

Abstract
The total synthesis of the natural stilbene (+)-schweinfurthin G (8) has been accomplished through a sequence based on an efficient cationic cascade cyclization. This cascade process is initiated by Lewis acid promoted ring opening of an epoxide and terminated through a novel reaction with a phenolic oxygen “protected” as its MOM ether. Several Lewis acids have been examined for their ability to induce this new reaction, and BF3·Et2O was found to be the most effective. The only major by-product under these conditions was one where the expected secondary alcohol was found as its MOM ether derivative (e.g. 30). While this by-product could be converted to the original target compound through hydrolysis, it also could be employed as a protected alcohol to allow preparation of a benzylic phosphonate (43) without dehydration of the secondary alcohol. The resulting phosphonate was employed in a Horner-Wadsworth-Emmons condensation with an aldehyde representing the right half of the target compounds, an approach complementary to previous studies based on condensation of a right half phosphonate and a left half aldehyde.
Graphical Abstract
Introduction
Carbon–carbon bond formation is the central operation of organic synthesis, and reactions that accomplish this objective while bringing added complexity to the reaction product are particularly valuable. Consideration of the range of natural products generated in plants, animals, and fungi through cationic cascade cyclizations makes clear that this process has abundant potential.1 However, to date synthetic chemistry based on cationic cascades has been able to recapture only a fraction of the varied structures produced through biosynthesis and even the “successful” reactions often afford complex mixtures and modest yields. The history of cationic cascade cyclizations is extensive with much attention directed towards transformations of epoxy-enes.2 There is considerably less precedence for cationic cascade cyclizations terminated by phenolic nucleophiles. Previous work in this particular arena utilized harsh conditions to achieve cyclization and often suffered from low yields.3
Our interest in cationic cascade cyclizations originated in an effort to prepare some natural hexahydroxanthenes. The tetracyclic schweinfurthins A (1) and B (2), along with a simpler stilbene named schweinfurthin C (3), were isolated from the plant Macaranga schweinfurthii and were reported to have an interesting profile of anti-cancer activity (Figure 1).4 A short time later, a fourth member of this family with a hydrated terminal olefin in the geranyl chain was isolated from the same plant and named schweinfurthin D (4).5 The tetracyclic schweinfurthins are closely related to the natural product vedelianin (5), which bears a prenyl group on the D–ring instead of the geranyl group of the first schweinfurthins and was isolated from M. vedeliana.6 Very recently several new hexahydroxanthene natural products that bear D-ring prenyl groups were isolated from M. alnifolia and named as schweinfurthins E–H (6–9).7
Figure 1.

Structures of the natural schweinfurthins and vedelianin.
While the biosynthesis of the tetracyclic schweinfurthins has not been studied, both the structure of the hexahydroxanthene system and the co-occurrence of schweinfurthin A and B with schweinfurthin C strongly suggest a sequence that includes a cationic cascade cyclization. A process initiated through opening of an epoxide and terminated by reaction with a phenol would easily explain the A/B/C-ring system of schweinfurthin F and G, while a more oxidized precursor could lead to the A-ring diol schweinfurthins through a parallel process. From the standpoint of chemical synthesis, a biomimetic route to the schweinfurthins would be attractive but past examples of cascade cyclizations have emphasized use of olefins, arenes, and hydroxyl groups for termination of the cascades.2 Phenols or protected forms of them have been less studied,3,8 but may still offer great opportunity because they may be reasonably stable to the reaction conditions and yet serve as potent nucleophiles. The available precedents for cascade cyclizations of MOM-protected phenols were not encouraging. A paper from Kitahara and coworkers9a reported treatment of an epoxide bearing a remote MOM ether with picric acid and then with methanolic HCl. The desired cyclization product was obtained, but this compound was isolated in just 2% yield and the loss of a second MOM ether under the reaction conditions makes it unclear if MOM cleavage occurred prior to or during the cyclization. A more recent study of the same substrate was unable to effect cyclization with either protic or Lewis acids, and found instead only inseparable mixtures.9b There is a precedent for chromane formation through a cyclization of an epoxide with a MOM-protected phenol,10 but whether this process could be extended to formation of a hexahydroxanthene was not known. In this paper, we describe the synthesis of two schweinfurthins, F (7) and G (8), and two schweinfurthin analogues, 3-deoxyschweinfurthin A (3dSA, 10, Figure 2) and 3-deoxyschweinfurthin B (3dSB, 11), via efficient cascade cyclizations mediated by BF3·Et2O.
Figure 2.

Structures of schweinfurthin analogues.
Results and Discussion
Our general, convergent approach to the schweinfurthins (12) takes advantage of a late stage Horner–Wadsworth–Emmons (HWE) condensation to provide the central stilbene as the penultimate step (Scheme 1). The utility of this strategy was demonstrated first in the synthesis of schweinfurthin C (3).11 The parallel coupling of a “left–half” aldehyde (13) and a “right–half” phosphonate (14) at this stage offers the opportunity to use common precursors in the synthesis of several schweinfurthins.11 The “left–half” aldehydes were envisioned to arise from phenol terminated cationic cascade cyclizations of enantiopure epoxides 15. The intermediate epoxides were expected to be available through Shi epoxidation12 of a geranyl derivative (16) of commercially available vanillin (17) or through Shi epoxidation of a geranyl ester and alkylation of a vanillin derivative (18).13 Several “right–half” phosphonates appropriate for this strategy are known from our previous work.11, 14, 15
Scheme 1.

Retrosynthetic analysis of schweinfurthins.
The current studies began with the known compound bromovanillin (18), available through direct bromination of commercially available vanillin (17), and readily converted to the geranylated epoxide 19 (Scheme 2).13 Vanillin is an especially attractive starting material for schweinfurthins with a C–ring methoxy group (e.g. schweinfurthin F), and conversion of epoxide 19 to an intermediate hexahydroxanthene has been reported using a Brönsted acid to induce cyclization.16 While that approach allowed preparation of some key targets, in the best cases the acid-catalyzed cyclization proceeds in just 30–40% yield. A Lewis acid mediated cascade cyclization would be attractive if it allowed use of milder reaction conditions and proceeded in better yield.
Scheme 2.

Synthesis of an epoxide for preparation of schweinfurthin F.
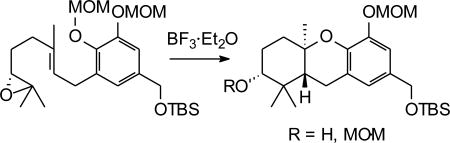
Studies on the use of Lewis acids to induce a cascade reaction were performed with epoxide 19 (Figure 3) to determine the conditions necessary to enact cyclization and formation of the desired tricycle (Table 1). Several Lewis acids were examined with varying conditions of reaction times and temperatures. Upon treatment with Ti(OiPr)4 no reaction was detected (Trials 1–3) and the starting epoxide could be recovered unchanged, suggesting that this Lewis acid was unable to enact cyclization in this system. In contrast, upon treatment with TiCl4 (Trial 4) or SnCl4 (Trial 5) no isolable compounds were obtained and no starting materials could be recovered.17 A reaction with MeAlCl2, which has been used in a number of other cationic cascades,18 gave the desired products in modest yield (Trial 6). The use of In(OTf)3 gave more promising results (Trials 7–10), including formation of significant amounts of the desired tricycle and merits further evaluation as a reagent to promote these cascade cyclizations.19 However, reactions with BF3·Et2O were inexpensive, convenient, and easily amenable to larger scales. They appeared to have great promise in providing tricycle 20, and thus this Lewis acid became our immediate focus.20 Even very brief exposure to this Lewis acid at low temperature (Trial 11) gave a yield comparable to the best obtained from reactions with protic acids. At slightly longer times (Trials 12 and 13), the yield of the tricyclic alcohol 20 improved to 64–68% and the major by-product was the tricyclic compound with an A-ring methoxymethyl (MOM) ether 21. Thus the total yield of hexahydroxanthene was ~75% under these conditions, or nearly double the best reported from the protic acid catalyzed reactions in similar systems.16 Further increases in reaction time did not lead to improved yields (Trials 14 and 15).
Figure 3.

Lewis acid mediated cyclization of epoxide 19.
Table 1.
Attempted cyclizations of epoxide 19.
| Trial # | Acid | Time (min.) | T (°C) | Distribution (by gc vs int std) |
||
|---|---|---|---|---|---|---|
| 19 | 20 | 21 | ||||
| 1 | Ti(OiPr)4 | 5 | −78 | 100 | 0 | 0 |
| 2 | Ti(OiPr)4 | 5 | rt | 100 | 0 | 0 |
| 3 | Ti(OiPr)4 | 1.5 | rt | 100 | 0 | 0 |
| 4 | TiCl4 | 5 | −78 | 0 | 0 | 0 |
| 5 | SnCl4 | 5 | −78 | 0 | 0 | 0 |
| 6 | CH3AlCl2 | 30 | −78 | 0 | 17a | 8a |
| 7 | In(OTf)3 | 1 | rt | 0 | 65 | 0 |
| 8 | In(OTf)3 | 5 | −78 | 55 | 29 | 0 |
| 9 | In(OTf)3 | 5 | 0 | 0 | 60 | 2 |
| 10 | In(OTf)3 | 8 | −35 | 14 | 36 | 3 |
| 11 | BF3·Et2O | 0.5 | −78 | 0 | 44 | 6 |
| 12 | BF3·Et2O | 1 | −78 | 0 | 64 | 11 |
| 13 | BF3·Et2O | 2 | −78 | 0 | 68a | 7a |
| 14 | BF3·Et2O | 4 | −78 | 0 | 49a | 11a |
| 15 | BF3·Et2O | 8 | −78 | 0 | 51 | 15 |
Isolated yields following column chromatography.
To determine if these conditions could be applied to other substrates, and to gain access to intermediates for more facile synthesis of hexahydroxanthenes with a C-ring phenol, aromatic compounds with two MOM groups were sought. Our synthesis of schweinfurthin G (8) and its analogue 3dSA (10) proceeded from bromovanillin (18) through treatment with aluminum (III) chloride as a standard demethylation21 to provide catechol 22 (Scheme 3). Protection of compound 22 as its MOM ether (23) followed by reduction of the aldehyde afforded the known benzylic alcohol 24.11 The resultant alcohol was masked as the silyl ether to give differentially protected bromide 25. Treatment of bromide 25 with n–BuLi enacted halogen metal exchange and subsequent reaction with geranyl bromide installed the geranyl chain to provide compound 26 in 86% yield. A Shi epoxidation12 of geranylated arene 26 was observed upon treatment with hydrogen peroxide in the presence of sugar catalyst 27 and acetonitrile in buffered solution to afford epoxide 28 in a modest yield but excellent enantioselectivity (vide infra). In addition this application of the Shi epoxidation generally results in recovery of 33–54% of the starting olefin so yields based on recovered starting material are very attractive. With epoxide 28 in hand, the stage was set for Lewis–acid mediated cascade cyclizations. Brief treatment with BF3·Et2O afforded tricycle 29 in a 52% yield along with tricycle 30 in 30% yield, representing very efficient formation of the tricyclic system. Removal of the silyl protecting group of tricycle 29 was successful using standard conditions and oxidation gave the new “left-half” aldehyde 32.
Scheme 3.

Synthesis of a C-5 MOM ether and its BF3·Et2O mediated cyclization.
Formation of the A-ring MOM ethers as by-products of the cyclization of both epoxides 19 and 28 was not expected. These products can be readily explained by formation of a formal CH3OCH2+ cation during the course of cyclization, and ultimate reaction of this cation with the nucleophilic oxygen of the C-2 hydroxyl group. However, the corresponding by-product from reaction of such a cation at the benzylic oxygen was not observed, so a more complex mechanism may be at play and further studies will be necessary to clarify this process.
Completion of the target compounds proceeded in a straightforward manner. Aldehyde 32 was employed for synthesis of schweinfurthin G (8) and 3dSA (10). In the first case, an HWE condensation of aldehyde 32 and known phosphonate 3314 provided the penultimate compound as stilbene 34. Acidic hydrolysis of the three MOM ethers yielded schweinfurthin G (8) in 57% yield (Scheme 4). This route represents the first total synthesis of schweinfurthin G and spectral data matched that of the isolated natural product. Analysis of the product by HPLC indicated an ee of ~94%, originating in the Shi epoxidation. A similar strategy was used for the synthesis of 3–deoxyschweinfurthin A (10). For this target aldehyde 32 underwent a smooth HWE condensation with known phosphonate 3511 to afford stilbene 36. Treatment of compound 36 with TsOH resulted in cleavage of the three MOM groups and provided the new schweinfurthin analogue 10 in a modest yield. Finally, standard cleavage of the silyl protecting group of tricycle 20 followed by MnO2 oxidation to the corresponding aldehyde represents a formal synthesis of both schweinfurthin F (7)12 and 3dSB (11).16 Synthesis of 3dSB through this BF3·Et2O mediated cyclization shows consistent transmission of stereochemical integrity from the epoxide through the cyclization, in parallel with our work using protic acids.16
Scheme 4.

Synthesis of schweinfurthin G and 3dSA.
In principle, the A-ring MOM compounds 21 and 30 could be hydrolyzed to the corresponding alcohols 20 and 29, and thus recycled into the desired stilbenes. Instead we chose to take advantage of these compounds as protected A-ring alcohols and use them in a different manner. Accordingly, compound 21 was treated with TBAF to afford the benzylic alcohol 37, and this alcohol was converted to the corresponding phosphonate through sequential treatment with mesyl chloride, NaI, and triethyl phosphite. In contrast, if the A-ring alcohol is left unprotected, attempted reactions with triethyl phosphite resulted in dehydration. An HWE condensation of phosphonate 38 with aldehyde 3922 (Scheme 5), readily available by MnO2 oxidation of the corresponding benzylic alcohol,11 gave the desired stilbene 40 in a modest yield (Scheme 5). Final hydrolysis of the three MOM groups by treatment with TsOH in methanol gave the desired 3dSB (11). Through a parallel sequence, compound 30 was converted to the benzylic alcohol 41 and then to phosphonate 42. In this case, the HWE condensation with aldehyde 39 gave stilbene 43 in a good yield, and final deprotection of the four MOM groups gave 3dSA (10) in 52% yield, along with a partial hydrolysis product (44) that retained the A-ring MOM group (33%). These experiments show that the adventitious formation of the A-ring MOM compound allows preparation of the benzylic phosphonate, and that condensation of this phosphonate “left half” with an aldehyde “right half” is a viable alternative to the reverse strategy (i.e. a phosphonate “right half” with an aldehyde “left half”) that has been employed in all previous studies. Although the yields have not been optimized and currently are somewhat lower for this approach than for the original strategy, this allows attractive flexibility in distribution of the number of steps between the two branches of the convergent synthesis.
Scheme 5.

Synthesis of hexahydroxanthene phosphonates en route to stilbenes.
In conclusion, the scope of Lewis acid mediated cascade cyclizations has been expanded to include two examples of efficient epoxy-ene cyclizations mediated by brief treatment with BF3·Et2O and terminated by reaction with a MOM-protected phenol. These cyclizations were used to provide the hexahydroxanthene core necessary for the synthesis of two schweinfurthin natural products (schweinfurthins F and G), and two parallel schweinfurthin analogues (3dSA and 3dSB). Further application of the use of Lewis acid mediated cascade cyclizations, particularly those terminated by oxygen nucleophiles, will be reported in due course.
Experimental Section
Benzylic silyl ethers 20 and 21
To a solution of epoxide 19 (320 mg, 0.66 mmol) in CH2Cl2 was added BF3·Et2O (0.35 mL, 2.8 mmol) at −78 °C. The resulting solution was allowed to stir 16 minutes and triethylamine (0.5 mL, 4 mmol) was added. After an additional 10 minutes, water (75 mL) was added, the phases were separated, and the aqueous phase was extracted with CH2Cl2. The combined organic phase was washed with water and brine, then dried (MgSO4), and concentrated. Final purification by flash chromatography (15:1 to 3:1 hexanes:EtOAc) afforded the tricyclic alcohol 20 (132 mg, 48%) as a clear oil with spectral data matching previously reported data.13,16 Also isolated as a clear oil (69 mg, 22%) was the A-ring methoxymethylether 21: [α]26.4D = +8.9° (c 0.03, CHCl3); 1H NMR δ 6.70 (s, 1H), 6.64 (s, 1H), 4.77 (d, J = 6.6 Hz, 1H), 4.64 (d, J = 6.6 Hz, 1H), 4.64 (s, 2H), 3.83 (s, 3H), 3.41 (s, 3H), 3.27 (dd, J = 11.3, 4.0 Hz, 1H), 2.70–2.67 (m, 2H), 2.13 (dt, J = 12.7, 3.2 Hz, 1H), 1.97 (dq, J = 13.0, 3.7 Hz, 1H), 1.85–1.52 (m, 3H), 1.24 (s, 3H), 1.08 (s, 3H), 0.95 (s, 9H), 0.90 (s, 3H), 0.11 (s, 6H); 13C NMR δ 148.6, 141.4, 132.3, 122.2, 119.0, 107.4, 96.1, 84.0, 76.4, 64.9, 55.8, 55.8, 47.0, 38.3, 37.5, 27.4, 26.0 (3C), 25.2, 23.1, 19.4, 18.4, 15.1, –5.2(2C); HRMS (EI+) m/z calcd for C26H44O5Si (M+) 464.2958, found 464.2944.
5–((2E)–3,7–Dimethyl–6–epoxy–octa–2–enyl) –3,4–bis(O-methoxymethyl)benzyloxy–tert–butyldimethylsilane (28)
To a solution of olefin 26 (689 mg, 1.4 mmol) in acetonitrile (0.4 mL), n–butanol (10 mL) and a buffer solution (5 mL, 2 M K2CO3, 0.4 mM EDTA) at 0 °C was added the sugar catalyst 27 (95 mg, 0.4 mmol). Hydrogen peroxide (0.85 mL, 30% solution, 8.3 mmol) was added dropwise to the cooled mixture and the reaction was stirred for 22 h. The reaction was quenched by addition of Na2SO3 and extracted with EtOAc. The combined organic extracts were washed with H2O and brine, dried (MgSO4), and concentrated in vacuo to afford a pale liquid. Final purification by flash column chromatography (8% EtOAc in hexanes) provided epoxide 28 (277 mg, 39%) as a colorless liquid: [α]26.4D = +0.56° (c 0.11, CHCl3); 1H NMR δ 6.98 (d, J = 1.8 Hz, 1H), 6.78 (d, J = 1.5 Hz, 1H), 5.36 (t, J = 7.2 Hz, 1H), 5.17 (s, 2H), 5.08 (s, 2H), 4.64 (s, 2H), 3.59 (s, 3H), 3.49 (s, 3H), 3.41 (d, J = 6.9 Hz, 2H), 2.71 (t, J = 6.3 Hz, 1H), 2.20–2.05 (m, 2H), 1.73 (s, 3H), 1.70–1.60 (m, 2H), 1.27 (s, 3H), 1.25 (s, 3H), 0.92 (s, 9H), 0.09 (s, 6H); 13C NMR δ 149.7, 143.6, 137.6, 135.5, 135.4, 123.3, 120.5, 112.2, 99.2, 95.3, 64.8, 64.3, 58.4, 57.6, 56.2, 36.5, 28.6, 27.6, 26.0 (3C), 25.0, 18.9, 18.5, 16.3, –5.1 (2C). Anal. Calcd for C27H46O6Si · ½ H2O: C, 64.38; H, 9.40. Found: C, 64.48; H, 9.35.
Tricycles 29 and 30
To a solution of epoxide 28 (355 mg, 0.7 mmol) in CH2Cl2 (60 mL) at –78 °C was added BF3·Et2O (0.36 mL, 2.8 mmol) dropwise. The reaction was stirred for 3.5 minutes and quenched by addition of triethylamine, treated with NH4CL (sat.), and extracted with CH2Cl2. The combined organics were washed with H2O and brine, dried (MgSO4), and concentrated in vacuo to give a yellow liquid. Final purification by flash column chromatography (15% EtOAc in hexanes) provided tricycles 29 (163 mg, 52%) and 30 (108 mg, 30%) as colorless oils. For compound 29: [α]26.4D = +38.7° (c 0.02, CHCl3); 1H NMR δ 6.91 (s, 1H), 6.75 (s, 1H), 5.17 (d, J = 6.6 Hz, 1H), 5.13 (d, J = 6.3 Hz, 1H), 4.61 (s, 2H), 3.50 (s, 3H), 3.39 (dd, J = 11.7, 3.1 Hz, 1H), 2.71 (s, 1H), 2.70–2.66 (m, 1H), 2.10–2.00 (m, 1H), 1.80–1.65 (m, 5H), 1.22 (s, 3H), 1.09 (s, 3H), 0.94 (s, 9H), 0.87 (s, 3H), 0.10 (s, 6H); 13C NMR δ 145.9, 143.1, 134.7, 123.1, 121.5, 114.4, 96.2, 78.3, 76.7, 65.0, 56.3, 47.0, 38.6, 38.0, 28.5, 27.6, 26.3 (3C), 23.5, 20.1, 18.7, 14.5, –4.9 (2C); HRMS (EI+) m/z calcd for C25H42O5Si (M+) 450.2802, found 450.2809. For compound 30: [α]26.4D = +9.7° (c 0.05, CHCl3); 1H NMR δ 6.91 (d, J = 1.8 Hz, 1H), 6.74 (d, J = 1.8 Hz, 1H), 5.17 (d, J = 6.6 Hz, 1H), 5.13 (d, J = 6.3 Hz, 1H), 4.77 (d, J = 6.9 Hz, 1H), 4.64 (d, J = 6.9 Hz, 1H), 4.62 (s, 2H), 3.50 (s, 3H), 3.41 (s, 3H), 3.25 (dd, J = 11.7, 4.2 Hz, 1H), 2.71 (s, 1H), 2.69–2.66 (m, 1H), 2.12–2.04 (m, 1H), 2.02–1.94 (m, 1H), 1.80–1.65 (m, 3H), 1.23 (s, 3H), 1.08 (s, 3H), 0.94 (s, 9H), 0.89 (s, 3H), 0.09 (s, 6H); 13C NMR δ 145.9, 143.1, 132.6, 123.0, 121.4, 114.3, 96.3, 96.1, 84.2, 76.6, 64.9, 56.2, 55.7, 47.2, 38.4, 37.8, 27.5, 26.1(3C), 25.4, 23.4, 19.9, 18.6, 15.2, −5.1 (2C); HRMS (EI+) m/z calcd for C27H46O6Si (M+) 494.3064, found 494.3057.
Tricyclic alcohol 31
To a solution of tricycle 29 (112 mg, 0.25 mmol) in THF at 0 °C was added TBAF (0.3 mL, 0.3 mmol, 1 M in THF). The reaction was stirred for 2 h and quenched with NH4Cl (sat.), extracted with EtOAc, and washed with H2O and brine. The combined organics were dried (MgSO4) and concentrated in vacuo to afford alcohol 31 (83 mg, 99%) as a white solid: [α]26.4D = +43.5° (c 0.01, CHCl3); 1H NMR δ 6.96 (d, J = 1.8 Hz, 1H), 6.81 (d, J = 1.8 Hz, 1H), 5.19 (d, J = 6.3 Hz, 1H), 5.15 (d, J = 6.6 Hz, 1H), 4.56 (s, 2H), 3.51 (s, 3H), 3.40 (dd, J = 11.7, 3.0 Hz, 1H), 2.72 (s, 1H), 2.70 (d, J = 3.0 Hz, 1H), 2.15–2.05 (m, 1H), 1.86–1.81 (m, 2H), 1.69–1.63 (m, 4H), 1.26 (s, 3H), 1.09 (s, 3H), 0.87 (s, 3H); 13C NMR δ 146.1, 143.7, 132.2, 123.4, 122.6, 115.1, 96.0, 78.1, 76.9, 65.4, 56.3, 46.8, 38.5, 37.9, 28.4, 27.4, 23.3, 20.0, 14.4; HRMS (EI+) m/z calcd for C19H28O5 (M+) 336.1937, found 336.1932.
Tricyclic Aldehyde 32
To a solution of alcohol 31 (19 mg, 0.06 mmol) in CH2Cl2 was added manganese dioxide (120 mg, 1.2 mmol). The resulting solution was stirred for 16.5 h and quenched by filtration through a celite pad. After the pad was rinsed with CH2Cl2 and EtOAc, the combined organic extracts were dried (MgSO4) and concentrated to afford aldehyde 32 (19 mg, 100%) as a white solid: [α]26.4D = +74.8° (c 0.02, CHCl3); 1H NMR δ 9.79 (s, 1H), 7.45 (d, J = 1.8 Hz, 1H), 7.35 (d, J = 1.8 Hz, 1H), 5.23 (d, J = 6.6 Hz, 1H), 5.19 (d, J = 6.6 Hz, 1H), 3.51 (s, 3H), 3.42 (dd, J = 11.7, 4.5 Hz, 1H), 2.79 (s, 1H), 2.77 (d, J = 4.5 Hz, 1H), 2.16–2.10 (m, 1H), 1.91–1.84 (m, 2H), 1.74–1.60 (m, 4H), 1.26 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 191.0, 150.1, 146.7, 128.9, 127.3, 123.5, 115.4, 95.8, 78.5, 77.9, 56.4, 46.6, 38.6, 37.7, 28.3, 27.4, 23.2, 20.2, 14.4. Anal. Calcd for C19H26O5: C, 68.24; H, 7.84. Found: C, 67.94; H, 8.07.
(2R,4aR,9aR)–Tri(O-methoxymethyl)schweinfurthin G (34)
To a suspension of NaH (42 mg, 1.04 mmol, 60% oil dispersion) and 15–crown–5 (0.01 mL, 0.05 mmol) in THF at 0 °C was added a solution of phosphonate 33 (33 mg, 0.08 mmol) and aldehyde 32 (20 mg, 0.06 mmol) in THF. After the reaction was allowed to warm to room temperature and stirred for 18 h, it was quenched by addition of H2O and extracted with EtOAc. The combined organic extracts were washed with H2O and brine, dried (MgSO4), concentrated in vacuo to a yellow liquid, and purified by flash column chromatography (2:1 hexanes/EtOAc) to afford stilbene 34 (18 mg, 50%) as a colorless oil: 1H NMR δ 7.12 (d, J = 2.1 Hz, 1H), 6.95–6.93 (m, 2H), 6.90 (s, 2H), 6.89 (s, 1H), 5.25–5.18 (m, 7H), 3.55 (s, 3H), 3.51 (s, 6H), 3.47–3.44 (m, 1H), 3.38 (d, J = 7.2 Hz, 2H), 2.74 (s, 1H), 2.71 (d, J = 3.0 Hz, 1H), 2.15–2.05 (m, 1H), 1.90–1.80 (m, 2H), 1.79 (s, 3H), 1.75–1.70 (m, 3H), 1.66 (s, 3H), 1.24 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 156.6 (2C), 146.9, 144.6, 137.4, 131.8, 129.8, 128.8, 127.4, 123.9, 123.5, 122.8, 120.1, 114.7, 106.7 (2C), 96.6, 95.2 (2C), 78.7, 77.8, 57.0, 56.8 (2C), 47.5, 39.1, 36.5, 30.0, 29.0, 26.6, 23.9, 23.5, 20.7, 18.6, 15.0; HRMS (EI+) m/z calcd for C35H48O8 (M+) 596.3351, found 596.3349.
(2R,4aR,9aR)–Schweinfurthin G (8)
To a solution of stilbene 34 (24 mg, 0.04 mmol) in MeOH was added TsOH (31 mg, 0.18 mmol) at room temperature and the solution was stirred for 19 h. The reaction was quenched by addition of NaHCO3 (sat.) and extracted with EtOAc. The combined extracts were washed with H2O and brine, dried (MgSO4), and concentrated in vacuo to afford a yellow oil. Final purification by flash column chromatography (3:2 hexanes/EtOAc) afforded stilbene 8 (11 mg, 57%) as a colorless oil: [α]26.4D = +58.4° (c 0.007, CH3OH); literature7 [α]22 = +32.4° (c 0.04, CH OH); both 1H and 13C NMR spectra matched that of the natural product;7 HRMS (EI+) m/z calcd for C29H36O5 (M+) 464.2563, found 464.2565. The HPLC analyses were performed on a Shimadzu LC-20AT instrument with a Chiralcel OD-H column. The enantiomeric excess for compound 8 was determined by elution with a 4:1 mixture of hexanes and 2-propanol. Retention times for the enantiomers of schweinfurthin G (8) were 15.2 min (3.1 %) and 17.9 min (96.9 %), respectively.
(2R,4aR,9aR)–Tri(O-methoxymethyl)–3–deoxyschweinfurthin A (36)
To a suspension of NaH (50 mg, 1.24 mmol, 60% oil dispersion) and 15–crown–5 (0.01 mL, 0.05 mmol) in THF at 0 °C was added a solution of phosphonate 35 (37 mg, 0.08 mmol) and aldehyde 32 (25 mg, 0.08 mmol) in THF. After the reaction was allowed to warm to room temperature and stirred for 20 h, it was quenched by addition of H2O and extracted with EtOAc. The combined organic extracts were washed with H2O and brine, dried (MgSO4), concentrated in vacuo to a yellow liquid, and purified by flash column chromatography (2:1 hexanes/EtOAc) to afford stilbene 36 (33 mg, 66%) as a colorless oil: [α]26.4D = +39.0 (c 0.02, CHCl3); 1H NMR δ 7.11 (d, J = 2.1 Hz, 1H), 6.96–6.94 (m, 2H), 6.90 (s, 2H), 6.87–6.86 (s, 1H), 5.23–5.19 (m, 7H), 5.07–5.04 (m, 1H), 3.54 (s, 3H), 3.51–3.49 (m, 7H), 3.39 (d, J = 6.9 Hz, 2H), 2.73 (s, 1H), 2.71 (d, J = 3.2 Hz, 1H), 2.15–2.05 (m, 3H), 2.00–1.80 (m, 5H), 1.79 (s, 3H), 1.75–1.70 (m, 2H), 1.64 (s, 3H), 1.57 (s, 3H), 1.25 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 156.0 (2C), 146.3, 144.0, 136.8, 134.8, 131.4, 129.2, 128.2, 126.8, 124.5, 123.3, 122.8, 122.2, 119.7, 113.7, 106.2 (2C), 96.1, 94.6 (2C), 78.1, 77.1, 56.4, 56.1 (2C), 46.9, 40.0, 38.5, 37.9, 28.4, 27.4, 26.9, 25.7, 23.4, 23.1, 20.1, 17.8, 16.2, 14.4; HRMS (EI+) m/z calcd for C40H56O8 (M+) 664.3975, found 664.3989.
(2R,4aR,9aR)–3–Deoxyschweinfurthin A (10)
To a solution of stilbene 36 (4.9 mg, 0.01 mmol) in MeOH was added TsOH (7.3 mg, 0.04 mmol) at room temperature and the solution was stirred for 24 h. The reaction was quenched by addition of NaHCO3 (sat.) and extracted with EtOAc. The combined extracts were washed with H2O and brine, dried (MgSO4), and concentrated in vacuo to afford a yellow oil. After final purification by flash column chromatography (3:2 hexanes/EtOAc), stilbene 10 (2.7 mg, 69%) was obtained as a colorless oil:: 1H NMR (CD3OD) δ 6.83 (d, J = 16.5 Hz, 1H), 6.81–6.80 (m, 1H), 6.73–6.72 (m, 1H), 6.71 (d, J = 16.2 Hz, 1H), 6.43 (s, 2H), 5.23 (t, J = 6.6 Hz, 1H), 5.06 (t, J = 6.9 Hz, 1H), 3.34–3.28 (m, 3H), 2.74–2.69 (m, 2H), 2.05–2.00 (m, 4H), 1.95–1.90 (m, 2H), 1.75 (s, 3H), 1.70–1.65 (m, 3H), 1.61 (s, 3H), 1.55 (s, 3H), 1.21 (s, 3H), 1.08 (s, 3H), 0.86 (s, 3H); 13C NMR (CD3OD) δ 157.3 (2C), 147.0, 142.2, 137.5, 134.8, 132.0, 131.0, 128.6, 127.5, 125.5, 124.6, 124.0, 120.4, 115.9, 111.1, 105.7 (2C), 78.7, 78.2, 48.6, 41.0, 39.5, 38.9, 29.0, 27.9, 27.8, 25.9, 24.0, 23.2, 20.2, 17.7, 16.3, 14.8; HRMS (EI+) m/z calcd for C34H44O5 (M+) 532.3189, found 532.3193.
Alcohol 37
To a solution of silyl ether 21 (39 mg, 0.08 mmol) in THF at 0 °C was added a solution of TBAF (0.10 mL, 0.10 mmol, 1 M in THF). The resulting solution was stirred for 4 h, quenched by addition of NH4Cl (sat.), and extracted with EtOAc. The organic extracts were washed with H2O and brine, dried (MgSO4), and concentrated in vacuo to give a yellow liquid. Final purification by flash column chromatography (40% EtOAc in hexanes) afforded alcohol 37 (20 mg, 68%) as a colorless oil: [α]26.4D = +34.6 (c 0.09, CHCl3); 1H NMR (CDCl3) δ 6.73 (d, J = 1.8 Hz, 1H), 6.70 (s, 1H), 4.77 (d, J = 6.6 Hz, 1H), 4.64 (d, J = 7.2 Hz, 1H), 4.57 (s, 2H), 3.84 (s, 3H), 3.41 (s, 3H), 3.26 (dd, J = 11.4, 4.2 Hz, 1H), 2.71–2.68 (m, 2H), 2.13 (dt, J = 12.0, 3.3 Hz, 1H), 2.02–1.94 (m, 2H), 1.85–1.75 (m, 2H), 1.24 (s, 3H), 1.07 (s, 3H), 0.89 (s, 3H); 13C NMR (CDCl3) δ 148.9, 142.2, 132.1, 122.7, 120.5, 108.5, 96.2, 84.1, 76.8, 65.5, 56.1, 55.7, 47.1, 38.3, 37.7, 27.5, 25.4, 23.2, 19.8, 15.2; HRMS (EI+) m/z calcd for C20H30O5 (M+) 350.2093, found 350.2095.
Alcohol 41
To a solution of silyl ether 30 (76 mg, 0.15 mmol) in THF at 0 °C was added a solution of TBAF (0.18 mL, 0.18 mmol, 1 M in THF). The resulting solution was stirred for 16 h, quenched by addition of NH4Cl (sat.), and extracted with EtOAc. The organic extracts were washed with H2O and brine, dried (MgSO4), and concentrated in vacuo to give a yellow liquid. Final purification by flash column chromatography (30% EtOAc in hexanes) afforded alcohol 41 (35 mg, 59%) as a colorless oil: [α]26.4D = +26.2 (c 0.03, CHCl3); 1H NMR (CDCl3) δ 6.95 (d, J = 1.8 Hz, 1H), 6.81–6.80 (m, 1H), 5.19 (d, J = 6.9 Hz, 1H), 5.15 (d, J = 6.3 Hz, 1H), 4.77 (d, J = 7.2 Hz, 1H), 4.64 (d, J = 6.6 Hz, 1H), 4.55 (s, 2H), 3.51 (s, 3H), 3.41 (s, 3H), 3.27 (dd, J = 11.4, 4.2 Hz, 1H), 2.71 (s, 1H), 2.68 (d, J = 4.2 Hz, 1H), 2.15–2.05 (m, 1H), 2.02–1.95 (m, 1H), 1.90–1.80 (br s, 1H), 1.75–1.65 (m, 3H), 1.23 (s, 3H), 1.08 (s, 3H), 0.89 (s, 3H); 13C NMR (CDCl3) δ 146.0, 143.8, 132.2, 123.4, 122.6, 114.9, 96.2, 95.9, 84.1, 76.8, 65.3, 56.3, 55.8, 47.1, 38.3, 37.7, 27.5, 25.3, 23.3, 19.9, 15.2; HRMS (EI+) m/z calcd for C21H32O6 (M+) 380.2199, found 380.2201.
Phosphonate 42
To a solution of alcohol 41 (38 mg, 0.10 mmol) in CH2Cl2 at 0 °C was added triethylamine (0.08 mL, 0.58 mmol). Methanesulfonyl chloride (0.05 mL, 0.64 mmol) was added dropwise and the solution was stirred for 5.5 h. The reaction was quenched by addition of H2O and extracted with CH2Cl2. The combined organic layers were washed with NH4Cl (sat.) and brine, dried (MgSO4) and concentrated in vacuo to a yellow oil, which was then dissolved in acetone and treated with sodium iodide (79 mg, 0.53 mmol) for 15 h. After the solvent was removed in vacuo, the red oily residue was extracted with EtOAc. The organic layers were washed with H2O, NaHCO3 (sat.) and then Na2S2O3 until the reddish color dissipated. The combined aqueous layers were extracted with EtOAc. The combined organic extracts were washed with brine, dried (MgSO4), and concentrated to a yellow oil which was subsequently dissolved in P(OEt)3 (0.08 mL, 0.47 mmol). This solution was heated for 7 h at 80 °C and then quenched by addition of H2O and extracted with EtOAc. The organic extracts were washed with H2O and brine, dried (MgSO4) and concentrated in vacuo to a yellow liquid. Final purification by flash column chromatography (20–100% EtOAc in hexanes) yielded phosphonate 42 (31 mg, 62%) as a colorless oil: [α]26.4D = +20.0 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 6.86 (s, 1H), 6.74 (s, 1H), 5.17 (d, J = 6.3 Hz, 1H), 5.13 (d, J = 6.6 Hz, 1H), 4.77 (d, J = 6.9 Hz, 1H), 4.64 (d, J = 6.9 Hz, 1H), 4.07–3.99 (m, 4H), 3.49 (s, 3H), 3.41 (s, 3H), 3.27 (dd, J = 11.4, 4.2 Hz, 1H), 3.03 (d, JPH = 21.3 Hz, 2H), 2.69 (s, 1H), 2.66 (d, J = 4.2 Hz, 1H), 2.10–2.05 (m, 1H), 2.02–1.95 (m, 1H), 1.75–1.65 (m, 3H), 1.27 (t, J = 6.9 Hz, 6H), 1.22 (s, 3H), 1.07 (s, 3H), 0.88 (s, 3H); 13C NMR (CDCl3) δ 145.9 (d, JCP = 3.2 Hz), 143.1, 124.9 (d, JCP = 7.6 Hz), 123.4 (d, JCP = 2.7 Hz), 122.2 (d, JCP = 9.2 Hz), 117.3 (d, JCP = 6.4 Hz), 96.2, 96.0, 84.1, 77.4, 62.2 (d, JCP = 6.9 Hz), 62.2 (d, JCP = 6.9 Hz), 56.2, 55.8, 47.0, 38.3, 37.8, 33.0 (d, JCP = 138.1 Hz), 27.5, 25.4, 23.2, 19.9, 16.5 (d, JCP = 6.2 Hz), 16.5 (d, JCP = 6.2 Hz), 15.2; 31P NMR δ 28.1; HRMS (EI+) m/z calcd for C25H41O8P (M+) 500.2541, found 500.2539.
(2R,4aR,9aR)–Tetra(O-methoxymethyl)–3–deoxyschweinfurthin A (43)
To a suspension of NaH (56 mg, 1.40 mmol, 60% oil dispersion) and 15–crown–5 (0.01 mL, 0.05 mmol) in THF at 0 °C was added a solution of phosphonate 42 (31 mg, 0.06 mmol) and aldehyde 39 (39 mg, 0.11 mmol) in THF. After the reaction was allowed to warm to room temperature and stirred for 16 h, it was quenched by addition of H2O and extracted with EtOAc. The organic extracts were washed with H2O and brine, dried (MgSO4), concentrated in vacuo to a yellow liquid, and purified by flash column chromatography (10–25% EtOAc in hexanes) to afford stilbene 43 (30 mg, 67%) as a colorless oil: [α]26.4D = +42.2 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 7.12 (d, J = 1.8 Hz, 1H), 6.95 (d, J = 1.5 Hz, 1H), 6.94–6.90 (m, 3H), 6.86 (d, J = 16.2 Hz, 1H), 5.25–5.18 (m, 7H), 5.07 (t, J = 6.9 Hz, 1H), 4.78 (d, J = 6.9 Hz, 1H), 4.65 (d, J = 6.6 Hz, 1H), 3.54 (s, 3H), 3.50 (s, 6H), 3.41 (s, 3H), 3.41–3.37 (m, 2H), 3.28 (dd, J = 11.4, 4.2 Hz, 1H), 2.73 (s, 1H), 2.70 (d, J = 4.2 Hz, 1H), 2.15–1.95 (m, 7H), 1.78 (s, 3H), 1.75–1.70 (m, 2H), 1.65 (s, 3H), 1.57 (s, 3H), 1.25 (s, 3H), 1.09 (s, 3H), 0.91 (s, 3H); 13C NMR (CDCl3) δ 156.1 (2C), 146.3, 144.0, 136.8, 134.8, 131.4, 129.2, 128.2, 126.7, 124.5, 123.3, 122.7, 122.2, 119.6, 113.6, 106.1 (2C), 96.3, 96.0, 94.6 (2C), 84.2, 77.1, 56.4, 56.1 (2C), 55.8, 47.2, 40.0, 38.4, 37.8, 27.7, 26.9, 25.8, 25.4, 23.3, 22.8, 20.0, 17.8, 16.2, 15.3; HRMS (EI+) m/z calcd for C42H60O9 (M+) 708.4237, found 708.4231.
(2R,4aR,9aR)–3–Deoxyschweinfurthin A (10)
To a solution of stilbene 43 (28 mg, 0.04 mmol) in MeOH was added TsOH (43 mg, 0.25 mmol) at room temperature and the solution was stirred for 136 h. The reaction was quenched by addition of NaHCO3 (sat.) and extracted with EtOAc. The organic extracts were washed with H2O and brine, dried (MgSO4), and concentrated in vacuo to afford a yellow oil. Final purification by flash column chromatography (60:40 hexanes/EtOAc) afforded the desired target 3dSA (10, 11 mg, 52%) as a yellow oil: [α]26.4D = +48.5 (c 0.006, CD3OD); with spectral data matching that reported above. Also isolated was a partially hydrolyzed analogue bearing an A-ring MOM ether (44, 7 mg, 33%) as a yellow oil: 1H NMR (CDCl3) δ 6.92 (d, J = 2.1 Hz, 1H), 6.87 (d, J = 16.8 Hz, 1H), 6.75 (d, J = 16.2 Hz, 1H), 6.75–6.74 (m, 1H), 6.56 (s, 2H), 5.68 (br s, 2H), 5.49 (br s, 1H), 5.28 (t, J = 6.6 Hz, 1H), 5.06 (t, J = 5.4 Hz, 1H), 4.78 (d, J = 6.6 Hz, 1H), 4.65 (d, J = 7.2 Hz, 1H), 3.45–3.42 (m, 2H), 3.42 (s, 3H), 3.29 (dd, J = 11.4, 3.6 Hz, 1H), 2.73–2.66 (m, 2H), 2.13–1.96 (m, 5H), 1.82 (s, 3H), 1.78–1.70 (m, 4H), 1.68 (s, 3H), 1.59 (s, 3H), 1.24 (s, 3H), 1.10 (s, 3H), 0.90 (s, 3H); 13C NMR (CDCl3) δ 155.5 (2C), 145.3, 139.1, 137.2, 132.2, 129.8, 128.5, 126.3, 123.9, 122.2, 121.7, 119.7, 113.0, 109.6, 106.2 (2C), 105.8, 96.3, 84.0, 78.0, 63.9, 55.8, 47.6, 39.9, 38.4, 37.7, 27.5, 26.5, 25.8, 25.3, 22.8, 20.3, 17.9, 16.3, 15.2; HRMS (EI+) m/z calcd for C20H30O5 (M+) 350.2093, found 350.2095.
Supplementary Material
Acknowledgments
Financial support from the Roy J. Carver Charitable Trust, the Children’s Tumor Foundation, the Predoctoral Training Program in the Pharmacological Sciences (2 T32 GM067795), and the UI Graduate College in the form of a Presidential Fellowship to NRM, is gratefully acknowledged.
Footnotes
Supporting Information.
Experimental paragraphs for compounds 25, 26, 38, 39, 40, and 11, along with complete 1H and 13C NMR spectra for compounds 8, 10, 21, 25, 26, 28–32, 34, and 36–44, can be found here. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For some recent reviews cf: Tang Y, Oppenheimer J, Song ZL, You LF, Zhang XJ, Hsung RP. Tetrahedron. 2006;62:10785–10813.de la Torre MC, Sierra MA. Angew. Chem.-Int. Edit. 2004;43:160–181. doi: 10.1002/anie.200200545.
- 2.Taylor SK. Org. Prep. Proced. Int. 1992;24:245–284. [Google Scholar]
- 3.(a) Barua AK, Banerjee SK, Basak A, Bose PK. J. Ind. Chem. Soc. 1976;53:638–639. [Google Scholar]; (b) Mechoulam R, Yagen B. Tetrahedron Lett. 1969:5349–5352. doi: 10.1016/s0040-4039(01)88961-9. [DOI] [PubMed] [Google Scholar]; (c) Stevens KL, Jurd L. Tetrahedron. 1976;32:665–668. [Google Scholar]; (d) de Alleluia IB, Braz Fo R, Gottlieb OR, Magalhaes EG, Marques R. Phytochemistry. 1978;17:517–521. [Google Scholar]; (e) Manners G, Jurd L, Stevens K. Tetrahedron. 1972;28:2949–2959. [Google Scholar]
- 4.Beutler JA, Shoemaker RH, Johnson T, Boyd MR. J. Nat. Prod. 1998;61:1509–1512. doi: 10.1021/np980208m. [DOI] [PubMed] [Google Scholar]
- 5.Beutler JA, Jato J, Cragg GM, Boyd MR. Nat. Prod. Lett. 2000;14:399–404. [Google Scholar]
- 6.Thoison O, Hnawia E, Guieritte-Voegelein F, Sevenet T. Phytochemistry. 1992;31:1439–1442. [Google Scholar]
- 7.Yoder BJ, Cao S, Norris A, Miller JS, Ratovoson F, Razafitsalama J, Andriantsiferana R, Rasamison VE, Kingston DGI. J. Nat. Prod. 2007;70:342–346. doi: 10.1021/np060484y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For a cyclization catalyzed by a non-racemic Lewis acid, see: Ishihara K, Ishibashi H, Yamamoto H. J. Am. Chem. Soc. 2002;124:3647–3655. doi: 10.1021/ja0124865.
- 9.(a) Kumanireng AS, Kato T, Kitahara Y. Chem. Lett. 1973:1045–1047. [Google Scholar]; (b) Begley MJ, Fish PV, Pattenden G. J. Chem. Soc. Perkin Trans. I. 1990:2263–2271. [Google Scholar]
- 10.Sato K, Nishimoto T, Tamoto K, Omote M, Ando A, Kumadaki I. Heterocycles. 2002;56:403–412. [Google Scholar]
- 11.Treadwell EM, Cermak SC, Wiemer DF. J. Org. Chem. 1999;64:8718–8723. [Google Scholar]
- 12.(a) Shi Y. Acc. Chem. Res. 2004;37:488–496. doi: 10.1021/ar030063x. [DOI] [PubMed] [Google Scholar]; (b) Shu L, Shi Y. Tetrahedron Lett. 1999;40:8721–8724. [Google Scholar]
- 13.Neighbors JD, Mente NR, Boss KD, Zehnder DW, II, Wiemer DF. Tetrahedron Lett. 2008;49:516–519. [Google Scholar]
- 14.Mente NR, Wiemer AJ, Neighbors JD, Beutler JA, Hohl RJ, Wiemer DF. Biorg. Med. Chem. Lett. 2007;17:911–915. doi: 10.1016/j.bmcl.2006.11.096. [DOI] [PubMed] [Google Scholar]
- 15.Neighbors JD, Salnikova MS, Wiemer DF. Tetrahedron Lett. 2005;46:1321–1324. [Google Scholar]
- 16.Neighbors JD, Beutler JA, Wiemer DF. J. Org. Chem. 2005;70:925–931. doi: 10.1021/jo048444r. [DOI] [PubMed] [Google Scholar]
- 17.(a) Yee NKN, Coates RM. J. Org. Chem. 1992;57:4598–4608. [Google Scholar]; (b) Corey EJ, Liu K. J. Am. Chem. Soc. 1997;119:9929–9930. [Google Scholar]
- 18.(a) Corey EJ, Sodeoka M. Tetrahedron Lett. 1991;32:7005–7008. [Google Scholar]; (b) Zhang JH, Corey EJ. Org. Lett. 2001;3:3215–3216. doi: 10.1021/ol016543a. [DOI] [PubMed] [Google Scholar]; (c) Taylor SK, Ivanovic M, Simons LJ, Davis MM. Tetrahedron: Asymmetry. 2003;14:743–747. [Google Scholar]
- 19.(a) Lacey JR, Anzalone PW, Duncan CM, Hackert MJ, Mohan RS. Tetrahedron Lett. 2005;46:8507–8511. [Google Scholar]; (b) Bogenstatter M, Limberg A, Overman LE, Tomasi AL. J. Am. Chem. Soc. 1999;121:12206–12207. [Google Scholar]
- 20.(a) Aggarwal VK, Bethel PA, Giles R. J. Chem. Soc.-Perkin Trans. 1999;1:3315. [Google Scholar]; (b) Nagamitsu T, Sunazuka T, Obata R, Tomoda H, Tanaka H, Harigaya Y, Omura S, Smith AB. J. Org. Chem. 1995;60:8126–8127. [Google Scholar]; (c) Smith AB, Kinsho T, Sunazuka T, Omura S. Tetrahedron Lett. 1996;37:6461–6464. [Google Scholar]; (d) Sen SE, Roach SL, Smith SM, Zhang YZ. Tetrahedron Lett. 1998;39:3969–3972. [Google Scholar]
- 21.Lange RG. J. Org. Chem. 1962;27:2037–2039. [Google Scholar]
- 22.Wang B, Zhang Y, Chen J, Zhang W, Song J, Del BU, Brown L, Miller G. PCT Int. Appl. 2003:161. CODEN: PIXXD2 WO 2003009807 A2 20030206 CAN 138:153318 AN 2003:97274. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.