

Abstract
We previously reported low sensitivity of the hepatitis B virus (HBV) ribonuclease H (RNaseH) enzyme to inhibition by N-hydroxyisoquinolinedione (HID) compounds. Subsequently, our biochemical RNaseH assay was found to have a high false negative rate for predicting HBV replication inhibition, leading to underestimation of the number of HIDs that inhibit HBV replication. Here, 39 HID compounds and structurally related polyoxygenated heterocycles (POH), N-hydroxypyridinediones (HPD), and flutimides were screened for inhibition of HBV replication in vitro. Inhibiting the HBV RNaseH preferentially blocks synthesis of the positive-polarity DNA strand and causes accumulation of RNA:DNA heteroduplexes. Eleven HIDs and one HPD preferentially inhibited HBV positive-polarity DNA strand accumulation. EC50s ranged from 0.69 μM to 19 μM with therapeutic indices from 2.4 – 71. Neither the HIDs nor the HPD had an effect on the ability of the polymerase to elongate DNA strands in capsids. HBV RNaseH inhibition by the HIDs was confirmed with an improved RNaseH assay and by detecting accumulation RNA:DNA heteroduplexes in HBV capsids from cells treated with a representative HID. Therefore, the HID scaffold is more promising for anti-HBV drug discovery than we originally reported, and the HPD scaffold may hold potential for antiviral development. The preliminary structure-activity relationship will guide optimization of the HID/HPDs as HBV inhibitors.
Keywords: Hepatitis B Virus, ribonuclease H, N-hydroxyisoquinolinediones, N-hydroxypyridinediones, polyoxygenated heterocycles, flutimide
1. Introduction
Hepatitis B virus (HBV) is a small, enveloped DNA virus that replicates by reverse transcription (Seeger et al., 2013; Summers and Mason, 1982; Tavis and Badtke, 2009). Despite the widespread availability of an excellent vaccine, up to 350 million people are chronically infected worldwide, resulting in more than 700,000 deaths annually, with most of those cases in sub-Saharan Africa and East Asia (Sorrell et al., 2009; Trépo et al., 2014). Current treatments in the USA are limited to two forms of interferon alpha and five nucleos(t)ide analogs (Trépo et al., 2014). The nucleos(t)ide analogs target the viral DNA polymerase and suppress HBV viremia by 4–5 log10 or more (Marcellin et al., 2008; van Bommel et al., 2010). However, HBV replication persists during treatment in part due to incomplete inhibition of viral replication (Cox and Tillmann, 2011; Kwon and Lok, 2011). While current nucleos(t)ide analog treatments are highly effective at suppressing viremia to very low levels, they cure the infection in only a small percentage of patients and must be used indefinitely. Therefore, there is an urgent need for new anti-HBV therapies.
The HBV polymerase protein that replicates the HBV genome has two enzymatic activities. The reverse transcriptase synthesizes the viral DNA from an RNA template, and then the ribonuclease H (RNaseH) degrades the RNA template (Seeger et al., 2013; Tavis and Badtke, 2009). Nuclos(t)ide analog drugs inhibit DNA synthesis by the viral reverse transcriptase, but no drugs exist that target the RNaseH, primarily due to challenges in development of screening assays. Blocking the RNaseH causes accumulation of RNA:DNA heteroduplexes, rendering the negative-polarity DNA strand useless as a template for subsequent positive-polarity strand synthesis. Therefore, suppressing the RNaseH activity blocks both transmission of functional HBV genomes to the nucleus to replenish the pool of viral covalently closed circular DNA (the template for all HBV RNAs), and secretion of infectious virus. We recently developed a low throughput RNaseH inhibitor screening pipeline and identified more than 60 compounds that inhibit the HBV RNaseH in the low micromolar range and below (Cai et al., 2014; Hu et al., 2013; Lu et al., 2015; Tavis et al., 2013).
Our initial screens for HBV RNaseH inhibitors focused on compound classes known to inhibit the HIV RNaseH because the two enzymes belong to the nucleotidyl transferase superfamily, which includes the HBV RNaseH, the human immunodeficiency virus (HIV) RNaseH, the HIV integrase, and the human RNaseH1 (huRNaseH1) (Dyda et al., 1994; Lima et al., 2001; Nowotny, 2009; Tavis et al., 2013; Yang and Steitz, 1995). The HIV and HBV RNaseH domains share 23% sequence homology at the amino acid level and both enzymes contain an active site with four conserved carboxylates that bind two essential Mg++ ions (Nowotny et al., 2005). N-hydroxyisoquinolinedione (HID) compounds inhibit the HIV RNaseH in vitro by binding to the two Mg++ ions in the RNaseH active site (Billamboz et al., 2008; Billamboz et al., 2011a; Billamboz et al., 2011b; Billamboz et al., 2016; Billamboz et al., 2013; Desimmie et al., 2013; Hang et al., 2004; Klumpp et al., 2003; Suchaud et al., 2014).
We previously found that one of 16 HIDs tested inhibited the HBV RNaseH in a oligonucleotide-directed RNA cleavage assay and blocked HBV replication in cells (Cai et al., 2014). It was subsequently determined that the oligonucleotide-directed RNA cleavage assay employed in this screen has a high false negative rate in predicting inhibitors of HBV replication in culture. Therefore, we expanded our evaluation of the HIDs and assessed N-hydroxypyridinedione (HPD), flutimide, and related polyoxygenated heterocycle (POH) compounds for efficacy against HBV replication and for activity in an improved biochemical RNaseH assay. We found the HID scaffold is more promising for anti-HBV drug discovery than we originally concluded, and that the HPD scaffold also holds potential for antiviral development.
2. Materials and methods
2.1 Compound acquisition and synthesis
Compound structures and chemical names are in Supplemental Fig. 1 and Table 1. Compounds #1, 41–45, 78–91, 138–140, and 190 were reported previously (Cai et al., 2014). Compounds #236–241 were synthesized as previously reported (Zoidis et al., 2016). Compounds #128, 132, 191, 197, 198, 204, 206, 208, 211, and 217 were purchased commercially. All compounds used in this study were ≥95% pure. The compounds were dissolved at 10 mM in DMSO and stored in opaque tubes at −80°C.
Table 1.
| Compound # |
Formal Name |
Compound Structure | Derivative series (a) |
Inhibition of HBV RNaseH |
HBV replication inhibition (EC50,μ3M) (e) |
HSV-1 suppression (log10 at 5 μM) (f) |
Inhibition of huRNaseH1 (b) |
Toxicity in HepDES19 (g) | TI | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Oligo- nucleotide directed cleavage assay (b, c) |
Molecular Beacon cleavage assay (d) |
MTS | NR | LDH | ||||||||
| 208 | Sun B8155 |
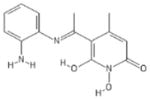
|
HPD | + | + | 0.69 ± 0.2 | − | − | 15 ± 7 | 13 ± 1 | 12 ± 0.1 | 22 |
| 211 | Rilopirox |
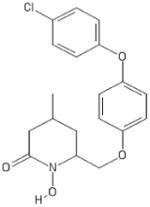
|
HPD | − | − | − | − | − | ||||
| 86 | VS42 |
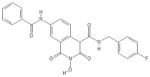
|
HID | − | − | 1.4 ± 0.3 | − | + | 99 ± 2 | 52 ± 11 | >100 | 71 |
| 83 | MB88 |
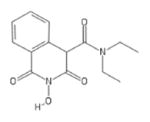
|
HID | − | + | 2.3 ± 1.6 | − | + | 100± 35 | >100 | >100 | 43 |
| 88 | VS45 |
|
HID | − | − | 2.5 ± 0.5 | − | + | 40 ± 6 | 26 ± 2 | 57 ± 6 | 16 |
| 89 | VS51 |
|
HID | − | − | 2.6 ± 0.8 | − | + | 28 ± 8 | 19 ± 3 | 25 ± 4 | 11 |
| 1 | TRC 939800 |
|
HID | + | + | 4.2 ± 1.4 (b) | − | + | 75 ± 24 | 81 ± 4 | 79 ± 12 | 18 |
| 81 | MB4 |
|
HID | − | + | 4.4 ± 3.0 | − | + | 57 ± 16 | 31 ± 7 | 24 ± 5 | 13 |
| 91 | MB106 |
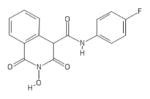
|
HID | − | − | 4.6 ± 1.0 | − | + | 11 ± 4 | 33 ± 3 | 58 ± 22 | 2.4 |
| 87 | VS55 |
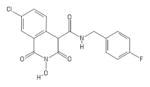
|
HID | − | − | 4.5 ± 3.7 | − | + | 37 ± 17 | 24 ± 1 | 52 ± 15 | 8.2 |
| 85 | MB103 |
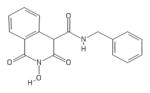
|
HID | − | − | 6.5 ± 1.5 | − | + | 29 ± 12 | 67 ± 13 | >100 | 4.5 |
| 84 | MB105 |
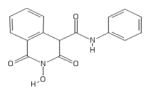
|
HID | − | − | 6.5 ± 4.5 | − | + | 23 ± 9 | 32 ± 8 | 37 ± 15 | 3.5 |
| 90 | MB104 |
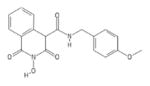
|
HID | − | − | 19 ± 0.9 | − | + | 88 ± 17 | >100 | >100 | 4.6 |
| 82 | MB71 |
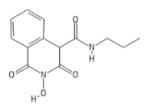
|
HID | − | − | − | + | |||||
| 78 | MB1 |
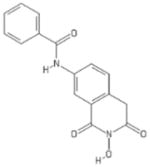
|
HID | − | − | − | + | |||||
| 79 | MB2 |
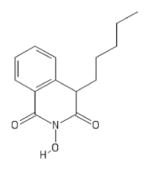
|
HID | − | − | − | − | |||||
| 80 | MB3 |
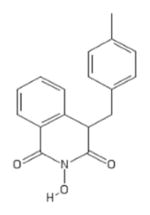
|
HID | − | − | − | − | |||||
| 41 | TRC C432800 (Ciclopirox) |
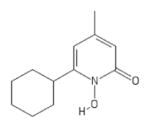
|
POH | − | − | 5.0 | + | |||||
| 42 | Labotest 72543251 |
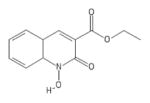
|
POH | − | − | − | + | |||||
| 43 | Sigma PH008969 |
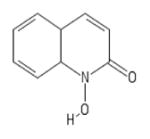
|
POH | − | − | − | − | |||||
| 44 | Labotest 12243782 |
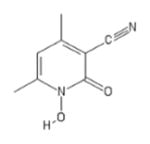
|
POH | − | − | − | − | |||||
| 45 | TCI America H1040 |
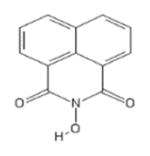
|
HID | − | − | − | − | |||||
| 128 | Aldrichselect CNC_ID100615760 |
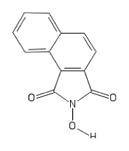
|
POH | − | − | − | − | |||||
| 132 | Aldrichselect CNC_ID 389306767 |
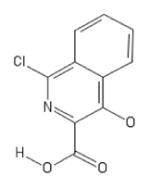
|
POH | − | − | |||||||
| 138 | Sigma H53704 |
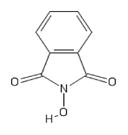
|
POH | − | − | − | − | − | ||||
| 139 | Sigma 130672 |
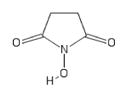
|
POH | − | − | − | ||||||
| 140 | Chembridge 6325462 |
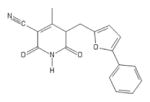
|
POH | − | − | − | + | |||||
| 191 | Piroctone olamine |
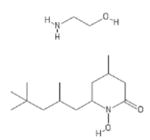
|
POH | − | − | 4.6 | − | |||||
| 197 | Benzoylene urea |
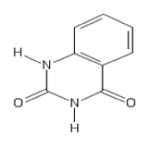
|
POH | − | − | − | − | |||||
| 198 | 2,3-dihydroxy-quinoxaline |
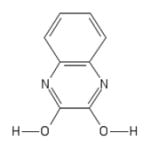
|
POH | − | − | − | − | |||||
| 204 | AK-830/13217043 |
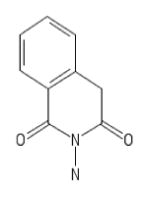
|
POH | − | − | − | + | |||||
| 206 | AJ-333/25006202 |
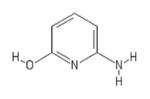
|
POH | − | − | − | + | |||||
| 217 | Visas M Lab 444035142 |
|
POH | − | − | − | + | |||||
| 236 | ZF4 |
|
FLT | − | − | − | − | |||||
| 237 | ZF13 |
|
FLT | − | − | − | ||||||
| 238 | ZF18 |
|
FLT | − | − | − | ||||||
| 239 | ZF19 |
|
FLT | − | − | − | ||||||
| 240 | ZF24 |
|
FLT | − | − | − | + | |||||
| 241 | ZF29 |
|
FLT | − | − | − | ||||||
N-hydroxyisoquinolinedione (HID), polyoxygenated heterocycles (POH), N-hydroxypyridinediones (HPD), and flutimides (FLT)
Compounds 1–91, and 138–140 previously reported in (Cai et al., 2014)
(+), Dose-dependent inhibition at ≤100 μM; (−), No inhibition detected at 100 μM
(+), Dose-dependent inhibition at ≤500 μM; (−), No inhibition detected at 500 μM
(−), No inhibition detected at 20 μM
(−), ≤1 log10 suppression of plaque forming units at 5 μM
CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (MTS), neutral red retention assay (NR), lactate dehydrogenase release (LDH)
2.2 Expression and purification of HBV RNaseH and huRNaseH1
HuRNaseH1 and HBV RNaseH were expressed in Escherichia coli and purified by nickel-affinity chromatography as described (Villa et al., 2016).
2.3 RNaseH assays
The oligonucleotide-directed RNA cleavage assay was reported previously (Hu et al., 2013; Tavis et al., 2013). Briefly, a 32P-labeled RNA was combined with a DNA oligonucleotide and the RNA:DNA substrate was incubated in the presence of the RNaseH and test compounds in 50 mM tris pH 8.0, 190 mM NaCl, 5 mM MgCl2, 3.5 mM DTT, 0.05% NP40, 6% glycerol, and 1% DMSO at 42°C for 90 minutes. The products were reso lved by gel electrophoresis and detected by audioradioagraphy. Inhibition was qualitatively determined as a dose-dependent reduction in the amount of substrate degraded in the reaction.
Inhibition of HBV RNaseH was also evaluated using a molecular beacon fluorescence assay originally developed for the HIV enzyme (Chen et al., 2008). Purified HBV RNaseH (2.1 μg) was added to RNaseH buffer (50 mM HEPES pH 8.0, NaCl 100 mM, TCEP 2 mM, Tween 20 0.05%), an DNA/RNA heteroduplex substrate (25 nM), and 20 units of RNaseOut in the presence of 0 to 500 μM of the inhibitors in a final concentration of 5% DMSO in a 100 μL reaction. The substrate is a hairpin DNA oligonucleotide with a 5′ fluorescein reporter and a 3′ black hole quencher annealed to a complementary RNA oligonucleotide. The reaction was initiated by adding 5 mM Mg++, and fluorescence was monitored at 37°C with a Synergy 4 96-well plate reader. Inhibition was qualitatively determined as a dose-dependent reduction in the rate of substrate degradation.
2.4 Cells and cell culture
HepDES19 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) with 1 μg/mL tetracycline. Tetracycline was removed to induce expression of HBV. Test compounds were applied to the cells in the presence of 1% DMSO.
Vero cells were maintained in DMEM supplemented with 3% newborn calf serum, 3% bovine growth serum, 2 mM L-glutamine, and P/S. Test compounds were applied to the cells in the presence of 0.05% DMSO. The herpes simplex virus 1 (HSV-1) strain used for screening was a de-identified clinical isolate from the Saint Louis University Hospital passaged once in culture. Virus titers were determined as previously described (Knipe and Spang, 1982; Morrison and Knipe, 1996).
2.5 HBV replication inhibition assay
HBV replication inhibition was determined using HepDES19 cells as previously described (Cai et al., 2014). Briefly, HepDES19 were seeded in 12-well plates at 2 × 105 cells per well in the absence of tetracycline. Test compound was applied to cells 48 hours after removal of tetracycline. Cells were lysed 3 days after compound addition, and nonencapsidated nucleic acids were digested with micrococcal nuclease as described (Hu et al., 2013). HBV DNA was purified from capsids using a QIAamp pathogen minikit with proteinase K digestion extended to overnight at 37°C. TaqMan PCR was perfo rmed for 40 cycles with an annealing temperature of 60°C. The primers and probe (IDT Inc.) for the plus-polarity DNA strand were 5′CATGAACAAGAGATGATTAGGCAGAG3′, 5′GGAGGCTGTAGGCATAAATTGG3′, and 5′/56-FAM/CTGCGCACC/ZEN/AGCACCATGCA/3IABkFQ. The primers and probe for the minus-polarity DNA strand were 5′GCAGATGAGAAGGCACAGA3′, 5′CTTCTCCGTCTGCCGTT3′, and 5′/56-FAM/AGTCCGCGT/ZEN/AAAGAGAGGTGCG/3IABkFQ. The effective concentration 50% (EC50) values were calculated with GraphPad Prism using the four-parameter log(inhibitor)-versus-response algorithm with the bottom value set to zero.
2.6 HSV-1 replication inhibition assay
Vero cells were plated in 24-well plates and infected with HSV-1 at a multiplicity of infection (MOI) of 0.1 as previously described (Tavis et al., 2014). Compounds and virus were diluted in phosphate buffered saline (PBS) containing 2% newborn calf serum and 2 mM L-glutamine so that the final concentration of compound was 5 μM. The cells were incubated at 37°C for 1 hour with the virus-containing inoculum, then the inoculum was removed and the wells were washed once in PBS. Compound diluted to 5 μM in supplemented DMEM was added and cells were incubated at 37°C for an addit ional 23 hours. The plates were then microscopically inspected for cytopathic effect (CPE) or toxicity and then frozen at −80°C. Virus titers for wells with limited CPE compared to DMSO vehicle-treated controls were then determined by plaque assay on Vero cells. Each experiment was repeated at least once.
2.7 Cytotoxicity assays
HepDES19 cells were seeded at 1 × 104 cells per well in a 96 well plate in the absence of tetracycline. The test compounds were applied in triplicate to the cells 48 hours later and the cells were incubated for 72 hours. Cell viability was measured using the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega). [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] (MTS) and phenazine methosulfate (PMS) solution at 2 mg/mL MTS and 0.043 mg/mL PMS was added to the medium, the cells were incubated for two hours at 37°C, and absorbance was measured at 490 nm. Neutral red uptake was measured as described (Repetto et al., 2008). Neutral red was added to the media at a final concentration of 40 μg/mL. Cells were washed with PBS and incubated with neutral red medium for two hours at 37°C. The neutral red medium was removed and the cells were washed with PBS. Neutral red destain solution (50% ethanol, 49% deionized water, 1% glacial acetic acid) was added, the plate was shaken rapidly for 10 minutes, and absorbance was measured at 540 nm. Lactate dehydrogenase (LDH) release was measured using the LDH-cytotoxicity colorimetric assay kit (BioVision). Cells were lysed with 1% Triton-X to provide the high control and DMSO vehicle treatment was the low control. Supernatants were collected and transferred to a fresh 96 well plate, and LDH reaction mixture containing the catalyst and dye solution was added. The reaction was incubated for 15–20 minutes at room temperature in a dark box and absorbance was measured at 490 nm. Percent cytotoxicity was calculated as [(test sample – low control)/(high control – low control)] × 100. The cytotoxic concentration 50% (CC50) values were calculated with GraphPad Prism by using the four-parameter variable-response log(inhibitor)-versus-response algorithm with the bottom value set to zero.
2.8 Core particle isolation and endogenous polymerase reaction
HBV cores were harvested from HepDES19 cells grown in the absence of tetracycline for 20 days using polyethylene glycol 8000 precipitation (Guo et al., 2003). Cells were rinsed in PBS and lysed in 1 mL core lysis buffer with 2 μL of protease inhibitor cocktail (Sigma) at room temperature for 10 minutes. Cell lysates were collected and 10 mM CaCl2 was added. The lysate was centrifuged for 5 minutes at 21,000 g, the supernatant was incubated with 150 U of micrococcal nuclease at 37°C for 60 minutes, and then 27 mM EDTA was added to stop the reaction. 333 μL of 26% PEG8000 was added to precipitate core particles and incubated at 4°C for 6 hours. The samples were centrifuged at 21,000 g for 20 minutes at 4°C. The supernatant was removed and the pellet was resuspended in 250 μL of 10 mM Tris buffer pH 7.5. Cores were stored at −80°C.
The endogenous polymerase reaction was done using a modified procedure from (Nguyen et al., 2007). 50 μL of cores were incubated with 4 mM CaCl2 and 15 U of micrococcal nuclease at 37°C for 30 minutes. The reaction was terminated with 5.7 mM EGTA. The EPR reaction contained 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 0.1% NP-40, 0.1% β-mercaptoethanol, 150 mM NaCl, 5.7 μM each dTTP, dGTP, dATP, 1 μL of [α32P]dCTP (10 μ Ci) and test compounds at 4X EC50 or 1% DMSO as a vehicle control. The reaction was incubated at 37°C for 7 hours and terminated by adding EDTA to 10 mM. Nucleic acids were purified by incubating the cores overnight with proteinase K (0.5 mg/mL) and 1.5% SDS at 37°C, followed by phenol-chloroform extraction and ethanol precipitation. These nucleic acids were dissolved in tris-EDTA buffer, resolved on a 0.8% agarose gel, transferred to a HyBondN membrane, and detected by autoradiography. Band intensities for each experiment were quantified using Image J and percent inhibition was calculated relative to the DMSO-treated reactions. Statistical significance of variation in band intensities was evaluated using a one-way ANOVA with a Dunnett post hoc analysis, with p = 0.05 indicating significance.
2.9 Detection of capsid-associated RNA:DNA heteroduplexes
HepDES19 cells were plated in 100 mm dishes without tetracycline and incubated for 72 hours in the absence of tetracycline before addition of compound #89 at 4X EC50 (10.4 μM) and the negative control compound #138 at 30 μM; compound-containing media was replaced every three days. HBV nucleic acids were purified seven days after compound addition. HBV core particles were isolated by sedimentation through a sucrose cushion as described (Tavis et al., 1998). Cores were treated overnight with proteinase K (0.5 mg/mL) and 1.5% SDS at 37°C. Nucleic acids were purified by phenol-chloroform extraction and ethanol precipitation. The nucleic acids were split into two pools. One pool was treated with RNaseA (1.5 μg/μL) at 37°C for 30 minutes and the other was mock-treated. The nucleic acids were resolved by electrophoresis on a 0.8% agarose gel and detected by Southern analysis using a 32P-labeled full-length HBV DNA probe to detect both the plus-polarity and minus-polarity HBV DNA strands by autoradiography or phosphorimage analysis.
3. Results
3.1 Activity in the oligonucleotide-directed RNA cleavage assay
Effects on activity of purified HBV RNaseH by compounds #1, 41–45, 78–91, 138–140, and 190 (Table 1) was reported previously (Cai et al., 2014). We examined the HBV RNaseH inhibition activity of the 16 previously untested compounds using an oligonucleotide-directed cleavage assay. Compounds were incubated with the HBV RNaseH, a 32P-labeled RNA substrate plus a complementary DNA oligonucleotide and compounds at 100, 60, and 20 μM and the amount of the RNA cleavage products was measured following resolution on an acrylamide gel. Of these 16 compounds, only compound #208 inhibited RNA cleavage by the HBV RNaseH (Table 1)
3.2 HBV replication inhibition and cytotoxicity
Thirty-nine HID, HPD, flutimide, and related POH compounds (compound structures in Table 1 and Supplemental Fig. 1) were screened for their ability to inhibit HBV replication in HepDES19 cells. HepDES19 cells are HepG2 cells that carry a stably transfected HBV genotype D genome under control of a tetracycline-repressible promoter (Guo et al., 2007). The cells were treated with compound for three days and capsid-associated nucleic acids were quantified by strand-preferential quantitative PCR (q-PCR) because inhibition of the HBV RNaseH blocks positive-polarity DNA strand synthesis (Cai et al., 2014; Gerelsaikhan et al., 1996; Hu et al., 2013; Tavis et al., 2013). Inhibition was calculated as the amount of plus-polarity DNA relative to the DMSO treated controls. Qualitative screens at 20 μM identified 12 compounds that preferentially inhibited plus-polarity DNA synthesis, 11 HIDs and one HPD (Table 1). None of the flutimide or other POH compounds inhibited HBV replication. EC50 values for suppression of both the plus- and minus-polarity DNA strands were then determined for all active compounds (Table 1 and representative data in Fig. 1). All of the active compounds preferentially inhibited positive-polarity DNA accumulation.
Figure 1. Representative replication inhibition and cytotoxicity experiments.

Replication inhibition by HIDs and an HPD was measured against an HBV genotype D isolate in HepDES19 cells and plotted against compound concentration (log10[M]). Cytotoxicity was assessed by MTS, NR, and LDH assays and plotted against compound concentration (log10[M]). EC50 experiments were done with a single replicate per condition and CC50 assays were each conducted in triplicate. EC50 values were calculated based on the decline of the plus-polarity DNA strand; positive-polarity DNAs are in black; negative-polarity DNAs are in grey. EC50 and CC50 values are reported as the average of two or three independent experiments ± one standard deviation.
These preliminary hits were then screened for cytotoxicity using the MTS assay to evaluate the possibility that loss of HBV DNA in the q-PCR assay was due to cell death. CC50 values were ranged from 11 to >100 μM (Table 1). EC50 values were then calculated for the 12 hits by treating the cells replicating HBV with a wide range of compound concentrations. EC50 values ranged from 0.69 to 18 μM (Table 1), with #208 having the lowest EC50 value of 0.69 μM ± 0.2. Fig. 1 shows example CC50 and EC50 data for compounds #208, 86, 88, and 89. A therapeutic index (TI) (CC50(MTS))/EC50) comparing the CC50 calculated by MTS assay and the EC50 was determined for each compound; TI values ranged from 2.4 to 71.
To further evaluate potential cytotoxicity of these hits, we determined the effect of the compounds on uptake of neutral red (NR) by the lysosomes and lactate dehydrogenase (LDH) release into the supernatant due to cell membrane permeability. The CC50 values for the LDH assay ranged from 12 to >100 μM and the CC50 values for the NR assay were 13 to >100 μM (Table 1 and Fig. 1). Overall, the TI values indicate that cytotoxicity was not the cause of antiviral efficacy, although it may have contributed to the apparent efficacy for #84, 85, 90, and 91, which all have TI values below 5.
3.3 Activity in an improved RNaseH assay
The lack of sensitivity in the oligonucleotide directed RNA cleavage assay led us to improve the purification of the recombinant RNaseH (Villa et al., 2016) and develop an alternative RNaseH assay that employs a molecular beacon (Chen et al., 2008). In this assay, a hairpin DNA oligonucleotide labeled with fluorescein on one end and a quencher on the other is held in a linear conformation by annealing to a complementary RNA; cleavage of the RNA causes the DNA to fold into a hairpin, suppressing fluorescence (Fig. 2A). Control reactions with 0.98 μg each the wild-type HBV RNaseH and an enzymatically inactive active site mutant (D702A/E731A) validated that loss in fluorescence in these reactions is due to action of the HBV RNaseH (Fig. 2B).
Figure 2. Effects of the compounds on purified HBV RNaseH.

(Panel A) HBV RNaseH inhibition was determined using a molecular beacon RNaseH assay in which quenching of fluorescence from the RNaseH substrate is measured following removal of the RNA strand by the RNaseH, causing the folding of the DNA strand into a hairpin. (Panel B) Activity of wild-type and an active site knockout mutant (D702A/E731A) HBV RNaseHs. (Panels C to H) Fluorescence intensity from representative RNaseH reactions incubated for 60 min. in the presence of 0 to 500 μM of compounds #1, 81, 83, 138, 208, and 211.
Compounds #1, 81, 83, and 208 inhibited reactions containing 2.1 μg purified HBV RNaseH in the molecular beacon assay, and negative control compounds #138 and #211 that cannot coordinate the active site Mg++ ions had no effect on the rate of substrate degradation compared to the DMSO vehicle control (Fig 2. and Table 1). This molecular beacon RNaseH assay is more sensitive than the oligonucleotide directed RNA cleavage assay at detecting HBV replication inhibitors. However, it still underestimates the degree of inhibition compared to activity seen in cells, and hence the assay at present is used only for qualitative interpretation.
3.4 Inhibition of the human RNaseH1
Inhibition of purified huRNaseH1 by compounds #1, 41–45, 78–91, 138–140, and 190 (Table 1) was reported previously (Cai et al., 2014). We examined the huRNaseH1 inhibition activity of the previously untested compounds using an oligonucleotide-directed cleavage assay (Cai et al., 2014). Compounds were incubated with the huRNaseH1, a 32P-labeled RNA substrate plus a complementary DNA oligonucleotide and compounds at 100, 60, and 20 μM. Compounds #204, 206, 217, and 240 detectably inhibited the huRNaseH1 (Table 1).
3.5 HSV-1 replication inhibition
Thirty-seven compounds were counter-screened for efficacy against HSV-1 to evaluate specificity of the inhibitors and to help determine if the efficacy against HBV replication may be due to an unsuspected indirect effect on the cells. Vero cells infected with HSV-1 at an MOI of 0.1 were treated with 5 μM of compound for 24 hours. Inhibition was calculated as log10 suppression of plaque forming units compared with DMSO vehicle control. Of the tested compounds, only compounds #41 and 191 inhibited HSV-1 by more than 1 log10.
3.6 Effect of the compounds on HBV DNA elongation
Compounds #1 (Cai et al., 2014), 81, 83, and 208 inhibited the HBV RNaseH in biochemical assays (Table 1). However, the 8 other HBV replication inhibitors did not suppress the HBV RNaseH in the biochemical assays, leading to questions about their mechanism of action. Anti-RNaseH activity is implied from their preferential suppression of the plus-polarity DNA strand (Fig. 1), but strand-preferential inhibition could also result from inhibiting the polymerase also. This is because positive-polarity DNA synthesis can be suppressed both directly by inhibiting the RNaseH enzyme and indirectly by reducing the amount of negative-polarity DNA strand that templates the positive-polarity strand, whereas minus-polarity DNA synthesis can only be suppressed by chain termination. To determine if the efficacy of these compounds was due to inhibition of the HBV polymerase, capsids isolated from HepDES19 cells were tested in the endogenous polymerase reaction (EPR) to measure DNA chain elongation by the HBV polymerase along the HBV nucleic acids within the capsids. Isolated HBV capsids were supplied with dATP, dTTP, dGTP, and [α32P]dCTP in the presence of inhibitors at 4X their EC50 values; negative control compounds #138 and #211 were tested at 30 μM. 1% DMSO was used as a vehicle control, and ddTTP (5.7 μM) was the positive control polymerase inhibitor. Samples were incubated at 37°C for 7 hours to permit DNA chain elongation by the encapsidated HBV polymerase. Nucleic acids were purified, resolved on a 0.8% agarose gel and detected by autoradiography. DNAs from HBV capsids treated with the compounds incorporated 32P similarly to DMSO control (Fig. 3). Minor variations were observed in signal intensities among the HID and HPD treated samples, but these variations were not consistent between multiple replicate experiments and none of them were statistically significant. In contrast, the chain terminator ddTTP strongly inhibited 32P incorporation compared to the DMSO-treated samples in all assays (p ≤ 0.001). Therefore, these HIDs and HPDs do not inhibit HBV DNA polymerization inside capsids, further implicating RNaseH inhibition as their mechanism of action.
Figure 3. Effects of the compounds on HBV DNA elongation.
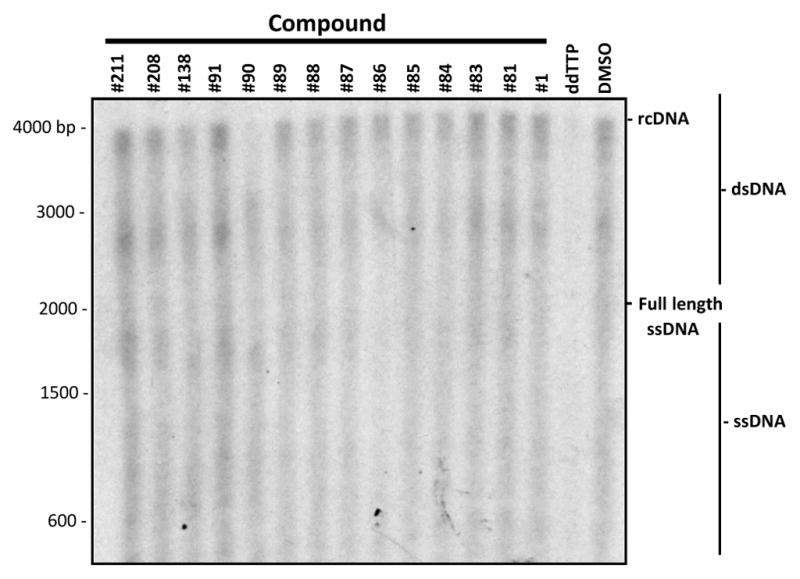
Isolated HBV capsids were incubated with radiolabeled dNTPs and compounds at 4X their EC50s to measure DNA elongation by the encapsidated viral reverse transcriptase in an EPR reactions. Nucleic acids were harvested following the EPR reaction, resolved by electrophoresis, and detected by autoradiography.
3.7 Detection of capsid-associated RNA:DNA heteroduplexes
To confirm that the strand preferential inhibition seen by the compounds was due to RNaseH inhibition, accumulation of RNA:DNA heteroduplexes in cells treated with the HID compound #89, which is inactive in both biochemical RNaseH assays, was detected by Southern blotting. Inhibiting the RNaseH blocks synthesis of the positive-polarity DNA strand by causing accumulation of RNA:DNA heteroduplexes (Cai et al., 2014; Gerelsaikhan et al., 1996; Hu et al., 2013; Tavis et al., 2013). RNA:DNA heteroduplexes migrate as double-stranded products, but degradation of the RNA strand with exogenous RNase causes the DNAs to migrate as single-stranded species. To test for accumulation of RNA:DNA heteroduplexes, HepDES19 cells replicating HBV were treated with 4X EC50 (10.4 μM) of compound #89 and 30 μM compound #138 (negative control). HBV cores were harvested after seven days of compound treatment. Capsid-associated nucleic acids were purified and split into two pools; one pool was treated with RNaseA and the other pool was mock treated. These nucleic acids were resolved by gel electrophoresis and detected by Southern analysis using a full-length double-stranded HBV 32P-labeled probe.
The DMSO vehicle-treated control revealed the expected accumulation of relaxed circular DNAs (rcDNA), shorter double-stranded species, and a smear of single-stranded products of varying lengths. Exogenous RNase treatment did not impact migration of these species (Fig. 4). Compound #138 was used as negative control as it does not inhibit HBV replication (Table 1), and migration of DNAs from cells treated with #138 was unaltered by exogenous RNase treatment. In contrast, treatment with 4X EC50 of compound #89 caused the accumulation of truncated double-stranded species that migrated below the rcDNA in the DMSO control samples, plus accumulation of a smear of single-stranded species. Treatment with exogenous RNase to remove RNA from the sample resulted in faster-migrating single-stranded DNAs (Fig. 4). This indicates that compound #89 caused RNA:DNA heteroduplexes to accumulate by inhibiting the HBV RNaseH.
Figure 4. Detection of capsid-associated RNA:DNA heteroduplexes.
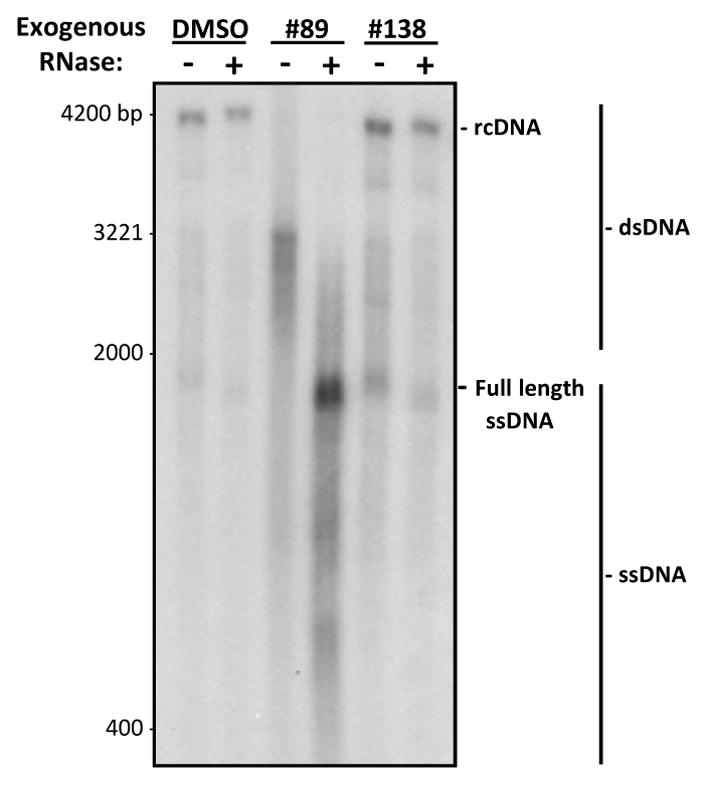
HepDES19 cells replicating HBV were treated with 4X EC50 of the HID compound #89, 30 μM of the inactive compound #138, or with DMSO as a vehicle control. HBV capsids were harvested and encapsidated nucleic acids were purified. The nucleic acids were split into two pools, one that was mock treated and the other which was treated with RNaseA. Nucleic acids were resolved by electrophoresis and HBV sequences were detected by Southern analysis. RNA:DNA heteroduplex accumulation was determined as a collapse in migration patterns in the RNaseA-treated samples. rcDNA, relaxed circular DNA; DS DNA, double-stranded DNAs; ssDNA, single-stranded DNAs.
4. Discussion
We previously reported that compound #1, an HID, inhibits both the HBV RNaseH activity and HBV genomic replication (Cai et al., 2014). We concluded that the HID scaffold was not very promising for anti-HBV drug development because only one of 11 HIDs inhibited the HBV RNaseH in the oligonucleotide-directed RNA cleavage assay. However, further evaluation of this scaffold against HBV replication revealed a lack of sensitivity in the biochemical RNaseH assay for predicting inhibitors of HBV replication. Therefore, we screened 39 HIDs, HPDs, flutimides, and related POHs for their ability to inhibit HBV replication. Here, we identified 11 new inhibitors of HBV replication, 10 HIDs and one HPD, with EC50 values ranging from 0.69 to 19 μM and TI values of 2.4 to 71.
To improve sensitivity of the RNaseH assay, we improved the recombinant RNaseH purification (Villa et al., 2016) and developed an alternative assay that employs a molecular beacon (Chen et al., 2008). Using the molecular beacon assay, we identified four compounds as RNaseH inhibitors because they reduced the rate of substrate degradation with a dose-dependent pattern. Two of these inhibitors (#81 and 83) were previously reported as negative in the oligonucleotide-directed RNA cleavage assay. Although this newer RNaseH assay is more sensitive, it still under-reports the number of HBV replication inhibitors and is currently used only for qualitative assessment.
One possible explanation for the high false negative rate in ability of the biochemical assay to predict inhibitors of HBV replication is that the compounds may be metabolized to an active form upon entry to the cell. However, this is unlikely to be the only cause because compound #208 is stable in mouse liver microsomes (unpublished data). The compounds could become concentrated in cells, causing the increased efficacy in replication inhibition assays. However, this is also unlikely to be the only cause because the concentration would need to reach at least 50 to 100-fold higher in the cells to account for the discrepancy between the two assays. Lastly, the native conformation of the RNaseH protein is likely different inside the cell when compared to the recombinant enzyme used in the biochemical assay. The RNaseH is only one domain of the multifunctional viral polymerase, which exists as a larger protein complex associated with host chaperones (Hu and Seeger, 1996). In our biochemical assay, we use a purified fragment of the enzyme that contains only the RNaseH and lacks the chaperones. Therefore, we favor the possibility that the compounds have a higher affinity for the native enzyme complex than our recombinant protein and are therefore more active inside cells.
All compounds that inhibited HBV replication were screened in the MTS assay to measure mitochondrial function and had CC50 values ranging from 11 to >100 μM (Table 1). To determine the breadth of cytotoxicity of these hits, we expanded the cytotoxicity screens to include lysosomal function using the neutral red release assay and cell membrane permeability using the LDH release assay. Those compounds with low CC50 values in the MTS assay also had significant cytotoxicity in the neutral red and LDH release assays, and compounds with little evidence of cytotoxicity in the MTS assay were also fairly nontoxic in the other two assays (Table 1 and Fig. 1). The concordance among the assays in this initial cytotoxicity panel implies that these assays may all be measuring secondary cytotoxic effects on the cells rather than primary cytotoxic events. Regardless, a good estimation of the compounds’ adverse cellular effects can be obtained from the MTS assay alone.
Specificity of these compounds was evaluated by counter-screening for activity against the huRNaseH1. The huRNaseH1 is a potential off-target host enzyme for these compounds, and inhibition of the huRNaseH1 could cause significant toxicity during the long-term drug exposure likely to be required for curative anti-HBV therapy. All 11 HIDs inhibited the huRNaseH1 in qualitative screens at 20 μM (Cai et al., 2014). However, the HPD #208, did not inhibit the huRNaseH1 at 100 μM (Table 1). These data demonstrate a need to expand selectivity for the HBV RNaseH over the huRNaseH1, but the high selectivity of #208 indicates that improved selectivity is possible.
We also counter-screened 37 compounds for efficacy against HSV-1. None of the 12 inhibitors of HBV replication inhibited HSV replication by more than 1 log10 at 5 μM (Table 1). Only compounds #41 and 191, which are both negative against HBV replication, inhibited HSV-1 by >3 log10. Previous suppression levels observed for strong HSV-1 inhibitors in this assay were >5 log10 at 5 μM (Tavis et al., 2014). This implies that there is little to no cross-reactivity of the compounds with HSV-1, and consequently increases our confidence that these compounds are not working by some unsuspected indirect effect on the cell.
HIDs have been tested extensively for activity against the HIV RNaseH and were found to be potent inhibitors in vitro against the purified enzyme (Billamboz et al., 2008; Billamboz et al., 2011a; Billamboz et al., 2011b; Billamboz et al., 2016; Billamboz et al., 2013; Desimmie et al., 2013; Hang et al., 2004; Klumpp et al., 2003; Suchaud et al., 2014). However, efficacy of these compounds against HIV replication is significantly lower (Billamboz et al., 2008; Billamboz et al., 2011a; Billamboz et al., 2011b; Billamboz et al., 2016; Billamboz et al., 2013; Chung et al., 2011; Fuji et al., 2009; Himmel et al., 2009; Kirschberg et al., 2009; Su et al., 2010; Suchaud et al., 2014). In comparison, the HIDs inhibited the HBV RNaseH in vitro poorly, but several HIDs inhibited HBV replication well in culture (Table 1). Compound #81 inhibits HIV replication with an EC50 of 13.4 μM (Billamboz et al., 2011b) and HBV replication at 4.4 μM, a 3-fold improvement. Compound #86 inhibits HIV replication at >21μM (Suchaud et al., 2014) but inhibited HBV replication at 1.4 μM, a 15-fold improvement. In general, inhibition of HBV replication by HIDs is more potent than that of HIV replication despite the HIDs being better inhibitors of HIV RNaseH in the biochemical assays. The ability of the HIDs to inhibit both HIV and HBV indicates they may be useful for the treatment of both HIV and HBV. However, it is too early to determine if development of a dual specificity inhibitor such as Tenofovir may be feasible.
Our primary screening results and EC50 curves show strong preferential inhibition of the plus-polarity DNA strand (Fig. 1), which is a hallmark of HBV RNaseH inhibition (Hu et al., 2013; Tavis et al., 2013). However, this strand preferentiality could also result from inhibition of DNA elongation by the HBV polymerase because positive-polarity DNA synthesis can be suppressed both directly by inhibiting the enzyme and indirectly by reducing the amount of negative-polarity DNA strand that templates the positive-polarity strand. To rule out the possibility that observed strand preferentiality is caused by inhibition of the HBV polymerase rather than RNaseH inhibition, DNA elongation by the native HBV polymerase within viral capsids was measured (Fig. 3). The positive control, the DNA chain terminator ddTTP, reduced DNA elongation by ~90% (p < 0.001). None of the HBV inhibitors nor the two negative control compounds (#138 and 211) significantly altered incorporation of [α32P]dCTP into the growing DNA chains. Therefore, the strand preferentiality seen in replication inhibition assays with the HID and HPD compounds is not a result of inhibiting the HBV polymerase. To confirm that an HID inhibitors can target the HBV RNaseH, we characterized the effects that compound #89 had on HBV DNAs in capsids. Despite being negative in the biochemical assay (Table 1), compound #89 prevented formation of rcDNA and caused the accumulation of RNA:DNA heteroduplexes (Fig. 4), demonstrating that it is an RNaseH inhibitor. Together, inhibition of purified HBV RNaseH by compounds #1, 81, 83, and 208 (Table 1), the preferential inhibition of the positive-polarity DNA strand by all active compounds (Fig. 1), the failure of all active compounds to inhibit elongation of DNAs by the HBV polymerase within capsids (Fig. 3), and the accumulation of RNA:DNA heteroduplexes in cultures treated with #89 indicates that the HID and HPD compounds inhibit HBV replication primarily by suppressing the viral RNaseH.
These data permit us to generate a preliminary structure activity relationship (SAR). The six-membered nitrogenous HPD ring appears to be the minimal pharmacophore because it is shared by all active compounds (Fig. 5A). Addition of a second six-membered ring bridging positions five and six of the HPD ring creates the HID scaffold. Both the HPD and HID scaffolds have an oxygen trident at positions R1-R3 which is essential for their activity. The loss of any one of these oxygens results in inactive compounds. For example, compound #204 (HID) lacks the oxygen at R2 and is inactive, and compound #211 (HPD) lacks the oxygen at R3 and is inactive (Fig. 5B). This is consistent with the known mechanism by which the HIDs inhibit the HIV RNaseH (Billamboz et al., 2008; Billamboz et al., 2011a; Billamboz et al., 2011b; Billamboz et al., 2016; Billamboz et al., 2013; Desimmie et al., 2013; Hang et al., 2004; Klumpp et al., 2003; Suchaud et al., 2014). Compounds that contain a five-membered ring (#138 and #139) are also inactive, presumably due to an inability to coordinate the Mg++ ions at the active site of the HBV RNaseH. This failure could be due to inappropriate bond angles and/or to the lack of a relatively acidic hydrogen at R4 of the HID ring. Substitutions by alkyl groups at R4 (compounds #79 and # 80) are not tolerated, whereas including a carbonyl group (ester or amide functions) at R4, which increases the acidity of the hydrogen linked to carbon 4 and therefore the ability to coordinate the Mg++ ions (Billamboz et al., 2011b), followed by large hydrophobic groups (aryl or alkaryl) is tolerated. Lastly the addition of a third ring condensed in positions R6 and R7 and a second nitrogen on the HID scaffold to create the flutimides is not tolerated (Fig. 5B). Six flutimides carrying this third ring and second nitrogen were tested and none of them were active against HBV replication or the HBV RNaseH. This preliminary SAR is being used to guide a hit-to-lead medicinal chemistry campaign.
Figure 5. Preliminary SAR.
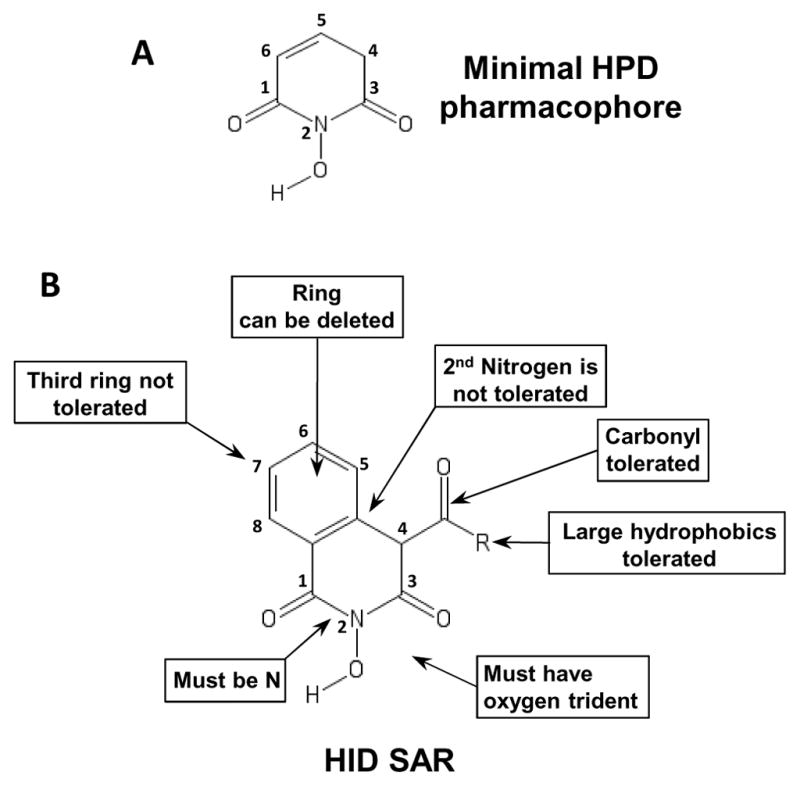
(Panel A) The minimal pharmacophore share by all active compounds. (Panel B) Preliminary SAR for the HID compounds.
These studies indicate that the HIDs are more promising than previously reported (Cai et al., 2014) and that the HPD scaffold holds potential for anti-HBV drug development. Future studies will need to focus on minimizing the cytotoxicity of these compounds while improving efficacy and improving selectivity for the HBV enzyme over huRNaseH1.
Supplementary Material
Structures of the compounds used in this study.
Highlights.
HID and HPD compounds inhibit HBV replication with low micromolar EC50 values.
HID compounds inhibit HBV replication by blocking the viral RNaseH and they do not inhibit the viral reverse transcriptase.
The HID and HPD compounds may be promising scaffolds for anti-HBV drug development.
Acknowledgments
This work was supported by grant R01 AI104494 to JET and a seed grant from the Saint Louis University Cancer Center. The funding sources had no role in the study design, data interpretation, or publication of the results. Synthesis of compounds #78-91 was financially supported by grants from le Ministère de l’Enseignement Supérieur et de la Recherche Française and l’Agence Nationale de la Recherche contre le Sida (ANRS).
We thank Nathan Ponzar, Maureen Donlin, Kaelin Bernier, and Bindi Patel for technical assistance.
Abbreviations
- HBV
hepatitis B virus
- RNaseH
ribonuclease H
- huRNaseH1
human ribonuclease H 1
- HIV
human immunodeficiency virus
- HSV-1
herpes simplex virus 1
- HID
2-hydroxyisoquinoline-1,3(2H,4H)-diones
- HPD
N-hydroxypyridinediones
- POH
polyoxygenated heterocycles
- FLT
flutimides
- DMSO
dimethyl sulfoxide
- PBS
phosphate buffered saline
- IC50
50% inhibitory concentration
- EC50
50% effective concentration
- CC50
50% cytotoxic concentration
- TI
therapeutic index
- SAR
structure–activity relationship
- DMEM
Dulbecco’s modified Eagle’s medium
- P/S
penicillin streptomycin
- FBS
fetal bovine serum
- MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt]
- PMS
phenazine methosulfate
- LDH
lactate dehydrogenase
- NR
neutral red
- MOI
multiplicity of infection
- rcDNA
relaxed circular DNA
- q-PCR
quantitative polymerase chain reaction
Footnotes
Conflict of interest
JET, FB, and PC are inventors on patent applications covering the use of the HIDs and HPDs for therapeutic uses as HBV RNaseH inhibitors, and JET receives research support from Arbutus BioPharma. LM and JET are inventors on patent applications covering the use of compounds #191 and #41 against HSV-1.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Billamboz M, Bailly F, Barreca ML, De LL, Mouscadet JF, Calmels C, Andreola ML, Witvrouw M, Christ F, Debyser Z, Cotelle P. Design, synthesis, and biological evaluation of a series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. J Med Chem. 2008;51:7717–7730. doi: 10.1021/jm8007085. [DOI] [PubMed] [Google Scholar]
- Billamboz M, Bailly F, Lion C, Calmels C, Andreola ML, Witvrouw M, Christ F, Debyser Z, De Luca L, Chimirri A, Cotelle P. 2-hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: influence of the alkylation of position 4. European journal of medicinal chemistry. 2011a;46:535–546. doi: 10.1016/j.ejmech.2010.11.033. [DOI] [PubMed] [Google Scholar]
- Billamboz M, Bailly F, Lion C, Touati N, Vezin H, Calmels C, Andreola ML, Christ F, Debyser Z, Cotelle P. Magnesium chelating 2-hydroxyisoquinoline-1,3(2H,4H)-diones, as inhibitors of HIV-1 integrase and/or the HIV-1 reverse transcriptase ribonuclease H domain: discovery of a novel selective inhibitor of the ribonuclease H function. J Med Chem. 2011b;54:1812–1824. doi: 10.1021/jm1014692. [DOI] [PubMed] [Google Scholar]
- Billamboz M, Suchaud V, Bailly F, Lion C, Andreola ML, Christ F, Debyser Z, Cotelle P. 2-hydroxyisoquinoline-1, 3 (2H, 4H)-diones (HIDs) as human immunodeficiency virus type 1 integrase inhibitors: Influence of the alkylcarboxamide substitution of position 4. European journal of medicinal chemistry. 2016;117:256–268. doi: 10.1016/j.ejmech.2016.03.083. [DOI] [PubMed] [Google Scholar]
- Billamboz M, Suchaud V, Bailly F, Lion C, Demeulemeester J, Calmels C, Andreola ML, Christ F, Debyser Z, Cotelle P. 4-Substituted 2-hydroxyisoquinoline-1,3(2H,4H)-diones, as a novel class of HIV-1 integrase inhibitors. ACS Med Chem Lett. 2013;4:606–611. doi: 10.1021/ml400009t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai CW, Lomonosova E, Moran EA, Cheng X, Patel KB, Bailly F, Cotelle P, Meyers MJ, Tavis JE. Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res. 2014;108:48–55. doi: 10.1016/j.antiviral.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang CJ, Wu Y, Conlon P, Kim Y, Lin H, Tan W. Light-Switching Excimer Beacon Assays For Ribonuclease H Kinetic Study. ChemBioChem. 2008;9:355–359. doi: 10.1002/cbic.200700542. [DOI] [PubMed] [Google Scholar]
- Chung S, Himmel DM, Jiang JK, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SF. Synthesis, activity, and structural analysis of novel alpha-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. Journal of medicinal chemistry. 2011;54:4462–4473. doi: 10.1021/jm2000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox N, Tillmann H. Emerging pipeline drugs for hepatitis B infection. Expert Opin Emerg Drugs. 2011;16:713–729. doi: 10.1517/14728214.2011.646260. [DOI] [PubMed] [Google Scholar]
- Desimmie BA, Demeulemeester J, Suchaud V, Taltynov O, Billamboz M, Lion C, Bailly F, Strelkov SV, Debyser Z, Cotelle P, Christ F. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones (HIDs), novel inhibitors of HIV integrase with a high barrier to resistance. ACS chemical biology. 2013;8:1187–1194. doi: 10.1021/cb4000426. [DOI] [PubMed] [Google Scholar]
- Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- Fuji H, Urano E, Futahashi Y, Hamatake M, Tatsumi J, Hoshino T, Morikawa Y, Yamamoto N, Komano J. Derivatives of 5-nitro-furan-2-carboxylic acid carbamoylmethyl ester inhibit RNase H activity associated with HIV-1 reverse transcriptase. J Med Chem. 2009;52:1380–1387. doi: 10.1021/jm801071m. [DOI] [PubMed] [Google Scholar]
- Gerelsaikhan T, Tavis JE, Bruss V. Hepatitis B Virus Nucleocapsid Envelopment Does Not Occur without Genomic DNA Synthesis. J Virol. 1996;70:4269–4274. doi: 10.1128/jvi.70.7.4269-4274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol. 2007;81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. Conditional replication of duck hepatitis B virus in hepatoma cells. Journal of virology. 2003;77:1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang JQ, Rajendran S, Yang Y, Li Y, In PW, Overton H, Parkes KE, Cammack N, Martin JA, Klumpp K. Activity of the isolated HIV RNase H domain and specific inhibition by N-hydroxyimides. Biochem Biophys Res Commun. 2004;317:321–329. doi: 10.1016/j.bbrc.2004.03.061. [DOI] [PubMed] [Google Scholar]
- Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, Clark AD, Jr, Ryan K, Hickey MJ, Love RA, Hughes SH, Bergqvist S, Arnold E. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure. 2009;17:1625–1635. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Seeger C. Expression and Characterization of Hepadnavirus Reverse Transcriptases. Methods in Enzymology. 1996;275:195–208. doi: 10.1016/s0076-6879(96)75013-9. [DOI] [PubMed] [Google Scholar]
- Hu Y, Cheng X, Cao F, Huang A, Tavis JE. beta-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res. 2013;99:221–229. doi: 10.1016/j.antiviral.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Kirschberg TA, Balakrishnan M, Squires NH, Barnes T, Brendza KM, Chen X, Eisenberg EJ, Jin W, Kutty N, Leavitt S, Liclican A, Liu Q, Liu X, Mak J, Perry JK, Wang M, Watkins WJ, Lansdon EB. RNase H active site inhibitors of human immunodeficiency virus type 1 reverse transcriptase: design, biochemical activity, and structural information. J Med Chem. 2009;52:5781–5784. doi: 10.1021/jm900597q. [DOI] [PubMed] [Google Scholar]
- Klumpp K, Hang JQ, Rajendran S, Yang Y, Derosier A, Wong KI, Overton H, Parkes KE, Cammack N, Martin JA. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003;31:6852–6859. doi: 10.1093/nar/gkg881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Spang AE. Definition of a series of stages in the association of two herpesviral proteins with the cell nucleus. Journal of virology. 1982;43:314–324. doi: 10.1128/jvi.43.1.314-324.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H, Lok AS. Hepatitis B therapy. Nat Rev Gastroenterol Hepatol. 2011;8:275–284. doi: 10.1038/nrgastro.2011.33. [DOI] [PubMed] [Google Scholar]
- Lima WF, Wu H, Crooke ST. Human RNases H. Methods Enzymol. 2001;341:430–440. doi: 10.1016/s0076-6879(01)41168-2. [DOI] [PubMed] [Google Scholar]
- Lu G, Lomonosova E, Cheng X, Moran EA, Meyers MJ, Le Grice SF, Thomas CJ, Jiang JK, Meck C, Hirsch DR, D’Erasmo MP, Suyabatmaz DM, Murelli RP, Tavis JE. Hydroxylated Tropolones Inhibit Hepatitis B Virus Replication by Blocking the Viral Ribonuclease H Activity. Antimicrob Agents Chemother. 2015;59:1070–1079. doi: 10.1128/AAC.04617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellin P, Heathcote EJ, Buti M, Gane E, De Man RA, Krastev Z, Germanidis G, Lee SS, Flisiak R, Kaita K, Manns M, Kotzev I, Tchernev K, Buggisch P, Weilert F, Kurdas OO, Shiffman ML, Trinh H, Washington MK, Sorbel J, Anderson J, Snow-Lampart A, Mondou E, Quinn J, Rousseau F. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359:2442–2455. doi: 10.1056/NEJMoa0802878. [DOI] [PubMed] [Google Scholar]
- Morrison LA, Knipe DM. Mechanisms of immunization with a replication-defective mutant of herpes simplex virus 1. Virology. 1996;220:402–413. doi: 10.1006/viro.1996.0328. [DOI] [PubMed] [Google Scholar]
- Nguyen DH, Gummuluru S, Hu J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. Journal of virology. 2007;81:4465–4472. doi: 10.1128/JVI.02510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M. Retroviral integrase superfamily: the structural perspective. EMBO Rep. 2009;10:144–151. doi: 10.1038/embor.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nature protocols. 2008;3:1125–1131. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- Seeger C, Zoulim F, Mason WS. Hepadnaviruses. In: Knipe DM, Howley P, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2977–3029. [Google Scholar]
- Seeger C, Zoulim F, Mason WS. Hepadnaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6. Lippincott Williams & Wilkins; Philadelphia PA: 2013. pp. 2185–2221. HBV Ribonuclease H inhibition by N-hydroxyisoquinolinediones. [Google Scholar]
- Sorrell MF, Belongia EA, Costa J, Gareen IF, Grem JL, Inadomi JM, Kern ER, McHugh JA, Petersen GM, Rein MF, Strader DB, Trotter HT. National Institutes of Health Consensus Development Conference Statement: management of hepatitis B. Ann Intern Med. 2009;150:104–110. doi: 10.7326/0003-4819-150-2-200901200-00100. [DOI] [PubMed] [Google Scholar]
- Su HP, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J Virol. 2010;84:7625–7633. doi: 10.1128/JVI.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchaud V, Bailly F, Lion C, Calmels C, Andreola ML, Christ F, Debyser Z, Cotelle P. Investigation of a novel series of 2-hydroxyisoquinoline-1,3-(2H,4H)-diones (HIDs) as human immunodeficiency virus type 1 integrase inhibitors. J Med Chem. 2014;57:4640–4660. doi: 10.1021/jm500109z. [DOI] [PubMed] [Google Scholar]
- Summers J, Mason WS. Replication of the Genome of a Hepatitis B-Like Virus by Reverse Transcription of an RNA Intermediate. Cell. 1982;29:403–415. doi: 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- Tavis JE, Badtke MP. Hepadnaviral Genomic Replication. In: Cameron CE, Götte M, Raney KD, editors. Viral Genome Replication. Springer Science+Business Media, LLC; New York: 2009. pp. 129–143. [Google Scholar]
- Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. The hepatitis B virus ribonuclease h is sensitive to inhibitors of the human immunodeficiency virus ribonuclease h and integrase enzymes. PLoS pathogens. 2013;9:e1003125. doi: 10.1371/journal.ppat.1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Massey B, Gong Y. The Duck Hepatitis B Virus Polymerase Is Activated by Its RNA Packaging Signal, Epsilon. J Virol. 1998;72:5789–5796. doi: 10.1128/jvi.72.7.5789-5796.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Wang H, Tollefson AE, Ying B, Korom M, Cheng X, Cao F, Davis KL, Wold WS, Morrison LA. Inhibitors of nucleotidyl transferase superfamily enzymes suppress herpes simplex virus replication. Antimicrob Agents Chemother. 2014;58:7451–7461. doi: 10.1128/AAC.03875-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trépo C, Chan HL, Lok A. Hepatitis B virus infection. The Lancet. 2014;384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- van Bommel F, De Man RA, Wedemeyer H, Deterding K, Petersen J, Buggisch P, Erhardt A, Huppe D, Stein K, Trojan J, Sarrazin C, Bocher WO, Spengler U, Wasmuth HE, Reinders JG, Moller B, Rhode P, Feucht HH, Wiedenmann B, Berg T. Long-term efficacy of tenofovir monotherapy for hepatitis B virus-monoinfected patients after failure of nucleoside/nucleotide analogues. Hepatology. 2010;51:73–80. doi: 10.1002/hep.23246. [DOI] [PubMed] [Google Scholar]
- Villa JA, Pike DP, Patel KB, Lomonosova E, Lu G, Abdulqader R, Tavis JE. Purification and enzymatic characterization of the hepatitis B virus ribonuclease H, a new target for antiviral inhibitors. Antiviral Research. 2016;132:186–195. doi: 10.1016/j.antiviral.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Steitz TA. Recombining the structures of HIV integrase, RuvC and RNase H. Structure. 1995;3:131–134. doi: 10.1016/s0969-2126(01)00142-3. [DOI] [PubMed] [Google Scholar]
- Zoidis G, Giannakopoulou E, Stevaert A, Frakolaki E, Myrianthopoulos V, Fytas G, Mavromara P, Mikros E, Bartenschlager R, Vassilaki N. Novel indole–flutimide heterocycles with activity against influenza PA endonuclease and hepatitis C virus. MedChemComm. 2016;7:447–456. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structures of the compounds used in this study.