. G. Nat. Chem. 2011, 3, 894. 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]; d Molander G. A.; Wisniewski S. R. J. Am. Chem. Soc. 2012, 134, 16856. 10.1021/ja307861n. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Li L.; Zhao S.; Joshi-Pangu A.; Diane M.; Biscoe M. R. J. Am. Chem. Soc. 2014, 136, 14027. 10.1021/ja508815w. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lou Y.; Cao P.; Jia T.; Zhang Y.; Wang M.; Liao J. Angew. Chem., Int. Ed. 2015, 54, 12134. 10.1002/anie.201505926. [DOI] [PubMed] [Google Scholar]
- a Bonet A.; Odachowski M.; Leonori D.; Essafi S.; Aggarwal V. K. Nat. Chem. 2014, 6, 584. 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]; b Odachowski M.; Bonet A.; Essafi S.; Conti-Ramsden P.; Harvey J. N.; Leonori D.; Aggarwal V. K. J. Am. Chem. Soc. 2016, 138, 9521. 10.1021/jacs.6b03963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llaveria J.; Leonori D.; Aggarwal V. K. J. Am. Chem. Soc. 2015, 137, 10958. 10.1021/jacs.5b07842. [DOI] [PubMed] [Google Scholar]
- Panda S.; Coffin A.; Nguyen Q. N.; Tantillo D. J.; Ready J. M. Angew. Chem., Int. Ed. 2016, 55, 2205. 10.1002/anie.201510027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review, see:Sandford C.; Aggarwal V. K. Chem. Commun. 2017, 53, 5481. 10.1039/C7CC01254C. [DOI] [PubMed] [Google Scholar]
- For a related reaction of 1,2-migration using a π-allyl-Pd complex as the trigger, see:Panda S.; Ready J. M. J. Am. Chem. Soc. 2017, 139, 6038. 10.1021/jacs.7b01410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bubnov Y. N. Pure Appl. Chem. 1987, 59, 895. 10.1351/pac198759070895. [DOI] [Google Scholar]; b Bubnov Y. N.; Gurskii M. E.; Gridnev I. D.; Ignatenko A. V.; Ustynyuk Y. A.; Mstislavsky V. I. J. Organomet. Chem. 1992, 424, 127. 10.1016/0022-328X(92)83142-5. [DOI] [Google Scholar]
- Lombardo M.; Morganti S.; Tozzi M.; Trombini C. Eur. J. Org. Chem. 2002, 2002, 2823. 10.1002/1099-0690(200208)2002:16<2823::AID-EJOC2823>3.0.CO;2-J. [DOI] [Google Scholar]
- van der Mei F. W.; Miyamoto H.; Silverio D. L.; Hoveyda A. H. Angew. Chem., Int. Ed. 2016, 55, 4701. 10.1002/anie.201600546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer G. W.; Brown H. C. J. Organomet. Chem. 1977, 132, 9. [Google Scholar]
- Hancock K. G.; Kramer J. D. J. Organomet. Chem. 1974, 64, C29. 10.1016/S0022-328X(00)87905-X. [DOI] [Google Scholar]
- a Kitazawa Y.; Takita R.; Yoshida K.; Muranaka A.; Matsubara S.; Uchiyama M. J. Org. Chem. 2017, 82, 1931. 10.1021/acs.joc.6b02677. [DOI] [PubMed] [Google Scholar]; b Fujiki K.; Ikeda S.; Kobayashi H.; Mori A.; Nagira A.; Nie J.; Sonoda T.; Yagupolskii Y. Chem. Lett. 2000, 29, 62. 10.1246/cl.2000.62. [DOI] [Google Scholar]
- Because of 1H NMR signal overlap, determination of the dr of the crude product was complicated; therefore, the dr of isolated product after flash chromatography is reported.
- Dumeunier R.; Jaeckh S. Chimia 2014, 68, 522. 10.2533/chimia.2014.522. [DOI] [PubMed] [Google Scholar]
- Ramachandran P. V.; Gagare P. D.; Nicponski D. R.. Allylborons. In Comprehensive Organic Synthesis, 2nd ed.; Knochel P., Molander G. A., Eds.; Elsevier: Oxford, 2014; Vol. 2; p 1. [Google Scholar]
- Hesse M. J.; Butts C. P.; Willis C. L.; Aggarwal V. K. Angew. Chem., Int. Ed. 2012, 51, 12444. 10.1002/anie.201207312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.