

ABSTRACT
Porphyromonas gingivalis, a major etiologic agent of periodontitis, has been reported to induce the expansion of myeloid-derived suppressor cells (MDSC); however, little is known regarding the subpopulations of MDSC expanded by P. gingivalis infection. Flow cytometry was used to evaluate bone marrow and spleen cells from mice infected with P. gingivalis and controls for surface expression of CD11b, Ly6G, and Ly6C. To characterize the phenotype of MDSC subpopulations induced by infection, cells were sorted based on the differential expression of Ly6G and Ly6C. Moreover, since MDSC are suppressors of T cell immune activity, we determined the effect of the induced subpopulations of MDSC on the proliferative response of OVA-specific CD4+ T cells. Lastly, the plasticity of MDSC to differentiate into osteoclasts was assessed by staining for tartrate-resistant acid phosphatase activity. P. gingivalis infection induced the expansion of three subpopulations of MDSC (Ly6G++ Ly6C+, Ly6G+ Ly6C++, and Ly6G+ Ly6C+); however, only CD11b+ Ly6G+ Ly6C++-expressing cells exerted a significant suppressive effect on T cell proliferation. Inhibition of proliferative responses required T cell-MDSC contact and was mediated by inducible nitric oxide synthase and cationic amino acid transporter 2 via gamma interferon. Furthermore, only the CD11b+ Ly6G+ Ly6C++ subpopulation of MDSC induced by P. gingivalis infection was able to differentiate into osteoclasts. Thus, the inflammatory response induced by P. gingivalis infection promotes the expansion of immune-suppressive cells and consequently the development of regulatory inhibitors that curtail the host response. Moreover, monocytic MDSC have the plasticity to differentiate into OC, thus perhaps contributing to the OC pool in states of periodontal disease.
KEYWORDS: periodontitis, Porphyromonas gingivalis, MDSC
INTRODUCTION
Periodontitis is an infectious inflammatory process that affects the periodontium. The continuous inflammation characteristic of this disease underlies its chronic inflammatory nature, ultimately destroying the supportive tissues of teeth (1, 2). Current thought suggests that the development of periodontitis is associated with a shift from a balanced microbial community to an imbalanced one, with the ability to undermine the host response and allowing the persistence of pathogens in the inflammatory milieu (3, 4). Within the oral microflora, Porphyromonas gingivalis is considered a key pathogen able to exert an influence on the microbial environment and, consequently, in concert with other periodontal microorganisms, initiate and promote periodontitis (5, 6). P. gingivalis has a number of virulence factors by which it can escape or dampen host immunity, alter cytokine production, and affect the cell signaling mechanisms (7–10). Interestingly, studies in mice infected with P. gingivalis have shown a downregulation of more than 1,000 genes modulating CD4 and CD8 activation and function, suggesting the suppression of these cells (11). Thus, although P. gingivalis can induce an inflammatory response, the resulting inflammation likely contributes to the characteristic chronicity of infection. While most studies have focused on understanding interactions of immune cells and P. gingivalis in the periodontium, little information is available on the effect P. gingivalis exerts systemically on the host immune response. This is most relevant, as P. gingivalis is able to disseminate from local sites of infection to the circulation and to distal sites (12, 13). Furthermore, purified T and B cells from infected human periodontal tissues express mainly memory phenotype (14, 15), and antigen-specific T cells can migrate from the circulation to the periodontium (16). Thus, exposure and priming of T cells and other immune cells likely occur systemically in the blood and/or in secondary lymphoid organs. Moreover, there is significant epidemiological evidence of associations between this bacterium and systemic disorders, where P. gingivalis infection does not cause the pathological condition but aggravates the severity of systemic diseases (17–21).
Cytokines released systemically in states of immune stress induce the expansion of myeloid-derived suppressor cells (MDSC) generated from bone marrow (BM) hematopoietic precursors. Under healthy conditions, the majority of MDSC reside in the BM and differentiate into mature granulocytes, macrophages, or dendritic cells involved in regulating hyperactive or autoimmune responses, whereas a small proportion of MDSC are found in blood and spleen. In response to inflammation and infection, MDSC rapidly expand without differentiating into mature cells and enter lymphoid organs and peripheral tissues (22). Upon constant antigenic stimulation, as in chronic infections, expansion and accumulation of MDSC are significantly increased. This has been observed in parasitic (23), bacterial (24), and viral (25) infections. Systemic infection of mice with P. gingivalis using a chamber model of chronic periodontitis has also been shown to induce the expansion of MDSC (26). MDSC have a striking ability to suppress immune responses. They can suppress effector T cells directly by depriving them of essential nutrients or indirectly via the recruitment of T regulatory cells (22, 27, 28). MDSC can also modulate the response of other immune cells (29, 30). Thus, increasing evidence suggests that MDSC postpone host pathogen clearance and contribute to the critical balance between pathogen eradication and pathogenicity (23, 25). It is currently known that MDSC represent a heterogeneous population of cells, and that the diverse subpopulations have differential biological functions; however, characterization of these subpopulations is required to enable precise therapeutic targeting (22, 31). MDSC not only exert powerful immune regulation on immune cells but also differentiate into osteoclast (OC) progenitors (32, 33). This cell plasticity has biological significance; hence, MDSC may contribute not only to the immune inhibition observed in periodontal disease and with P. gingivalis infection but also to the increased number and activity of OC seen in chronic periodontitis (34).
In mice, MDSC are identified by the expression of the myeloid markers CD11b and Gr-1. Gr-1+ cells are granulocytic and monocytic cells characterized by the expression of CD11b+ Ly6G+ Ly6Clow or CD11b+ Ly6G− Ly6Chi phenotype, respectively (35). Interestingly, it has been reported that monocytic MDSC are the main type of cells involved in chronic infections (36). Here, we report the phenotype of MDSC subpopulations induced by P. gingivalis systemic infection, the ability of MDSC to suppress T cell-proliferative responses, the mechanisms mediating such an effect, and the plasticity of MDSC to differentiate into OC. Our findings reveal, for the first time, that systemic infection with P. gingivalis induces the expansion of three subpopulations of MDSC, as suggested by the differential expression of Ly6G and Ly6C. Of these, only one subpopulation was significantly suppressive of CD4+ T cell-proliferative responses and was able to differentiate into OC. Furthermore, nitric oxide synthase (NOS2; iNOS) and gamma interferon (IFN-γ), induced upon infection with P. gingivalis, are critical inflammatory factors conducive to the inhibition of T cell activity.
RESULTS
Infection with P. gingivalis induces the expansion of MDSC.
Previous studies have shown that MDSC play a critical role in inflammatory immune responses resulting from bacterial infections (37, 38). Furthermore, infection with P. gingivalis has been shown to expand the Gr-1+ Mac-1+ (CD11b/CD18) cell population (26). The epitope Gr-1 is present on Ly6G and Ly6C glycoproteins, encoded by separate genes and expressed on granulocytic and monocytic cells, and the differential expression of these molecules can differentiate MDSC into separate subpopulations (22, 39). However, it is not known if MDSC differentially express Ly6G and Ly6C as a result of P. gingivalis infection. Therefore, C57BL/6 mice were P. gingivalis infected or left uninfected (i.e., phosphate-buffered saline [PBS] treatment) using microosmotic pumps, which allowed the daily delivery of bacteria or PBS for 14 days (Fig. 1). On day 14, mice were sacrificed and BM and spleen cells from infected and noninfected control mice were assessed for the expression of Ly6G and Ly6C by flow cytometry. The cells were gated on CD11b+ expression and then further delineated based on the expression of Ly6G and Ly6C surface antigens (Fig. 2A and B). A significant increase in the percentage of MDSC expressing these surface molecules was observed in the BM and spleen cells derived from infected mice compared to the noninfected control (Fig. 2C).
FIG 1.
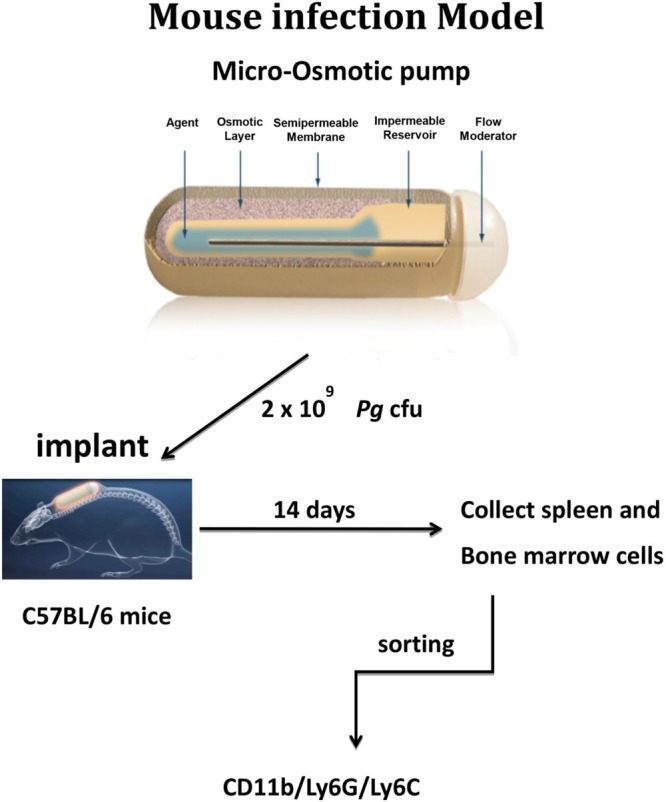
Infection model. A suspension of freshly harvested P. gingivalis was loaded into the microosmotic pumps (100 μl/pump) at a concentration of 2 × 1010 bacterial cells/ml (∼2 × 109 CFU). Micropumps filled with PBS served as controls. Pumps were then implanted into the dorsolumbar region of mice and left undisturbed for 14 days according to the manufacturer's instructions. Microosmotic pumps delivered bacteria or PBS daily at a continuous and controlled rate (0.25 μl/h).
FIG 2.
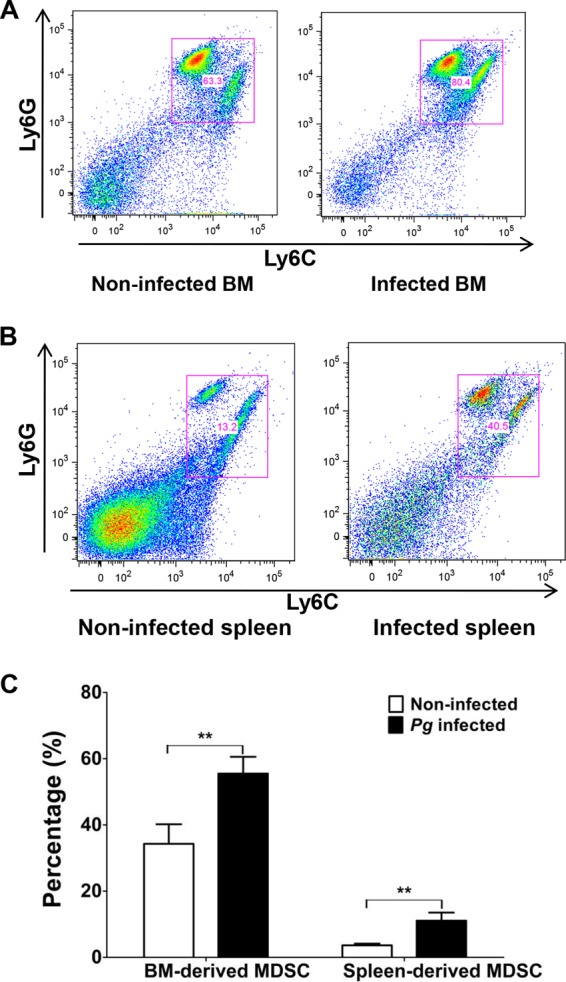
Expansion of MDSC by P. gingivalis infection. A suspension of P. gingivalis was loaded into the microosmotic pumps (100 μl/pump) at a concentration of 2 × 1010 bacterial cells/ml. Micropumps filled with PBS served as controls. Pumps were then implanted into the dorsolumbar region of mice and left undisturbed for 14 days according to the manufacturer's instructions. At the end of 14 days, mice were sacrificed and BM and spleen cells from infected and noninfected mice were harvested and stained for CD11b (FITC), Ly6G (APC), and Ly6C (PE), characteristic markers of MDSC. For flow cytometry, cells were gated on the CD11b+ population and then analyzed for Ly6G+ and Ly6C+ cells. (A and B) Data expressed as dot plots showing expression of Ly6G+ and Ly6C+ cells in BM (A) and spleens (B) of infected and noninfected control mice. (C) Graph representing percentages of Ly6G+ and Ly6C+ cells. Data are expressed as the means ± standard errors of the means (SEM) from three independent experiments. Pg, P. gingivalis. **, P < 0.01.
Since Toll-like receptors are critical for the induction of innate and adaptive host immune responses (40, 41), we next assessed the roles of TLR2, TLR4, and MyD88 in the expansion of MDSC induced by P. gingivalis infection. The induced expansion of MDSC in infected wild-type (WT) mice was similarly observed in infected TLR2−/−, TLR4−/−, and MyD88−/− mice (not shown). These results suggest that the expansion of MDSC is not contingent on TLR2, TLR4, or MyD88 signaling.
We next determined the phenotype of the subpopulations of MDSC expanded upon P. gingivalis infection. Using the infection model described above, BM and spleen cells were obtained from infected and noninfected control mice, and the gated CD11b+ cells were sorted into different subpopulations of MDSC based on their differential expression of Ly6G and Ly6C. Infection with P. gingivalis resulted in the expansion of three subpopulations of MDSC, specifically, Ly6G++ Ly6C+, Ly6G+ Ly6C++, and Ly6G+ Ly6C+ (Fig. 3A and B). It is worth noting that the percentages of the three subpopulations of MDSC identified by sorting and derived from either the BM or spleen of P. gingivalis-infected mice were significantly higher than those seen with cells derived from noninfected control mice (Fig. 3C), suggesting that infection with P. gingivalis induces the overall expansion of MDSC.
FIG 3.

Subpopulations of P. gingivalis-induced MDSC differentially express Ly6G and Ly6C surface antigens. A suspension of P. gingivalis was loaded into the microosmotic pumps (100 μl/pump) at a concentration of 2 × 1010 bacterial cells/ml. Micropumps filled with PBS served as controls. Pumps were then implanted into the dorsolumbar region of mice and left undisturbed for 14 days according to the manufacturer's instructions. At the end of 14 days, mice were sacrificed and BM and spleen cells from infected and noninfected mice were harvested and stained for CD11b (FITC), Ly6G (APC), and Ly6C (PE). For flow cytometry, cells were gated on the CD11b+ population and then sorted into subpopulations. Data are expressed as dot plots of cells showing Ly6G++ Ly6C+, Ly6G+ Ly6C++, and Ly6G+ Ly6C+ subpopulations of cells from the BM (A) and spleens (B) of infected and noninfected control mice. (C) Graph showing the percentages of Ly6G++ Ly6C+, Ly6G+ Ly6C++, and Ly6G+ Ly6C+ MDSC subpopulations derived from the BM and spleens of infected and noninfected control mice. Data are expressed as the means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
MDSC inhibit CD4+ T cell proliferation.
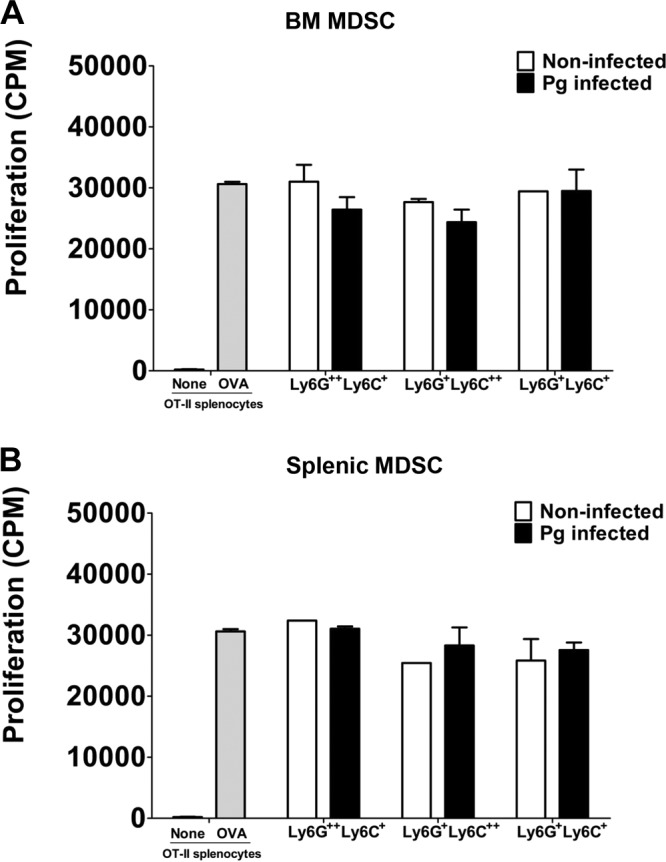
MDSC are well known for their ability to inhibit T-cell proliferation under pathological conditions, especially in chronic infections where expansion of MDSC is most notable (22, 36). Therefore, we wanted to determine if the sorted subpopulation of MDSC induced by P. gingivalis infection exerted an immunosuppressive effect on CD4+ T cell-proliferative responses. Each of the three subpopulations of MDSC derived from the BM and spleens of infected and noninfected control mice were cultured with T cell receptor-transgenic OT-II spleen cells and stimulated with OVA323–339 peptide for 72 h. The cultures were pulsed for the last 24 h with [3H]thymidine, and the proliferative activity was expressed as the amount of [3H]thymidine uptake by CD4+ proliferating T cells (counts per minute). Our results showed that CD11b+ Ly6G+ Ly6C++-expressing cells derived from BM (P < 0.01) and from spleen (P < 0.001) significantly inhibited the proliferation of CD4+ T cells compared to controls, i.e., OT-II spleen cells stimulated with OVA peptide (Fig. 4A and B). However, when the inhibitory effect of this subpopulation of MDSC from P. gingivalis-infected mice was compared to that from noninfected mice, BM-derived MDSC (Fig. 4A) obtained from infected mice showed a significantly (P < 0.01) greater inhibition of CD4+ T cell proliferation than that observed with splenic MDSC (P < 0.05) (Fig. 4B). These results suggest that while naturally occurring MDSC expressing CD11b+ Ly6G+ Ly6C++ have an ability to inhibit T cell proliferation, infection with P. gingivalis exacerbates the inhibitory effect exerted by this specific subpopulation. Interestingly, BM CD11b+ Ly6G++ Ly6C+ MDSC derived from infected or noninfected mice (Fig. 4A) showed significant (P < 0.05) inhibitory activity compared to controls, but no difference was seen between the infected and noninfected groups. A similar significant inhibitory effect was observed with splenic CD11b+ Ly6G++ Ly6C+ MDSC obtained from noninfected mice but not from infected mice compared to controls, and no difference was seen in the inhibitory activity between the infected and noninfected groups (Fig. 4B). Furthermore, the subpopulation of MDSC expressing CD11b+ Ly6G+ Ly6C+ derived from BM, but not from the spleen of noninfected mice, significantly (*, P < 0.05) inhibited CD4+ T cell-proliferative responses. As seen with CD11b+ Ly6G++ Ly6C+ cells, no statistical difference was observed between the infected and noninfected subpopulation of CD11b+ Ly6G+ Ly6C+ MDSC derived from BM and spleen (Fig. 4A and B). These data suggest that there is a differential regulation in the inhibitory activity of MDSC expressing the phenotype CD11b+ Ly6G++ Ly6C+ or CD11b+ Ly6G+ Ly6C+ compared to the subpopulation of MDSC expressing CD11b+ Ly6G+ Ly6C++ surface molecules. Our findings further suggest that the three subpopulations of MDSC can have a suppressive effect on CD4+ T cell proliferation regardless of P. gingivalis infection.
FIG 4.
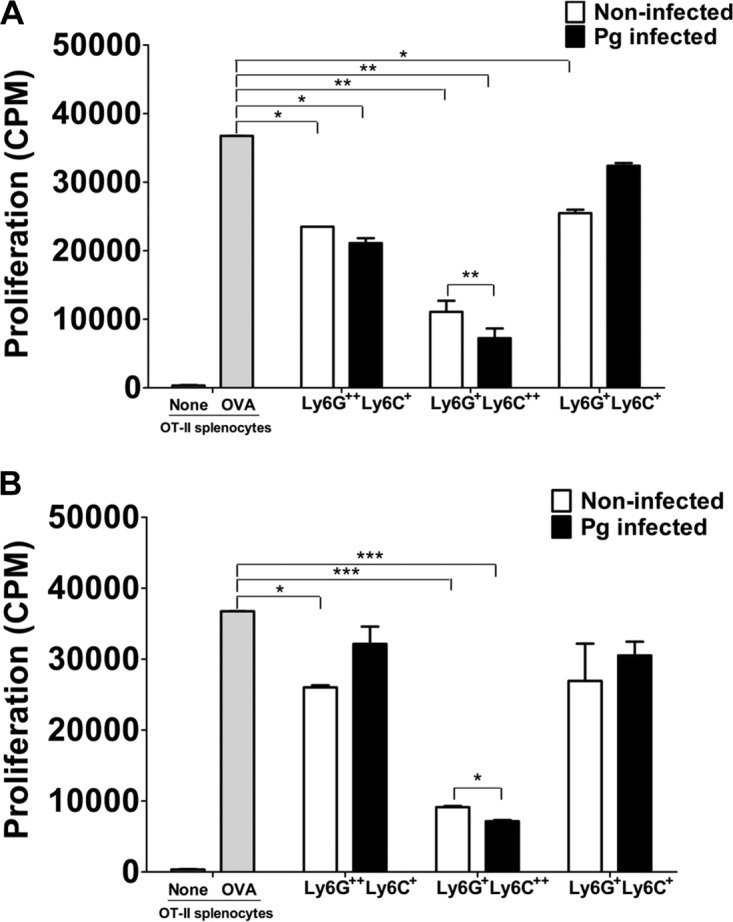
Immunosuppressive activity of T cell proliferation by sorted MDSC. A suspension of P. gingivalis was loaded into the microosmotic pumps (100 μl/pump) at a concentration of 2 × 1010 bacterial cells/ml. Micropumps filled with PBS served as controls. Pumps were then implanted into the dorsolumbar region of mice and left undisturbed for 14 days according to the manufacturer's instructions. At the end of 14 days, mice were sacrificed and BM and spleen cells from infected and noninfected mice were harvested and sorted into subpopulations of MDSC expressing the Ly6G++ Ly6C+, Ly6G+ Ly6C++, and Ly6G+ Ly6C+ phenotypes. The subpopulations of MDSC from BM (A) and spleens (B) of P. gingivalis-infected or noninfected control mice were then cocultured with spleen cells from OT-II mice and stimulated with the OVA323–339 peptide for 72 h. The cultures were pulsed with [3H]thymidine during the last 24 h to assess CD4+ T cell proliferation. Control cultures included OT-II spleen cells with or without the OVA323–339 peptide. Proliferative responses are a measure of [3H]thymidine uptake and are expressed as counts per minute (CPM). Data represent the means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Since inflammation is critical for the suppressive effect of MDSC (42, 43), we next determined the level of the inflammatory cytokine IFN-γ. Spleen cells from OT-II mice were cocultured with subpopulations of BM-derived MDSC, and the level of IFN-γ produced was assessed by enzyme-linked immunosorbent assay (ELISA). Significantly higher levels of IFN-γ were detected in supernatants of cocultures of OT-II spleen cells with each of the subpopulations of MDSC than in supernatants from cultures of OT-II spleen cells alone stimulated with the OVA peptide (Fig. 5). However, supernatants from cocultures containing CD11b+ Ly6G+ Ly6C++ MDSC contained significantly (P < 0.05) higher levels of IFN-γ when the cells were derived from P. gingivalis-infected mice compared to noninfected controls (Fig. 5). Moreover, supernatants from cultures with CD11b+ Ly6G+ Ly6C++ MDSC had significantly (P < 0.001) higher levels of IFN-γ than supernatants from cultures containing the other subpopulations of MDSC derived from P. gingivalis-infected mice (Fig. 5). It is noteworthy that although CD11b+ Ly6G+ Ly6C++ MDSC were the main players in the inhibition of CD4+ T cell proliferation, this subpopulation of MDSC was also able to promote the induction of the highest level of IFN-γ, especially when these cells were obtained from P. gingivalis-infected mice. Results with spleen-derived MDSC were similar to those seen with BM-derived MDSC (not shown).
FIG 5.
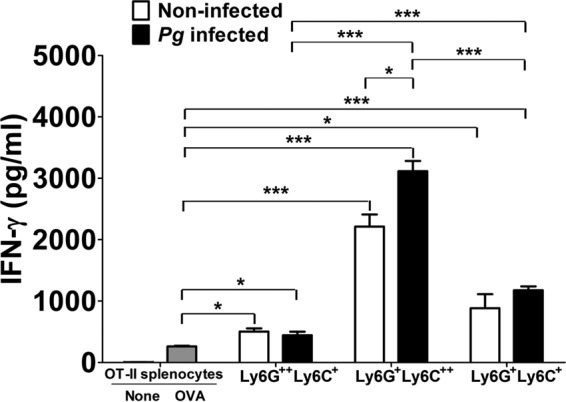
Cytokine analysis. Supernatants from cocultures of spleen cells from OT-II mice and sorted subpopulations of MDSC obtained from the BM of infected and noninfected mice were harvested after 72 h of incubation in the presence or absence of the OVA323–339 peptide. The level of IFN-γ in the culture supernatants was determined by ELISA according to the manufacturer's instructions. Data represent the means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Nos2 and Cat2 mRNA are highly expressed in CD11b+ Ly6G+ Ly6C++ MDSC, and inhibition of CD4+ T cell proliferation is dependent on NOS2 and IFN-γ.
An important factor involved in the regulation of immunity is the cationic amino acid l-arginine. MDSC can affect T cell-proliferative responses by l-arginine depletion through NOS2 and Arg1 enzymatic activity (44, 45). In the presence of IFN-γ, MDSC metabolize l-arginine by increasing nitric oxide (NO) production via NOS2 induction (46, 47), and in the presence of interleukin-4 (IL-4) or IL-13, MDSC regulate l-arginine depletion via Arg1 activity (48, 49). Importantly, regardless of the cytokine environment resulting in the corresponding regulation of l-arginine metabolism that consequently affects T cell-proliferative responses, the suppressive activity of MDSC also requires contact between T cells and MDSC (44). l-Arginine is present in the extracellular microenvironment, and thus transport systems are critical for l-arginine uptake. Studies have shown that CAT2 is the main l-arginine transporter in various cells, including MDSC (50). Moreover, CAT2 has been shown to be important in the establishment of efficient cell-mediated immunity (51), and MDSC recruited to sites of inflammation and tumor growth express increased amounts of Cat2 (50). Production of IFN-γ is characteristic of a number of microbial infections, including infection with P. gingivalis (52–55), and an inflammatory milieu has been shown to be important for the suppressive activity of MDSC (42, 43). Therefore, we wanted to determine if P. gingivalis-induced MDSC exhibited increased expression of Cat2 and NOS2 mRNA. Since we had detected IFN-γ in cocultures of MDSC and OT-II spleen cells, we also wanted to determine if IFN-γ played a role in the observed suppressive effect of MDSC on T cell proliferation. Due to the low yield of splenic MDSC, we only used sorted subpopulations of MDSC from the BM of infected and noninfected mice to carry out these quantitative, real-time PCR studies. For these studies, subpopulations of MDSC were cultured in the presence or absence of P. gingivalis for 48 h and then assessed for Nos2 and Cat2 mRNA expression. The greatest increase in Cat2 mRNA expression occurred in BM-derived CD11b+ Ly6G+ Ly6C++ cells obtained from infected mice and restimulated in vitro with P. gingivalis (Fig. 6A). Interestingly, the level of Cat2 mRNA was low in CD11b+ Ly6G++ Ly6C+ cells under all experimental conditions compared with the other two subpopulations of MDSC (Fig. 6A). While an increased level of Cat2 mRNA expression was observed in CD11b+ Ly6G+ Ly6C+ MDSC obtained from infected mice restimulated in vitro, the increase was only slightly higher than the level seen in cells from noninfected mice that were not stimulated in vitro with P. gingivalis (Fig. 6A). These results suggest that Cat2 mRNA expression is differentially regulated in MDSC subpopulations, and that antigen exposure is important for the increased expression of Cat2 mRNA only in CD11b+ Ly6G+ Ly6C++ MDSC, even when cells were derived from noninfected mice but exposed to P. gingivalis in vitro.
FIG 6.
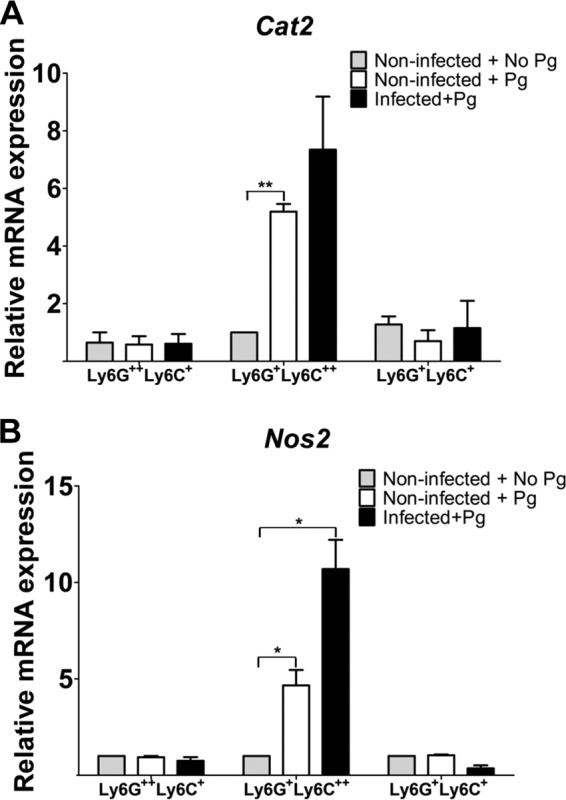
Expression of Nos2 and Cat2 mRNA in MDSC. Sorted subpopulations of MDSC from the BM of infected and noninfected mice were cultured in the presence or absence of P. gingivalis for 48 h. Total RNA was isolated, and the presence of mRNA for Cat2 (A) and Nos2 (B) was assessed by real-time PCR using SYBR green. HPRT was used as the endogenous control. Data represent the means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01.
Assessment of Nos2 mRNA also revealed that CD11b+ Ly6G+ Ly6C++ MDSC exhibited the highest level of Nos2 expression when cells were derived from infected mice and restimulated with P. gingivalis in vitro (Fig. 6B). Some increase in the level of Nos2 mRNA expression was seen in CD11b+ Ly6G+ Ly6C++ MDSC from noninfected mice stimulated in vitro with P. gingivalis compared to those not stimulated in vitro, although the increase was about 2-fold lower than that seen in CD11b+ Ly6G+ Ly6C++ MDSC obtained from infected mice and restimulated in vitro (Fig. 6B). Only a low level of Nos2 mRNA expression was detected in CD11b+ Ly6G+ Ly6C+ and CD11b+ Ly6G++ Ly6C+ MDSC under all experimental conditions. These subpopulations of MDSC showed a slight decrease in the level of Nos2 mRNA expression in cells derived from infected mice and restimulated with P. gingivalis in vitro compared to that seen with cells from noninfected mice (Fig. 6B). Although the relative level of Cat2 mRNA expression is lower than that of Nos2 expression, the results suggest that there is a correspondence in the increased expression of Nos2 and Cat2 mRNA mainly in CD11b+ Ly6G+ Ly6C++ MDSC obtained from P. gingivalis-infected mice and restimulated with P. gingivalis in vitro (Fig. 6B).
We next determined if MDSC-mediated inhibition of CD4+ T cell-proliferative responses was dependent on NOS2 and IFN-γ, especially that exerted by CD11b+ Ly6G+ Ly6C++-expressing cells. Subpopulations of MDSC obtained from the BM and spleens of infected and noninfected control mice were cocultured with spleen cells derived from OT-II mice to assess the proliferative response of CD4+ T cells upon stimulation with OVA323–339 peptide as described above. However, in this experimental series, inhibitors of NOS2 (1400W) and IFN-γ (IFN-γ receptor 1 blocking antibody) were added to or left out of cocultures of cells. Controls included OT-II spleen cells with or without OVA peptide and with or without the corresponding inhibitors. The inhibitory effect of the BM-derived CD11b+ Ly6G+ Ly6C++ MDSC from P. gingivalis-infected mice on the proliferative activity of CD4+ T cells was significantly ablated (P < 0.001) in the presence of the NOS2 inhibitor (Fig. 7A). Moreover, this subpopulation of MDSC, when obtained from noninfected P. gingivalis mice, also showed a significant (P < 0.05) reduction in its ability to suppress the proliferative activity of CD4+ T cells in the presence of the NOS2 inhibitor (Fig. 7A). Furthermore, the ability of the spleen-derived CD11b+ Ly6G+ Ly6C++ MDSC to suppress the proliferative responses of CD4 T cells was also ablated by the NOS2 inhibitor (Fig. 7B). Finally, no significant inhibitory effect was observed for the other subpopulations of MDSC (Fig. 7A and B). Interestingly, in the presence of the IFN-γ blocking antibody, we observed results similar to those obtained in the presence of the NOS2 inhibitor (Fig. 7C and D). These results suggest that inhibition of CD4+ T cell proliferation by CD11b+ Ly6G+ Ly6C++ MDSC involves the presence of the inflammatory cytokine IFN-γ and the consequent induction of NOS2 (45, 53).
FIG 7.

Inhibition of NOS2 and IFN-γ prevents the suppression of OVA-specific CD4+ T cell-proliferative responses by MDSC. Sorted subpopulations of MDSC from the BM and spleens of infected and noninfected mice were cocultured with spleen cells obtained from OT-II mice and stimulated with the OVA323–339 peptide for 72 h. The inhibitor of NOS2 (1400W) was added to some cocultures of BM (A) or spleen (B) MDSC and OT-II-derived spleen cells. Controls included OT-II-derived spleen cells left unstimulated or stimulated with the OVA peptide in the presence or absence of the NOS2 inhibitor. The IFN-γ-blocking antibody was added to other cocultures of BM (C) or spleen (D) MDSC and OT-II-derived spleen cells. Controls included OT-II-derived spleen cells stimulated with the OVA peptide or left unstimulated in the presence or absence of the IFN-γ blocking antibody. [3H]thymidine was added to cocultures during the last 24 h of incubation for the assessment of proliferative responses. Proliferative responses are a measure of [3H]thymidine uptake and expressed as counts per minute (CPM). Data represent the means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Cell-cell contact is necessary for the suppressive activity of MDSC.
Inhibition of T cell proliferation by MDSC often involves cell-cell contact not dependent on major histocompatibility complex (MHC) and mediated by mechanisms that involve, for instance, depletion of l-arginine (48, 53). Thus, we next determined if cell-cell contact played a role in the inhibition of CD4+ T cell proliferation by MDSC subpopulations induced by P. gingivalis. Each of the three subpopulations of MDSC was cultured separately on top of the filter membranes of Transwell inserts (0.4-μm pores) at a concentration of 2 × 105 cells. The spleen cells derived from OT-II mice were cultured in the lower wells of the plates at a 1:1 ratio. The OVA323–339 peptide was used to stimulate the proliferative response of CD4+ T cells for 72 h, and the cells were pulsed with [3H]thymidine for the last 24 h for the assessment of CD4+ T cell proliferation. Our findings revealed that the capacity to suppress CD4+ T cell proliferation by all subpopulations of MDSC derived from BM (Fig. 8A) or spleen (Fig. 8B), and especially that exerted by CD11b+ Ly6G+ Ly6C++-expressing cells, was lost when T cell-MDSC contact was prevented (Fig. 8A and B). These studies suggest that cell-cell contact is critical for the suppression of CD4+ T cell-proliferative responses by subpopulations of MDSC derived from noninfected and, in particular, P. gingivalis-infected mice.
FIG 8.
MDSC-mediated suppression of T cell proliferation is contact dependent. Sorted subpopulations of MDSC were obtained from the BM (A) and spleens (B) of P. gingivalis-infected or noninfected mice and cultured in Transwell inserts, whereas OT-II-derived splenic cells were cultured in the corresponding plate wells at a 1:1 ratio in a modified MDSC suppression assay. Cultures were stimulated with the OVA323–339 peptide for 72 h. Control cultures included OT-II spleen cells cultured with or without the OVA323–339 peptide. [3H]thymidine was added to all cultures during the last 24 h of incubation. Proliferative responses are a measure of [3H]thymidine uptake and expressed as counts per minute (CPM). Data represent the means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Differentiation of MDSC into OC.
Murine CD11b+ cells from BM or spleens can become osteoclasts (OC), and human circulating CD14+ monocytes can differentiate into OC (56). It has also been shown that MDSC can give rise to OC (33, 57). Therefore, in order to determine if the P. gingivalis-induced subpopulations of MDSC have the ability of differentiate into OC, sorted MDSC derived from the BM or spleens of P. gingivalis-infected and noninfected control mice were suspended in α-10 medium and plated in 24-well plates at 5 × 104 cells/well in the presence or absence of macrophage colony-stimulating factor (M-CSF) and RANKL. After 4 or 5 days, multinuclear OC were readily observed in cultures of CD11b+ Ly6G+ Ly6C++ MDSC derived from the BM and spleen, respectively, whereas very few multinuclear OC were observed in cultures of CD11b+ Ly6G+ Ly6C+ or CD11b+ Ly6G++ Ly6C+ MDSC (Fig. 9A and B). To further confirm these observations, we next assessed the cells for the expression of tartrate-resistant acid phosphatase (TRAP), a specific marker of OC. As can be seen, CD11b+ Ly6G+ Ly6C++-expressing MDSC were indeed TRAP+ (Fig. 9C and D). Importantly, a significantly higher number of TRAP+ multinuclear osteoclasts differentiated from MDSC derived from P. gingivalis-infected mice compared to the number of TRAP+ multinuclear osteoclasts in cultures of MDSC derived from noninfected controls (Fig. 9A and B). These findings suggest that CD11b+ Ly6G+ Ly6C++-expressing MDSC have the plasticity to differentiate into OC, and as such they may constitute a source of OC involved in the destruction of bone upon P. gingivalis infection.
FIG 9.

Subpopulation of MDSC has the ability to differentiate into OC. Sorted subpopulations of MDSC obtained from the BM and spleens of P. gingivalis-infected or noninfected mice were cultured in the presence of M-CSF. Cells were stimulated with RANKL for 4 to 5 days and then stained for TRAP activity. Osteoclasts derived from BM MDSC were detected by day 4 (A), whereas OC derived from splenic MDSC differentiated into OC by day 5 (B). A representative area of the cultures from each condition at ×40 magnification is shown. The number of TRAP-positive multinucleated cells (TRAP+ MNC) in cultures of the different subpopulations of MDSC from BM (C) and spleen (D) was counted on days 4 and 5, respectively. Data are expressed as means ± SEM from three independent experiments. **, P < 0.01.
DISCUSSION
MDSC are one of the dominant immunosuppressive populations of cells that rapidly expand under infectious, inflammatory conditions, especially in chronic infections (23–26). Although P. gingivalis can induce inflammation, this inflammatory response is necessary and conducive to the eventual inhibition of immunity. By subverting the host response, P. gingivalis promotes a chronic, infectious, inflammatory state locally in the periodontium and the occurrence of pathological, systemic, inflammatory events (58–62). It is worth noting that P. gingivalis is able to disseminate from local sites of infection to the circulation and to distal sites (12, 13), and antigen-specific T cells can migrate from the circulation to the periodontium (16). Furthermore, T cells from infected human periodontal tissues express mainly a memory phenotype (14), suggesting that exposure and priming of T cells and other immune cells occur systemically in the blood and/or secondary lymphoid organs.
Since cytokines, induced systemically in states of immune stress such as infections, induce the expansion of MDSC, it is not surprising that chronic infection promoted by P. gingivalis, a key pathogen able to support the development of pathogenic microbiota by altering the equilibrium of commensal bacteria (3, 63), can result in the expansion of MDSC (26). Indeed, our findings are in line with those of Ezernitchi et al. (26), who demonstrated that P. gingivalis induced the expansion of Gr-1+ Mac-1+ (CD11b/CD18) cells. However, in the present study, using a micropump that constantly delivered P. gingivalis, thus reflecting a chronic infection, we show that P. gingivalis induces the expansion of three subpopulations of MDSC based on the differential expression of Ly6G and Ly6C. These two glycoproteins have the Gr-1 epitope, are encoded by separate genes, and are expressed in granulocytic and monocytic cells, respectively (22, 64). In mice, MDSC have been divided into granulocytic (Ly6Ghigh Ly6Clow) and monocytic (Ly6Glow Ly6Chigh) cells based on the coexpression of CD11b, Ly6G, and Ly6C, nuclear morphology, and T cell-suppressive functions. Our findings revealed that infection with P. gingivalis induced the expansion of CD11b+ Ly6G++ Ly6C+, CD11b+ Ly6G+ Ly6C++, and CD11b+ Ly6G+ Ly6C+ MDSC. Surprisingly, this expansion was not dependent on TLR2, TLR4, or MyD88 signaling. Studies by Delano et al. (65) demonstrated that expansion of CD11b+ Gr-1+ cells early in sepsis was dependent on MyD88 but not TLR4. However, if sepsis was prolonged, some expansion was observed in the absence of MyD88 signaling. It is known that P. gingivalis is a pathogen that can signal via TLR4 and TLR2, hence, perhaps the difference in the results of these studies can be explained by the longer infection period used in our studies and by the participation of compensatory mechanisms, thus involving signaling via different molecules.
Under healthy conditions, the majority of MDSC reside in the BM and differentiate into mature granulocytes, macrophages, or dendritic cells, but in response to inflammation, MDSC rapidly expand without differentiating into mature cells and enter lymphoid organs and peripheral tissues (42, 43, 53). Interestingly, although in this study all subpopulations of MDSC exerted an inhibitory effect on the proliferative activity of OVA-specific CD4+ T cells derived from OT-II mice, the most significant inhibition of T cell proliferation was seen with the subpopulation of MDSC expressing a CD11b+ Ly6G+ Ly6C++ phenotype, especially those derived from P. gingivalis-infected mice. These findings suggest that P. gingivalis infection affects, in particular, the monocytic subpopulation of MDSC. In the case of CD11b+ Ly6G++ Ly6C+ and CD11b+ Ly6G+ Ly6C+ subpopulations, the MDSC from infected mice did not exhibit more inhibitory activity than those from noninfected mice. In fact, for these two subpopulations, it was the MDSC from the noninfected mice that exhibited slightly more inhibition of CD4+ T cell proliferation. Furthermore, the same subpopulation of MDSC from different tissues did not necessarily show the same inhibitory effect, suggesting that MDSC with the same phenotype have different properties, perhaps due to the influence of the specific environment of the BM or spleen. While the granulocytic population of MDSC is characterized by the surface expression of Ly6Ghigh Ly6Clow, cells expressing CD11b+ Ly6G+ Ly6C+ surface molecules detected in the present work may represent MDSC that are in transition to become granulocytic or monocytic cells. Whether further exposure to bacteria could push the cells to express a particular phenotype will have to be determined in future studies.
Surprisingly, cocultures of OT-II-derived spleen cells and CD11b+ Ly6G+ Ly6C++ MDSC showed a highly significant increase in the production of IFN-γ compared to the cocultures with the other MDSC subpopulations, and the increase was higher when cells were derived from P. gingivalis-infected than noninfected mice. It should be noted that the IFN-γ detected was not produced by the MDSC based on P. gingivalis stimulation of the MDSC (not shown). However, one of the main cytokines produced by the MDSC in our hands (not shown) and those of others (66, 67) was IL-1, a cytokine shown to act directly on CD4+ T cells and enhance their activation (68). IFN-γ stimulates and enhances the production of IL-1 (69, 70), thus possibly creating a self-feeding activation circle between MDSC and the OVA-specific CD4+ T cells. Strikingly, the monocytic MDSC were specifically affected, suggesting that the CD11b+ Ly6G+ Ly6C++ MDSC were the main effector cells within the expanded MDSC subpopulations resulting from P. gingivalis infection.
It is well known that IFN-γ induces the upregulation of NOS2 (iNOS), which catalyzes the synthesis of NO, a factor that is necessary and sufficient to inhibit T cell-proliferative responses. In line with findings of other investigators (53, 71, 72), our studies showed that inhibition of NOS2 and of IFN-γ signaling abrogated the suppressive effect of P. gingivalis-induced MDSC on CD4+ T cell proliferation. These observations indirectly suggest that secretion of NO requires surface and soluble signals, such as NOS2 and IFN-γ from activated T cells, factors that contribute to immune suppression. Our results further demonstrated the importance of cell-to-cell contact for the inhibition of OVA-specific CD4+ T cell-proliferative responses. Indeed, different suppressive mechanisms seem to be used by mouse and human MDSC, including cell-to-cell contact (67, 73). Thus, P. gingivalis-induced MDSC can inhibit T cell-proliferative responses by cell-to-cell contact in a manner that does not involve MHC antigens and also can use NOS2 induced by IFN-γ to produce NO and consequently suppress T cell activity (44, 53). It is important to keep in mind that while in the present study we showed the importance of IFN-γ in the indirect inhibition of T cell-proliferative responses, other cytokines, such as IL-13, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-6, also have been shown to induce MDSC to suppress T cell activity (67, 74). Whether these cytokines also play a role in the inhibitory mechanisms generated as a result of P. gingivalis infection needs to be addressed in future studies.
In addition to the suppressive effect of MDSC on immune cells, we have demonstrated that MDSC can differentiate into OC cells, which is in line with findings of others (32, 33). However, it is noteworthy that in our investigations, only CD11b+ Ly6G+ Ly6C++ MDSC had the ability to differentiate into TRAP+ cells. Studies by Gao et al. (75) have demonstrated that in an inflammatory milieu, IFN-γ indirectly promotes OC differentiation. Although in our studies OC differentiation involved only purified MDSC cultured in the presence of M-CSF and stimulated with RANKL, it would be interesting in future studies to determine if blocking IFN-γ signaling in cocultures of CD4+ T cells and CD11b+ Ly6G+ Ly6C++ MDSC would cause OC differentiation to be ablated. The plasticity of MDSC has biological significance, especially in the context of P. gingivalis infection, due to its association with periodontal disease, since its hallmark is the loss of alveolar bone. Hence, MDSC may contribute to the development and establishment of the disease process not only by exerting immune suppression but also by contributing to the increased number and activity of OC seen in chronic periodontitis (34).
MATERIALS AND METHODS
Mice.
C57BL/6 wild-type (WT) mice, OT-II transgenic mice, and TLR2−/−, TLR4−/−, and MyD88−/− mice (on the C57BL/6 background) were bred and maintained within an environmentally controlled, pathogen-free animal facility at the University of Alabama at Birmingham (UAB). Female mice (8 to 10 weeks of age) were used in this study. All studies were done according to National Institutes of Health guidelines, and all protocols were approved by the UAB Institutional Animal Care and Use Committee.
Bacterial cultures.
P. gingivalis ATCC 33277 was cultured and maintained on enriched Trypticase soy agar plates containing Trypticase soy agar, 1% yeast extract, 5% defibrinated sheep blood, 5 μg/ml hemin, and 1 μg/ml menadione at 37°C in an anaerobic atmosphere of 10% H2, 5% CO2, and 85% N2 (76, 77). For the preparation of P. gingivalis for in vitro and in vivo infection studies, bacteria were harvested, centrifuged, and washed in PBS. The number of bacteria (CFU per milliliter) was determined by measuring the optical density at 600 nm and extrapolating using a standard curve (7, 78).
Infection model.
Female C57BL/6 mice (8 to 10 weeks of age) were anesthetized with a solution of ketamine (100 mg/kg of body weight) and xylazine (50 mg/kg). The back of each mouse was shaved and prepped with 10% betadine. Microosmotic pumps (model 1002; 1.5 cm in length, 0.6 cm in diameter, 0.4 g in weight; Alzet Osmotic Pumps, Cupertino, CA) were loaded with 100 μl of freshly harvested bacteria and then surgically implanted into the dorsolumbar region according to the manufacturer's instructions.
Bacteria were delivered daily for 14 days at a constant pumping rate of 0.25 μl/h and then sacrificed (Fig. 1).
Cell preparation.
Murine bone marrow (BM) cells were collected from the femurs and tibias as previously described (79–82). Single-cell suspensions were prepared by mechanically dispersing the BM through a 40-μm cell strainer. Erythrocytes were lysed using M-lysis buffer (R&D Systems, Minneapolis, MN), and BM cells were then washed twice with PBS and suspended in fluorescence-activated cell sorter (FACS) buffer (PBS containing 5% bovine serum albumin without sodium azide) at a concentration of 1 × 107 cells/ml. For the preparation of spleen cell suspensions, spleens were removed from mice, processed through a cell strainer, and washed twice with PBS. Cells were then suspended in FACS buffer at a concentration of 1 × 107 cells/ml.
Cell sorting.
Suspensions of BM and spleen cells derived from P. gingivalis-infected and noninfected controls were stained with anti-mouse CD11b-fluorescein isothiocyanate (FITC), Ly6C-phycoerythrin (PE), and Ly6G-allophycocyanin (APC) antibodies (eBioscience, San Diego, CA) for 30 min on ice in the dark. After washing the cells twice with FACS buffer, cells were sorted for the isolation of MDSC subpopulations using a BD FACS Aria ΙΙΙu cell sorter (BD Bioscience, San Jose, CA) (Fig. 1). The data were analyzed with BD FACSDiva software (BD Bioscience) and FlowJo software (TreeStar, San Carlos, CA). The sorted MDSC subpopulations were suspended in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, penicillin (50 U/ml), streptomycin (50 μg/ml), l-glutamine (2 mM), β-mercaptoethanol (50 μM), sodium pyruvate (1 mM), sodium bicarbonate (1.5 mg/ml), and HEPES (25 mM), counted, and plated according to the appropriate experimental conditions.
CD4+ T cell proliferation assay.
Single-spleen cell suspensions from OT-II mice were cultured in 96-well plates at a concentration of 2 × 105 cells/well in the presence or absence of the different sorted MDSC subpopulations at a 1:1 ratio. Cells were stimulated with the OVA323–339 peptide (2.5 μg/ml) for 72 h. Inhibition of NOS2 and of IFN-γ was carried out by the addition of the NOS2 inhibitor 1400W [N-(3-aminomethyl)benzylacetamidine, 2HCl; Millipore, Billerica, MA] at a concentration of 0.1 mM or by the addition of the IFN-γ receptor 1 (eBioscience) blocking antibody at a concentration of 5 μg/ml to the cocultures of MDSC and OT-II cells, respectively. The indicated concentration of the inhibitor and blocking antibody used was shown to be optimal in preliminary studies. The NOS2 inhibitor and the IFN-γ blocking antibody had no detrimental effect on cell viability, as determined by trypan blue exclusion. Controls included cultures of OT-II spleen cells or sorted MDSC alone stimulated with the OVA323–339 peptide or left unstimulated. Twenty-four hours prior to the termination of the assay, [3H]thymidine (0.5 μCi) (Perkin-Elmer, Waltham, MA) was added to each well. Cells were then harvested using a cell harvester (Brandel, Gaithersburg, MD), and the amount of [3H]thymidine taken up by OVA-specific CD4+ proliferating T cells was determined in a scintillation counter (Beckman, Fullerton, CA).
Cytokine production.
Spleen cells derived from OT-II mice were cultured in a 96-well plate at 2 × 105 cells/well in the presence or absence of MDSC subpopulations at a 1:1 ratio. Cells were stimulated with OVA323–339 peptide as described above, and the culture supernatants were harvested 72 h later and analyzed for the presence of IFN-γ by ELISA (eBioscience) according to the manufacturer's instructions.
Real-time quantitative PCR.
Total RNA was extracted from 106 cells of each sorted subpopulation of BM-derived MDSC to assess the expression of Nos2 (nitric oxide synthase) and Cat2 (cationic amino acid transporter 2) mRNA at 48 h using RNeasy minikits (Qiagen, Valencia, CA) according to the manufacturer's recommendations. cDNA was synthesized from 500 ng of total RNA by reverse transcription using QuantiTect RT kits (Qiagen). Real-time PCR was done using a LightCycler 480 (Roche Molecular Biochemicals) with a FastStart DNA Master SYBR green Ι reagent (Roche Applied Science). Relative quantities of the tested gene were normalized to hypoxanthine phosphoribosyltransferase (HPRT) mRNA. The normalized data were expressed using the comparative 2−ΔΔCT method.
Transwell assay.
In order to determine if cell contact is required for MDSC-mediated inhibition of T cell-proliferative responses, spleen cells obtained from OT-II mice (2 × 105 cells/well) were cultured in plate wells, whereas sorted MDSC subpopulations were cultured in Transwell inserts (0.4-μm pores; Corning, NY) at a 1:1 ratio, thus preventing direct CD4+ T cell-MDSC contact. Cells were either left unstimulated or stimulated with the OVA323–339 peptide for 72 h, and the proliferative responses of CD4+ T cells were determined by [3H]thymidine uptake as described above.
Differentiation of MDSC into OC.
Spleen and BM cells obtained from P. gingivalis-infected or noninfected mice were sorted into the different MDSC subpopulations as described above. Each sorted MDSC subpopulation was cultured in 24-well plates (5 × 104 cells/well) in α-10 medium (α-minimum essential medium, 10% fetal bovine serum, 1 × penicillin-streptomycin) in the presence of M-CSF (50 ng/ml) and incubated in a humidified 5% CO2 atmosphere at 37°C. Cells were stimulated with RANKL (100 ng/ml), and the culture medium was replaced every 2 days. Cells were cultured for 4 to 5 days and stained for TRAP activity using a leukocyte acid phosphatase kit (Sigma-Aldrich, St. Louis, MO).
Statistical analysis.
Statistical significance was evaluated by analysis of variance and the Tukey's multiple-comparison test using the InStat program (GraphPad Software, San Diego, CA). Differences between groups were considered significant at P values of <0.05 (*), <0.01 (**), and <0.001 (***).
ACKNOWLEDGMENTS
We thank Gregory Harber for his technical assistance. We also thank Casey T. Weaver and Craig L. Maynard for providing the OT-II mice used in this study.
We have no conflicts of interest to declare.
L.S. was supported by training grant T90 DE022736 from the NIDCR.
REFERENCES
- 1.Van Dyke TE, Sheilesh D. 2005. Risk factors for periodontitis. J Int Acad Periodontol 7:3–7. [PMC free article] [PubMed] [Google Scholar]
- 2.Nussbaum G, Shapira L. 2011. How has neutrophil research improved our understanding of periodontal pathogenesis? J Clin Periodontol 38:49–59. doi: 10.1111/j.1600-051X.2010.01678.x. [DOI] [PubMed] [Google Scholar]
- 3.Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 27:409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darveau RP. 2010. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 5.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hajishengallis G, Darveau RP, Curtis MA. 2012. The keystone-pathogen hypothesis. Nat Rev Microbiol 10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katz J, Sambandam V, Wu JH, Michalek SM, Balkovetz DF. 2000. Characterization of Porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect Immun 68:1441–1449. doi: 10.1128/IAI.68.3.1441-1449.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oleksy A, Banbula A, Bugno M, Travis J, Potempa J. 2002. Proteolysis of interleukin-6 receptor (IL-6R) by Porphyromonas gingivalis cysteine proteinases (gingipains) inhibits interleukin-6-mediated cell activation. Microb Pathog 32:173–181. doi: 10.1006/mpat.2002.0491. [DOI] [PubMed] [Google Scholar]
- 9.Potempa J, Banbula A, Travis J. 2000. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000 24:153–192. doi: 10.1034/j.1600-0757.2000.2240108.x. [DOI] [PubMed] [Google Scholar]
- 10.Popadiak K, Potempa J, Riesbeck K, Blom AM. 2007. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol 178:7242–7250. doi: 10.4049/jimmunol.178.11.7242. [DOI] [PubMed] [Google Scholar]
- 11.Gemmell E, Drysdale KE, Seymour GJ. 2006. Gene expression in splenic CD4 and CD8 cells from BALB/c mice immunized with Porphyromonas gingivalis. J Periodontol 77:622–633. doi: 10.1902/jop.2006.050211. [DOI] [PubMed] [Google Scholar]
- 12.Boggess KA, Madianos PN, Preisser JS, Moise KJJ, Offenbacher S. 2005. Chronic maternal and fetal Porphyromonas gingivalis exposure during pregnancy in rabbits. Am J Obstet Gynecol 192:554–557. doi: 10.1016/j.ajog.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Hu SW, Huang HC, Lai YY, Lin YY. 2006. Transvascular dissemination of Porphyromonas gingivalis from a sequestered site is dependent upon activation of the kallikrein/kinin pathway. J Periodont Res 41:200–207. doi: 10.1111/j.1600-0765.2005.00858.x. [DOI] [PubMed] [Google Scholar]
- 14.Seymour GJ, Taubman MA, Eastcott JW, Gemmell E, Smith DJ. 1997. CD29 expression on CD4+ gingival lymphocytes supports migration of activated memory T lymphocytes to diseased periodontal tissue. Oral Microbiol Immunol 12:129–134. doi: 10.1111/j.1399-302X.1997.tb00368.x. [DOI] [PubMed] [Google Scholar]
- 15.Mahanonda R, Champaiboon C, Subbalekha K, Sa-Ard-Iam N, Rattanathammatada W, Thawanaphong S, Rerkyen P, Yoshimura F, Nagano K, Lang NP, Pichyangkul S. 2016. Human memory B cells in healthy gingiva, gingivitis, and periodontitis. J Immunol 197:715–725. doi: 10.4049/jimmunol.1600540. [DOI] [PubMed] [Google Scholar]
- 16.Kawai T, Shimauchi H, Eastcott JW, Smith DJ, Taubman MA. 1998. Antigen direction of specific T-cell clones into gingival tissues. Immunology 93:11–19. doi: 10.1046/j.1365-2567.1998.00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beck J, Garcia R, Heiss G, Vokonas PS, Offenbacher S. 1996. Periodontal disease and cardiovascular disease. J Periodontol 67:1123–1137. [DOI] [PubMed] [Google Scholar]
- 18.Craig RG. 2004. Inflammation, cardiovascular disease and destructive periodontal disease. The evolving role of the dental profession. N Y State Dent J 70:22–26. [PubMed] [Google Scholar]
- 19.Craig RG, Kotanko P, Kamer AR, Levin NW. 2007. Periodontal diseases–a modifiable source of systemic inflammation for the end-stage renal disease patient in haemodyalisis therapy? Nephrol Dial Transplant 22:312–315. doi: 10.1093/ndt/gfl604. [DOI] [PubMed] [Google Scholar]
- 20.Offenbacher S, Katz V, Fertik G, Collins J, Boyd D, Maynor G, McKaig R, Beck J. 1996. Periodontal infection as a possible risk factor for preterm low birth weight. J Periodontol 67:1103–1113. [DOI] [PubMed] [Google Scholar]
- 21.Scher JU, Bretz WA, Abramson SB. 2014. Periodontal disease and subgingival microbiota as contributors for rheumatoid arthritis pathogenesis: modifiable risk factors? Curr Opin Rheumatol 26:424–429. doi: 10.1097/BOR.0000000000000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabrilovich DI, Nagaraj S. 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Ginderachter JA, Beschin A, De Baetselier P, Raes G. 2010. Myeloid-derived suppressor cells in parasitic infections. Eur J Immunol 40:2976–2985. doi: 10.1002/eji.201040911. [DOI] [PubMed] [Google Scholar]
- 24.Martino A, Badell E, Abadie V, Balloy V, Chingnard M, Mistou MY, Combadiere B, Combadiere C, Winter N. 2010. Mycobacterium bovis bacillus Calmette-Guerin vaccination mobilizes innate myeloid-derived suppressor cells restraining in vivo T cell priming via IL-1R-dependent nitric oxide production. J Immunol 184:2038–2047. doi: 10.4049/jimmunol.0903348. [DOI] [PubMed] [Google Scholar]
- 25.Zhu J, Huang X, Yang Y. 2012. Myeloid-derived suppressor cells regulate natural killer cell response to adenovirus-mediated gene transfer. J Virol 86:13689–13696. doi: 10.1128/JVI.01595-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ezernitchi AV, Vaknin I, Cohen-Daniel L, Levy O, Manaster E, Halabi A, Pikarsky E, Shapira L, Baniyash M. 2006. TCR ζ down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J Immunol 177:4763–4772. doi: 10.4049/jimmunol.177.7.4763. [DOI] [PubMed] [Google Scholar]
- 27.Ostrand-Rosemberg S. 2010. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother 59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. 2010. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol 184:3106–3116. doi: 10.4049/jimmunol.0902661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. 2007. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179:977–983. [DOI] [PubMed] [Google Scholar]
- 30.Fortin C, Huang X, Yang Y. 2012. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J Immunol 189:1843–1849. doi: 10.4049/jimmunol.1200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schouppe E, De Baetselier P, Van Ginderachter JA, Sarukhan A. 2012. Instruction of myeloid cells by the tumor microenvironment: open questions on the dynamics and plasticity of different tumor-associated myeloid cell populations. Oncoimmunology 1:1135–1145. doi: 10.4161/onci.21566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhuang J, Zhang J, Lwin ST, Edwards JR, Edwards CM, Mundy GR, Yang X. 2012. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+ myeloid-derived suppressor cells. PLoS One 7:e48871. doi: 10.1371/journal.pone.0048871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sawant A, Deshane J, Jules J, Lee CM, Harris BA, Feng X, Ponnazhagan S. 2013. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res 73:672–682. doi: 10.1158/0008-5472.CAN-12-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cochran DL. 2008. Inflammation and bone loss in periodontal disease. J Periodontol 79:1569–1576. doi: 10.1902/jop.2008.080233. [DOI] [PubMed] [Google Scholar]
- 35.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. 2008. Subsets of myeloid-derived suppressor cells in tumor bearing mice. J Immunol 181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagaraj S, Youn JI, Gabrilovich DI. 2013. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol 191:17–23. doi: 10.4049/jimmunol.1300654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obregon-Henao A, Henao-Tamayo M, Orme IM, Ordway DJ. 2013. Gr1(int)CD11b+ myeloid-derived suppressor cells in Mycobacterium tuberculosis infection. PLoS One 8:e80669. doi: 10.1371/journal.pone.0080669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heim CE, Vidlak D, Scherr TD, Kozel JA, Holzapfel M, Muirhead DE, Kielian T. 2014. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J Immunol 192:3778–3792. doi: 10.4049/jimmunol.1303408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gabrilovich DI, Ostrand-Rosemberg S, Bronte V. 2012. Coordinated regulation of myeloid cells by tumors. Nat Rev Immunol 12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao X. 2016. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol 16:35–50. doi: 10.1038/nri.2015.8. [DOI] [PubMed] [Google Scholar]
- 41.Gerold G, Zychlinsky A, de Diego JL. 2007. What is the role of Toll-like receptors in bacterial infections? Semin Immunol 19:41–47. doi: 10.1016/j.smim.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 42.Bunt SK, Yang L, Shina P, Clements VK, Leips J, Ostrand-Rosemberg S. 2007. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67:10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosemberg S. 2006. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 44.Bronte V, Zanovello P. 2005. Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol 5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 45.Peranzoni E, Marigo I, Dolcetti L, Ugel S, Sonda N, Taschin E, Mantelli B, Bronte V, Zanovello P. 2007. Role of arginine metabolism in immunity and immunopathology. Immunobiology 212:795–812. doi: 10.1016/j.imbio.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 46.Atochina O, Daly-Engel T, Piskorska D, McGuire E, Harn DA. 2001. A schistosome-expressed immunomodulatory glycoconjugate expands peritoneal Gr1+ macrophages that suppress naive CD4+ T cell proliferation via an IFN-γ and nitric oxide-dependent mechanism. J Immunol 167:4293–4302. doi: 10.4049/jimmunol.167.8.4293. [DOI] [PubMed] [Google Scholar]
- 47.Cauley LS, Miller EE, Yen M, Swain SL. 2000. Superantigen-induced CD4 T cell tolerance mediated by myeloid cells and IFN-γ. J Immunol 165:6056–6066. doi: 10.4049/jimmunol.165.11.6056. [DOI] [PubMed] [Google Scholar]
- 48.Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. 2003. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol 24:301–305. doi: 10.1016/S1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- 49.Terabe M, Berzofsky JA. 2004. Immunoregulatory T cells in tumor immunity. Curr Opin Immunol 16:157–162. doi: 10.1016/j.coi.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 50.Cimen Bozkus C, Elzey BD, Crist SA, Ellies LG, Ratliff TL. 2015. Expression of cationic amino acid transporter 2 is required for myeloid-derived suppressor cell-mediated control of T cell immunity. J Immunol 195:5237–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson RW, Pesce JT, Ramalingam T, Wilson MS, White S, Cheever AW, Ricklefs SM, Porcella SF, Li L, Ellis LG, Wynn TA. 2008. Cationic amino acid transporter-2 regulates immunity by modulating arginase activity. PLoS Pathog 4:e1000023. doi: 10.1371/journal.ppat.1000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker PJ, Dixon M, Evans RT, Dufour L, Johnson E, Roopenian DC. 1999. CD4+ T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun 67:2804–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goni O, Alcaide P, Fresno M. 2002. Immunosuppression during acute Trypanosoma cruzi infection: involvement of Ly6G (Gr1+)CD11b+ immature myeloid suppressor cells. Int Immunol 14:1125–1134. doi: 10.1093/intimm/dxf076. [DOI] [PubMed] [Google Scholar]
- 54.Mencacci A, Montagnoli C, Bacci E, Cenci E, Pitzurra L, Spreca AK, Sharpe MAH, Romani L. 2002. CD80+ Gr-1+ myeloid cells inhibit development of antifungal Th1 immunity in mice with candidiasis. J Immunol 169:3180–3190. doi: 10.4049/jimmunol.169.6.3180. [DOI] [PubMed] [Google Scholar]
- 55.Ren H, Li Y, Jiang H, Du M. 2016. Interferon-gamma and Fas are involved in Porphyromonas gingivalis-induced apoptosis of human extravillous trophoblast derived HTR8/SVneo cells via ERK1/2 pathway. J Periodontol 28:1–15. [DOI] [PubMed] [Google Scholar]
- 56.Massey HM, Flanagan AM. 1999. Human osteoclasts derive from CD14− positive monocytes. Br J Hematol 106:167–170. doi: 10.1046/j.1365-2141.1999.01491.x. [DOI] [PubMed] [Google Scholar]
- 57.Zhang H, Huang Y, Wang S, Fu R, Guo C, Wang H, Zhao J, Gaskin F, Chen J, Yang N, Fu SM. 2015. Myeloid-derived suppressor cells contribute to bone erosion in collagen-induced arthritis by differentiating to osteoclasts. J Autoimmun 65:82–89. doi: 10.1016/j.jaut.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hajishengallis G, Lamont RJ. 2014. Breaking bad: manipulation of the host response by Porphyromonas gingivals. Eur J Immunol 44:328–338. doi: 10.1002/eji.201344202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. 2013. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J 7:1016–1025. doi: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. 2010. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol 6:727–730. doi: 10.1038/nrrheum.2010.139. [DOI] [PubMed] [Google Scholar]
- 61.Madianos PN, Bobetsis YA, Offenbacher S. 2013. Adverse pregnancy outcomes (APOs) and periodontal disease: pathogenic mechanisms. J Clin Periodontol 40:S170–S180. doi: 10.1111/jcpe.12082. [DOI] [PubMed] [Google Scholar]
- 62.Kozarov EV, Dorn BR, Shelburne CE, Dunn WAJ, Progulske-Fox A. 2005. A human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol 25:e17–. doi: 10.1161/01.ATV.0000155018.67835.1a. [DOI] [PubMed] [Google Scholar]
- 63.Whitmore SE, Lamont RJ. 2011. The pathogenic persona of community-associated oral streptococci. Mol Microbiol 81:305–314. doi: 10.1111/j.1365-2958.2011.07707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ribechini E, Greifenberg V, Sandwick S, Lutz MB. 2010. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol 199:273–281. doi: 10.1007/s00430-010-0151-4. [DOI] [PubMed] [Google Scholar]
- 65.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O'Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, Laface D, Heyworth PG, Clare-Salzler M, Moldawer LL. 2007. MyD88-dependent expansion of an immature Gr-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med 204:1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN. 2005. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1β-secreting cells. J Immunol 175:8200–8208. doi: 10.4049/jimmunol.175.12.8200. [DOI] [PubMed] [Google Scholar]
- 67.Lechner MG, Liebertz DJ, Epstein AL. 2010. Characterization of cytokine-induced myeloid-derived suppressor cells from normal peripheral blood mononuclear cells. J Immunol 185:2273–2284. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. 2009. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A 106:7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kakizaki Y, Kraft N, Atkins RC. 1993. Interferon gamma stimulates the secretion of IL-1 but not of IL-6, by glomerular mesangial cells. Clin Exp Immunol 91:521–525. doi: 10.1111/j.1365-2249.1993.tb05935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Danis VA, Kulesz AJ, Nelson DS, Brooks PM. 1990. Cytokine regulation of human monocyte interleukin-1 (IL-1) production in vitro. Enhancement of IL-1 production by interferon (IFN) gamma, tumor necrosis factor-alpha, IL-2 and IL-1, and inhibition of IFN-alpha. Clin Exp Immunol 80:435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manner CK, Nicholson B, MacLeod CL. 2003. CAT2 arginine transporter deficiency significantly reduces iNOS-mediated NO production in astrocytes. J Neurochem 85:476–482. doi: 10.1046/j.1471-4159.2003.01695.x. [DOI] [PubMed] [Google Scholar]
- 72.Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. 2002. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol 168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 73.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. 2008. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev 222:162–179. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 74.Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, Bicciato S, Bronte V. 2006. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Investig 116:2777–2790. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, Weitzmann MN, Pacifici R. 2007. IFN-γ stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Investig 117:122–132. doi: 10.1172/JCI30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Syed SA. 1980. Characteristics of Bacteroides asaccharolyticus from dental plaques of beagle dogs. J Clin Microbiol 11:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Syed SA, Loesche WJ. 1978. Bacteriology of human experimental gingivitis: effect of plaque age. Infect Immun 21:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Katz J, Ward DC, Michalek SM. 1996. Effect of host responses on the pathogenicity of strains of Porphyromonas gingivalis. Oral Microbiol Immunol 5:309–318. doi: 10.1111/j.1399-302X.1996.tb00187.x. [DOI] [PubMed] [Google Scholar]
- 79.Feng X, Novack DV, Faccio R, Ory DS, Aya K, Boyer MI, McHugh KP, Ross FP, Teitelbaum SL. 2001. A Glanzmann's mutation in beta 3 integrin specifically impairs osteoclast function. J Clin Investig 107:1137–1144. doi: 10.1172/JCI12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Katz J, Zhang P, Martin M, Vogel SN, Michalek SM. 2006. Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect Immun 74:2809–2816. doi: 10.1128/IAI.74.5.2809-2816.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang P, Liu J, Xu Q, Harber G, Feng X, Michalek SM, Katz J. 2011. TLR2-dependent modulation of osteoclastogenesis by Porphyromonas gingivalis through differential induction of NFATc1 and NF-kappaB. J Biol Chem 286:24159–24169. doi: 10.1074/jbc.M110.198085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen Z, Su L, Xu Q, Katz J, Michalek SM, Fan M, Feng X, Zhang P. 2015. IL-1R/TLR2 through MyD88 divergently modulates osteoclastogenesis through regulation of nuclear factor of activated T cells c1 (NFATc1) and B lymphocyte-induced maturation protein-1 (Blimp1). J Biol Chem 291:30163–30174. doi: 10.1074/jbc.M115.663518. [DOI] [PMC free article] [PubMed] [Google Scholar]