

Abstract
Sapoviruses (SaVs) belong to the Sapovirus genus, in the family Caliciviridae. They have been associated with gastroenteritis in humans and in pigs but not in other animals. In addition, some strains from pigs, chimpanzees and rodents show close sequence identity with human SaVs thereby suggesting the possibility of interspecies transmissions. Bats are known to be a major reservoir of zoonotic viruses, however, very little is known about the genetic diversity of SaVs in bats. To explore the genetic diversity of bat SaVs, fecal samples of Eidolon helvum and Epomophorus gambianus were treated according to the NetoVIR protocol and sequenced by Illumina technology. Nearly complete genome sequences of six highly divergent SaVs and one partial SaV (only VP1 region) were identified in Eidolon helvum and based on sequence identities and phylogenetic analysis, they potentially represent two novel genogroups, only distantly related to known SaVs. Furthermore, comparing these sequences with currently used screening primers and probes indicated that the novel SaVs would not be detected in routine epidemiological screening studies in humans in case an interspecies transmission would occur. Therefore, we designed and validated new primers that can detect both human and bat SaVs. In this study, we identified multiple novel bat SaVs, however, further epidemiological studies in humans are needed to unravel their potential role in gastroenteritis.
Keywords: bat, feces, interspecies transmission, new genogroups, sapovirus
INTRODUCTION
Sapovirus (SaV) particles were first detected in human diarrheic stool samples in 1976 in the United Kingdom using electron microscopy.1 Shortly thereafter they were recognized as a new gastroenteritis pathogen as reviewed by Oka and colleagues.2 Initially these viruses were known as 'typical human caliciviruses' or 'Sapporo-like' viruses. Since 2002, the International Committee on the Taxonomy of Viruses (ICTV) has assigned these viruses to the Sapporo virus species, Sapovirus genus, in the family Caliciviridae,3 together with four other genera: Norovirus, Lagovirus, Vesivirus and Nebovirus (www.ictvonline.org/). SaV particles are made up of a non-enveloped icosahedral capsid of 30–38 nm in diameter, with typical cup-shaped depressions on the surface.4 Inside the particle resides the positive-sense, single-stranded RNA genome of ~7.1–7.7 kb with a 3’- poly (A) tail. Generally, SaV genome contains two open reading frames (ORFs): ORF1 encodes for a large polyprotein containing the nonstructural proteins (helicase, VPg, protease, RNA-dependent-RNA-polymerase (RdRp)) followed by the major capsid protein, VP1; ORF2 is predicted to encode for the minor structural protein VP2.5 However, a third ORF has been found in some human6, 7, 8, 9, 10, 11 and bat12 SaV strains, although a known function is yet to be assigned. Until recently, SaVs were classified into five genogroups (GI–GV).13 With the exception of SaV GIII, SaV genogroups were further subdivided in one or multiple genotypes based on the full-length nucleotide (nt) sequence of VP1.2, 13 GI, GII, GIV and GV SaVs are known to infect humans, while GIII infects pigs.10 Recently, more SaVs have been detected in different animal species, and novel genogroups GVI–GXV were proposed on the bases that strains with ≥57% pairwise aa identity of VP1 should be in the same genogroup.14 A number of SaVs from animal hosts, such as chimpanzee15, pig16, 17, 18 and rodents19 have been found to cluster closely with human SaVs in GI, GV and GII, respectively. This clearly indicates the possibility of cross-species transmission of SaVs;20 however, this does not indicate the direction of the interspecies transmission. To further elucidate the zoonotic potential of SaVs, more human and animal samples need to be screened. Bats (order Chiroptera) constitute the second largest group of mammals after the order Rodentia21 and have recently been implicated as a reservoir of many different viruses including some highly pathogenic to humans.22 Little is known about the genetic diversity of SaVs in bats and currently bat SaVs have been described only in two studies conducted in China12 and Hungary.23 The bat SaVs found in these studies were highly divergent from other SaVs known thus far.
Here, we describe the nearly complete sequences of six highly divergent SaVs and one partial SaV obtained from the fecal samples of straw-colored fruit bats (Eidolon helvum) in Cameroon.
MATERIALS AND METHODS
Ethical authorization
Ethical authorization for the protocol and the use of animal samples was obtained from the Cameroon National Ethics Committee, Yaoundé. All animal experiments were performed in accordance with the Ministry’s National Ethics Committee guidelines. Administrative authorization (R.11/MINSANTE/SWR/RDPH/PS/290/157) was obtained from the Delegation of Public Health for South West Region, Cameroon.
Sample collection
Fecal samples were collected from straw-colored fruit bats (Eidolon helvum) between December 2013 and May 2014 by a previously described method developed by Donaldson et al.24 Briefly, bats were captured in three different regions (Lysoka, Muyuka and Limbe) of the South West Region of Cameroon using mist nets around fruit trees and around human dwellings. Captured bats were retrieved from the net traps and held in paper sacks for 10–15 min, allowing enough time for the production of fresh feces. Sterile disposable spatulas were used to retrieve feces from the paper sacks, and placed into tubes containing 1 mL of universal transport medium (COPAN Diagnostics, Brescia, Italy). Labeled samples were put in ice and then transferred to the Molecular and Cell Biology Laboratory, Biotechnology Unit, University of Buea, Cameroon and stored at −20 °C. Later on, they were shipped to the Laboratory of Clinical and Epidemiological Virology, Leuven, Belgium and stored at −80 °C. Each captured bat was assessed to determine species, weight (g), forearm length (mm), sex, reproductive state, and age (adult or juvenile). Parameters such as body temperature, activity, photophobia were not measured. All captured bats were then marked by hair clipping to facilitate identification of recaptures. Trained zoologists used morphological characteristics to determine the species of the bats before they were released. None of the captured animals showed physical signs of disease.
Sample preparation
Briefly, 25 pools (of two to five samples each) were made from 87 collected samples (85 from Eidolon helvum and two from Epomophorus gambianus) based on age, sex and location, and the pools were treated to enrich for viral particles using the NetoVIR protocol.25 Fecal suspensions (10% w/v in universal transport medium) were homogenized for 1 min at 3000 rpm with a MINILYS homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France) and filtered using a 0.8 μm PES filter (Sartorius, Goettingen, Germany). The filtrate was then treated with a cocktail of Benzonase (Millipore, Billerica, MA, USA) (Novagen, Madison, WI, USA) and Micrococcal Nuclease (New England Biolabs, Ipswich, MA, USA) at 37 °C for 2 h to digest free-floating nucleic acids. Samples were extracted using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions but without addition of carrier RNA to the lysis buffer. First and second strand synthesis and random PCR amplification for 17 cycles were performed using a slightly modified Whole Transcriptome Amplification (WTA2) Kit procedure (Sigma-Aldrich, St Louis, MO, USA). WTA2 products were purified with MSB Spin PCRapace spin columns (Stratec Biomedical, Birkenfeld, Germany) and the libraries were prepared for Illumina sequencing using a slightly modified version of the Nextera XT Library Preparation Kit (Illumina, San Diego, CA, USA). Sequencing of the samples was performed on a HiSeq 2500 platform (Illumina) for 300 cycles (2 × 150 bp paired ends).
Genomic and phylogenetic analysis
Raw reads were trimmed for quality and adapters using Trimmomatic26 and were de novo assembled into contigs using SPAdes.27 Contigs were then annotated using DIAMOND with the sensitive option28 using the non-redundant (nr) database of GenBank. ORFs were identified with ORF Finder analysis tools and the conserved motifs in the amino acid (aa) sequences were identified with HMMER29 and NCBI's conserved domain database (CDD).30 According to the International Calicivirus Conference Committee, a complete VP1 region sequence is required to designate new genogroups or genotypes, and strains in the same genogroups have ≥57% pairwise aa identity of VP1.13 Based on these complete gene sequence (at least 1602 nt), our novel sequences and 51 other complete reference gene sequences were aligned in MEGA631 using muscle and a phylogenetic tree generated using maximum-likelihood method, based on the LG+G+I+F substitution model (after testing for the best DNA/protein model in MEGA6). Sequences used in the phylogenetic analysis were representatives of the different genogroups and/or genotypes, complemented with all known bat SaVs, and SaVs which had been previously reported as potentially involved in zoonotic events.14, 19, 32, 33 Amino acid sequence pairwise distances were also calculated in MEGA6 using transition and transversion substitution models. Sequences from the novel SaVs were submitted to GenBank with the following accession numbers: KX759618–KX759624.
Primer design and validation
Degenerate screening primers (SaV4529F: CCN TCD GGV ATG CCW TTY AC and SaV5165R: TYR CCC TCC ATY WCA AAC AC) were designed based on an alignment of 26 human SaVs (representative members of all genogroups) and 12 bat SaVs. These primers were used to screen 12 SaV-positive samples (eight human samples and four bat samples) by PCR. The following PCR conditions were used: an initial reverse transcription step at 50 °C for 30 min was followed by a PCR activation step at 95 °C for 15 min, 39 cycles of amplification, and a final extension step for 10 min at 72 °C in a Biometra T3000 thermocycler (Biometra). The cycle conditions for denaturation, annealing and elongation were 94 °C for 45 s, 58 °C for 45 s and 72 °C for 1 min, respectively. PCR products were run in polyacrylamide gel, stained with EtBr and visualised under UV-light.
RESULTS
Fecal samples from apparently healthy straw-colored fruit bats were pooled (three to five samples per pool) based on sex age and location and sequenced by Illumina next-generation sequencing (NGS) technology. Fourteen out of the twenty-five pools contained SaV reads, ranging from 0.1% to 6.3% of the non-phage viral reads. All SaV-positive pools were from the straw-colored fruit bat (Eidolon helvum) while the lone pool of Gambian epauletted fruit bat (Epomophorus gambianus) contain no SaV reads. These positive pools were from both sexes and age groups and were identified in samples from two out of three localities (Limbe and Lysoka) (Details in Table 1). We were able to obtain two full-length genomes sequences (Limbe65 and Limbe899b); two near-complete genomes (Limbe900 and Limbe899a) with at least the full-length coding region; and two incomplete genomes (Limbe25 and Lysoka36) without a complete coding region. Also, a partial sequence of the major capsid gene (VP1) of Limbe894 was obtained. Bat SaVs Limbe65 and Limbe899b have sequence lengths of 7475 and 7474 nt, with a G+C content of 48.8% and 46.3%, respectively. Their genomes contain two non-overlapping ORFs flanked by 5’-UTRs of 9 nt and 3’-UTRs (52 nt and 51 nt, respectively) (Figures 1A and 1B). Both have an ORF1 of 6819 nt, encoding the nonstructural proteins and the major capsid protein VP1. The ORF2 (492 nt) encodes for a minor structural protein VP2. The other four nearly complete genomes (Figures 1C–1F) show similar genomic characteristics except that Lysoka36 (Figure 1D), has overlapping ORF1 and ORF2 sequences (an overlapping region of 102 nt). In addition, the two ORFs of SaVs Limbe25, Limbe900 and Limbe889a are located in the same reading frame, whereas those of Limbe65, Limbe899a and Limbe899b can be found in different frames.
Table 1. Number of SaV reads and percentages of total number of non-phage viral reads.
| Pool | Sex | Age | Location | # non-phage viral reads | # of sapovirus reads | % SaV reads | Other viruses in the pool |
|---|---|---|---|---|---|---|---|
| P1a | M | Adult | Lysoka | 7068 | 250 | 3.5 | 2, 9 and 12 |
| P2 | M | Adult | Lysoka | 71955 | 625 | 0.9 | 1, 9 and 11 |
| P3 | F | Adult | Lysoka | 81322 | 75 | 0.1 | 9 and 14 |
| P4 | F | Adult | Lysoka | 72953 | 0 | 0.0 | 4, 6, 7, 9, 12 and 15 |
| P5 | M | Adult | Moyuka | 1206 | 0 | 0.0 | 8, 9, and 16 |
| P6 | F | Adult | Moyuka | 450 | 0 | 0.0 | 9 |
| P7 | M | Adult | Limbe | 7156 | 0 | 0.0 | 4, 9, 11, 14 and 15 |
| P8 | M | Adult | Limbe | 52278 | 570 | 1.1 | 11, 14 and 15 |
| P9 | F | Adult | Limbe | 20866 | 224 | 1.1 | 2 and 4 |
| P10 | F | Adult | Limbe | 46295 | 56 | 0.1 | 2, 4, 6, 11 and 15 |
| P11a | M | Young | Limbe | 60856 | 3827 | 6.3 | 4, 6 and 15 |
| P12 | M | Young | Limbe | 787 | 0 | 0.0 | 2, 4, 6, 11 and 15 |
| P13 | M | Young | Limbe | 2016 | 0 | 0.0 | 4, 6 and 15 |
| P14a | F | Young | Limbe | 50567 | 452 | 0.9 | 2, 3, 12 and 17 |
| P14b | F | Young | Limbe | 188953 | 14068 | 7.4 | 2, 3, 12 and 17 |
| P15 | F | Young | Limbe | 108240 | 42 | 0.0 | 3, 12, 15 and 17 |
| P16c | F | Adult | Lysoka | 110542 | 0 | 0.0 | 3 and 9 |
| P17a | F | Adult | Lysoka | 217016 | 471 | 0.2 | 9, 11 and 14 |
| P18 | F | Adult | Lysoka | 1370 | 0 | 0.0 | 9, 11, 14, 16 and 17 |
| P19 | F | Adult | Moyuka | 2569 | 0 | 0.0 | 9, 11, 12, 14 and 17 |
| P20 | F | Adult | Limbe | 609 | 0 | 0.0 | 4, 6, 11, 14, 15, and 17 |
| P21 | F | Adult | Limbe | 2923 | 0 | 0.0 | 4, 6, 11, 14, 15 and 17 |
| P22d | F | Young | Limbe | 568732 | 7295 | 1.3 | 2 and 17 |
| P23a | M | Adult | Limbe | 620965 | 2563 | 0.4 | 4 |
| P24 | M | Young | Limbe | 1853 | 84 | 4.5 | 1, 2, 3, 5, 11, 14, 15 and 17 |
| P25 | F | Adult | Limbe | 15355 | 0 | 0.0 | 3, 4, 8, 11, 14, 15 and 17 |
Abbreviations: next-generation sequencing, NGS; sapovirus, SaV.
Pool from which SaV reads were described.
Pool on which NGS was repeated after pretreatment of the extracted viral nucleic acids with DNsase to obtain a relative enrichment of RNA virus (sapovirus) reads.
Pool of samples from Epomophorus gambianus, no SaV reads. Other viruses are presented with numbers from 1 to 17 as follows: 1=Astroviridae, 2=Caliciviridae, 3=Circoviridae, 4=Coronaviridae, 5=Hepeviridae, 6=Herpesviridae, 7=Nodaviridae, 8=Papillomaviridae, 9=Partitiviridae, 10=Paramyxoviridae, 11=Parvoviridae, 12=Picobirnaviridae, 14=Reoviridae, 15=Retroviridae, 16=Totiviridae, 17=Tymovirales.
Pool with two sapovirus strains.
Figure 1.

Genome organization of novel sapovirus strains from straw-colored fruit bats. (A, B) SaV strains Limbe65 and Limbe899b with non-overlapping ORF1 and ORF2 in different reading frames; (D) SaV strain Lysoka36 with an overlapping ORF1 and ORF2; (C, E, F) SaV strains Limbe25, Limbe899a and Limbe900 with non-overlapping ORF1 and ORF2 in the same reading frame.
Table 2 shows the characteristic aa motifs conserved in all caliciviruses: 2C-like NTPase motif 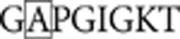 ; VPg motifs KGKTK and
; VPg motifs KGKTK and 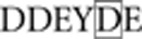 ; 3C-like protease motif GDCG; 3D-like RdRP motifs WKGL, KDEL, DYSKWDST, GLPSG and YGDD; and VP1 motif PPG. Only minor deviations were observed in the novel SaVs: in the 2C-like NTPase motif (
; 3C-like protease motif GDCG; 3D-like RdRP motifs WKGL, KDEL, DYSKWDST, GLPSG and YGDD; and VP1 motif PPG. Only minor deviations were observed in the novel SaVs: in the 2C-like NTPase motif (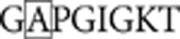 ), the A is replaced with either I or P and in the VPg motif (
), the A is replaced with either I or P and in the VPg motif (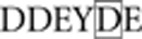 ), the last D is replaced by an E in all the novel sequences.
), the last D is replaced by an E in all the novel sequences.
Table 2. Motifs of functional proteins of newly described bat SaVs.

On the basis of the complete VP1 gene of all bat SaVs, human SaVs and animal SaVs closely related to human SaVs, the nt and aa similarity percentages of representative members of the different genogroups and/or genotypes were calculated (Supplementary Table S1). Hong Kong Hipposideros Pomona bat SaVs (TLC34, TLC39 and TLC58) identified by Tse et al12 shares 99%–100% aa identity among each other, are only 37%–42% similar to other SaVs, and were previously assigned to genotype XIV.14 On the other hand, two unpublished Chinese strains, GX2012 (from Rhinolophus sinicus) and JX2010 (from Myotis myotis), are only 50% similar to each other on the aa level while M63 (a Hungarian strain from Myotis daubentonii)23 shares 71% identity (67% nt identity) with JX2010. Therefore, the genotype numbers GXVI and GXVII were tentatively assigned to the strains GX2012, and JX2010 and M63, respectively. The sequences of the seven bat SaVs identified in Cameroon have an aa identity ranging from 31% to 41% (40%–49% nt identity) with all other SaVs and only 37%–41% (46%–48% nt identity) with other bat SaVs. On the basis of sequence similarity, the Cameroonian strains fall into two groups. The first group has two nearly identical pairs: Limbe65 and Limbe899b; Limbe 25 and Limbe 899a (99%–100% nt and aa identity) and between these pairs, there is an aa similarity of 77% (71% nt similarity). The rest of the strains (Limbe900, Lysoka36 and Limbe894) are more closely related to each other, with an aa similarity ranging from 61% to 78% (62%–72% nt similarity) among each other. These two groups share only 49%–54% aa (53%–57% nt) similarity among them. According to the classification scheme proposed by Oka et al,14 our bat SaVs represent two new genogroup within the Sapovirus genus. We tentatively placed them into genogroups GXVIII and GXVIX, respectively.
Phylogenetic analysis was performed using the aa sequences of the entire VP1 region of our sequences and 55 sequences representing genogroup and/or potential zoonotic strains. The human SaVs were found in four clusters, representing four genogroups (GI, GII, GIV and GV) while the porcine SaVs were found not only in genogroup GIII, but also in genogroups GV-GXI.
The phylogenetic analysis revealed a single monophyletic clade of bat SaVs, containing two major clusters (Figure 2). The first cluster contains SaV strains from Asia and Hungary divided over one established and two putative genogroups. Based on aa similarity, bat strains from Hong Kong (TLC39, TLC58 and TLC34) constitute genogroup GXIV. SaV strain GX2012 (a distantly related Chinese strain), is the lone strain in the putative genogroup XVI, while JX2010 (another Chinese strain) together with Hungarian strain M63 are found in the putative genogroups XVII. The African SaVs cluster contained the SaVs strains described in this paper. This cluster is further subdivided into two putative novel genogroups (Limbe65, Limbe899a, Limbe899b and Limbe25 on one hand and Limbe900, Limbe894 and Lysoka36 on the other hand). The phylogenetic tree based on the RdRP gene (Supplementary Figure S1) confirmed the observations based on the VP1 gene. Comparing these Cameroonian bat sequences to partial RdRP from African wildlife SaVs34 indicated only 46%–57% nt indentity, however, they were not included in the phylogenetic tree because only very short sequences (<300 nt) of the RdRPs were available in GenBank.
Figure 2.

Maximum-likelihood phylogenetic tree, based on a VP1 amino acid sequence alignment of Limbe65, Limbe899a, Limbe899b, Limbe25, Limbe900, Limbe894 and Lysoka36, and 55 other representative SaVs strains. Previously known bat SaVs are indicated with open triangles, whereas those described in this paper are indicated with filled triangle. Filled circles are strains with the potential history of cross between species. The numbers at the internal nodes represent the bootstrap probabilities (in percent), as determined for 1000 iterations. Only bootstrap values greater than 70% are shown. The scale bar indicates the genetic distance (amino acid substitutions per site).
Given the high genetic divergence of the bat SaVs described here and human SaVs, it is doubtful that the currently used human screening primers will also detect these newly found SaVs in case of interspecies transmission from bats to humans. To answer this question, we compared the frequently used screening primers (SaV124F, SaV1F, SaV5F and SaV1245R) and probes (SaV124TP and SaV5TP)35 with sequences of the novel bat strains (Table 3). It showed that the percentage similarity ranged from 25% to 66.7% for forward primers and 94.7% to 100% for the reverse primers. Considering the probes, the percentage similarity was only 42.9% and 57.1%. These primers and probes are unlikely to pick up SaVs from bats, and therefore we designed novel screening primers based on the RdRP segment of human and bat sequences, to screen both human and bat fecal samples positive for SaVs. The used samples contain human SaVs of genogroup GI.1, GI.3, GII.3, GII.5, GII.6, GIV and GV and the novel putative bat SaVs of genogroups GXVIII and GXIX. As expected, the novel primers detected both human and bat SaVs while the currently used primers detected only human SaVs (Figure 3).
Table 3. Nucleotide comparison between the primers sequences used for human sapovirus screening with the corresponding region of the newly identified bat sapovirus strains.

Figure 3.

Polyacrylamide gel electrophoresis of the PCR product amplified with novel primers (A) and currently used primers (B). Wells 1 and 14 or 15: DNA Molecular Weight Marker VIII; wells 2–9: human samples positive for SaV (genogroups GI.1, GI.3, GII.3, GII.5, GII.6, GIV and GV; wells 10–13: bat samples positive for SaV (novel putative genogroups GXVIII and GXIX).
DISCUSSION
SaV are known human gastroenteritis etiological agents and the identification of animal SaV strains closely related to human SaVs has raised awareness for the possibility of SaV interspecies transmissions. Due to an increasing interest in bats as a reservoir of zoonotic viruses and the fact that bat SaVs have only been sporadically reported, there is an eminent need to fully explore bat species for this virus and its zoonotic potential. Here, we collected and metagenomically screened 87 bat samples from fruit bats in Cameroon. SaV reads were present in samples from both juvenile and adult, as well as male and female bats, suggesting that viral presence does not seem to be restricted by either age or sex. We were able to obtain six nearly complete genomes and one capsid protein (VP1) of SaVs in Cameroonian bats, each of which encoded two ORFs. SaVs of genogroup GI, GIV, and GV have been reported to encode a third ORF overlapping the VP1 gene,10, 36 which was also detected in a previous study in bats, although their ORF3 did not overlap with the VP1 of ORF1.12 The fact that the third ORF was not identified in our novel strains and other animal SaVs18 points to the fact that the protein encoded by this ORF in bats and human GV strains might not be mandatory. To ascertain the function of the protein, cell culture propagation, functional and knockout studies are required. Furthermore, the GC content of the newly described bat SaVs falls within the typical GC range of the genus Sapovirus (49.0% to 53.6%). However, SaVs identified previously in Pomona roundleaf bat (Hipposideros pomona) showed a relatively high GC content (up to 60.2%).12 In addition, the overlap of ORF1 and ORF2 in the genome of Lysoka36 (Figure 1D), to the best of our knowledge is novel. This highlights the existence of a high genomic diversity of bat SaVs, although the true diversity is most likely much larger. Further studies are needed to better understand the SaV diversity in bats and other animals.
SaVs have been associated with gastroenteritis in people of all ages,37, 38 and in pigs,39 but not in other animals. It is not clear whether these viruses cause disease in bats, since the adult and young bats from which they were identified appeared healthy based on physical examination (for wounds or abnormalities on the body or wings) and no signs of white-nose syndrome were observed. This is not surprising as most bat viruses known today were discovered in apparently healthy bats, suggesting that they may have specific immune system or antiviral activity against virus infections,40 making them effective reservoirs of viruses.
These novel African SaVs clearly formed two phylogenetic clusters: the first cluster is made of Limbe65, Limbe899a, Limbe899b and Limbe25 and the second is made of Limbe900, Limbe894 and Lysoka36. Each cluster differs from the other and all other SaV genogroups by at least 40% aa identity and therefore constitute two new genogroups within the SaV genus, tentatively assigned as genogroups GXVIII and GXVIX, respectively. SaV genogroups GI, GII, GIV and GV infect humans while all other SaV genogroups (GIII and GVI–GXVII) have been identified in animals. Phylogenetically, some of the strains isolated from animals are related to human SaVs. Two SaVs (IJC09 and IJC04) isolated from chimpanzees in The Republic of Congo clustered closely with human G1 SaVs. Given that these chimps were living in close contact to humans (in the sanctuary) the authors suggested that there might have been a recent cross-species transmission of these viruses.15 Also three porcine SaV strains (TYMPo239, TYMPo031 and WG194D-1) and one California sea lion SaV were identified independently and are relatively closely related to human SaV GV.14, 32, 33 Lastly, based on the pairwise nt distances between the rodent strain Manhattan/Ro-SaV2 and the closest human SaVs (GII.1 to GII.7), it can be concluded that this rodent strain also belongs to the genogroup GII SaV. This suggests that these human and rat strains have a common origin, most likely reflecting an interspecies transmission event in a (distant) past.19 All these data from animal studies suggest that interspecies transmission of these viruses might have occurred in the past and as a consequence, raise the possibility of circulating animal SaVs strains in humans. Given that the bats we sampled live in closely proximity to humans in the region, there might have been an ample opportunity for such interspecies transmission to occur between bats and humans. However, most of the epidemiological SaVs studies described so far used conserved screening primers in PCR assay to identify and sequence the VP1 or VP1-RdRp region. Due to the nt divergence of bat SaVs observed here, even if there were zoonotic strains from bats circulating in humans, they might not be detected by the existing primers. Therefore, the novel primers presented here will be able to detect both zoonotic strains especially from bats. Further, to fully delineate the zoonotic capacity of these bat SaVs, in vitro studies in cell cultures would be very useful, as well as screening human samples around the same region for bat SaVs.
Acknowledgments
CKY was supported by the Interfaculty Council for Development Cooperation (IRO) from the KU Leuven. NC-N was supported by the Flanders Innovation & Entrepreneurship (VLAIO). The authors thank Prof Oka Tomoichiro (National Institute of Infectious Diseases, Department of Virology II, Japan) for reading through the manuscript and making valuable suggestions.
Footnotes
Supplementary Information for this article can be found on the Emerging Microbes & Infections website (http://www.nature.com/emi)
Supplementary Material
References
- Madeley CR, Cosgrove BP. Letter: Caliciviruses in man. Lancet 1976; 1: 199–200. [DOI] [PubMed] [Google Scholar]
- Oka T, Wang Q, Katayama K et al. Comprehensive review of human sapoviruses. Clin Microbiol Rev 2015; 28: 32–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MA. A summary of taxonomic changes recently approved by ICTV. Arch Virol 2002; 147: 1655–1663. [DOI] [PubMed] [Google Scholar]
- Madeley CR. Comparison of the features of astroviruses and caliciviruses seen in samples of feces by electron microscopy. J Infect Dis 1979; 139: 519–523. [DOI] [PubMed] [Google Scholar]
- Chang KO, Sosnovtsev SV, Belliot G et al. Reverse genetics system for porcine enteric calicivirus, a prototype sapovirus in the Caliciviridae. J Virol 2005; 79: 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata K, Hardy ME, Nakata S et al. Molecular characterization of morphologically typical human calicivirus Sapporo. Arch Virol 1997; 142: 1537–1552. [DOI] [PubMed] [Google Scholar]
- Liu BL, Clarke IN, Caul EO et al. Human enteric caliciviruses have a unique genome structure and are distinct from the Norwalk-like viruses. Arch Virol 1995; 140: 1345–1356. [DOI] [PubMed] [Google Scholar]
- Liu B, Clarke IN, Caul EO et al. The genomic 5' terminus of Manchester calicivirus. Virus Genes 1997; 15: 25–28. [DOI] [PubMed] [Google Scholar]
- Jiang X, Cubitt WD, Berke T et al. Sapporo-like human caliciviruses are genetically and antigenically diverse. Arch Virol 1997; 142: 1813–1827. [DOI] [PubMed] [Google Scholar]
- Farkas T, Zhong WM, Jing Y et al. Genetic diversity among sapoviruses. Arch Virol 2004; 149: 1309–1323. [DOI] [PubMed] [Google Scholar]
- Okada M, Yamashita Y, Oseto M et al. Genetic variability in the sapovirus capsid protein. Virus Genes 2006; 33: 157–161. [DOI] [PubMed] [Google Scholar]
- Tse H, Chan WM, Li KS et al. Discovery and genomic characterization of a novel bat sapovirus with unusual genomic features and phylogenetic position. PLoS One 2012; 7: e34987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Mori K, Iritani N et al. Human sapovirus classification based on complete capsid nucleotide sequences. Arch Virol 2012; 157: 349–352. [DOI] [PubMed] [Google Scholar]
- Oka T, Lu Z, Phan T et al. Genetic characterization and classification of human and animal sapoviruses. PloS One 2016; 11: e0156373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombo IM, Berthet N, Bouchier C et al. Characterization of a genogroup I sapovirus isolated from chimpanzees in the republic of congo. Genome Announc 2014; 2: e00680–14 s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella V, Lorusso E, Banyai K et al. Identification of a porcine calicivirus related genetically to human sapoviruses. J Clin Microbiol 2008; 46: 1907–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Homme Y, Brassard J, Ouardani M et al. Characterization of novel porcine sapoviruses. Arch Virol 2010; 155: 839–846. [DOI] [PubMed] [Google Scholar]
- Scheuer KA, Oka T, Hoet AE et al. Prevalence of porcine noroviruses, molecular characterization of emerging porcine sapoviruses from finisher swine in the United States, and unified classification scheme for sapoviruses. J Clin Microbiol 2013; 51: 2344–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth C, Bhat M, Firth MA et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio 2014; 5: e01933–01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar L, Di Martino B, Di Profio F et al. Detection and molecular characterization of sapoviruses in dogs. Infect Genet Evol 2016; 38: 8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudge C. The Variety of Life: A Survey and a Celebration of All the Creatures that Have Ever Lived. Oxford, UK: Oxford University Press, 2000. [Google Scholar]
- Rodhain E. [Bats and viruses: complex relationships]. Bull Soc Pathol Exot 2015; 108: 272–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemenesi G, Gellert A, Dallos B et al. Sequencing and molecular modeling identifies candidate members of Caliciviridae family in bats. Infect Genet Evol 2016; 41: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson EF, Haskew AN, Gates JE et al. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J Virol 2010; 84: 13004–13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conceicao-Neto N, Zeller M, Lefrere H et al. Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis. Sci Rep 2015; 5: 16532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012; 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015; 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 2011; 39: W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Derbyshire MK, Gonzales NR et al. CDD: NCBI's conserved domain database. Nucleic Acids Res 2015; 43: D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D et al. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 2013; 30: 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Saga Y, Iwai M et al. Frequent detection of noroviruses and sapoviruses in swine and high genetic diversity of porcine sapovirus in Japan during Fiscal Year 2008. J Clin Microbiol 2010; 48: 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shan T, Wang C et al. The fecal viral flora of California sea lions. J Virol 2011; 85: 9909–9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olarte-Castillo XA, Hofer H, Goller KV et al. Divergent sapovirus strains and infection prevalence in wild carnivores in the Serengeti ecosystem: a long-term study. PLoS One 2016; 11: e0163548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Katayama K, Hansman GS et al. Detection of human sapovirus by real-time reverse transcription-polymerase chain reaction. J Med Virol 2006; 78: 1347–1353. [DOI] [PubMed] [Google Scholar]
- Jiang X, Huang PW, Zhong WM et al. Design and evaluation of a primer pair that detects both Norwalk- and Sapporo-like caliciviruses by RT-PCR. J Virol Methods 1999; 83: 145–154. [DOI] [PubMed] [Google Scholar]
- Pang XL, Lee BE, Tyrrell GJ et al. Epidemiology and genotype analysis of sapovirus associated with gastroenteritis outbreaks in Alberta, Canada: 2004-2007. J Infect Dis 2009; 199: 547–551. [DOI] [PubMed] [Google Scholar]
- Hansman GS, Katayama K, Maneekarn N et al. Genetic diversity of norovirus and sapovirus in hospitalized infants with sporadic cases of acute gastroenteritis in Chiang Mai, Thailand. J Clin Microbiol 2004; 42: 1305–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn WT, Saif LJ, Moorhead PD. Pathogenesis of porcine enteric calicivirus-like virus in four-day-old gnotobiotic pigs. Am J Vet Res 1988; 49: 819–825. [PubMed] [Google Scholar]
- Shi Z. Bat and virus. Protein Cell 2010; 1: 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.