

Abstract
Introduction
Mantle cell lymphoma (MCL) is an incurable, often aggressive B-cell malignancy. Bortezomib (BTZ), the 20S proteasome inhibitor was originally developed and approved for treatment of relapsed refractory multiple myeloma, and subsequently approved for treatment of MCL. BTZ’s single-agent activity induces clinical responses in approximately one-third of relapsed MCL patients. BTZ-containing combination therapies have further improved the quality and duration of clinical responses compared to standard chemotherapies in previously untreated MCL patients.
Areas Covered
This review summarizes the discovery, mechanisms of -action and resistance, preclinical-clinical-developments, and FDA approval of BTZ for treatments of MCL.
Expert opinion
Preclinical MCL models demonstrated the apoptotic effect of BTZ through multiple mechanisms, as well as synergistic anti-MCL activity between BTZ and other chemotherapeutics. Single-agent and combinational clinical trials have validated the therapeutic potential of targeting the ubiquitin proteasome system (UPS) in MCL. However, inherent and acquired drug resistance remains a significant clinical problem and multiple potential mechanisms have been identified. Next-generation proteasome inhibitors with different pharmacodynamic properties from BTZ may partially address the issue of inherent resistance, with increased response rates noted in some diseases. In addition, upstream UPS components, e.g., E3 ligases or deubiquitinating enzymes, may also be targetable in MCL.
Keywords: ubiquitin–proteasome-system, bortezomib, cancer, drug-development, drug resistance, molecular-targeting, proteasome-inhibitors, mantle cell lymphoma, pre-clinical, clinical trials, targeted therapy
1. Introduction
Recognized by the World Health Organization as a distinct clinical entity in 1994, mantle cell lymphoma (MCL) possesses distinctive: clinical, biological, and molecular characteristics. MCL is considered a generally incurable B-cell malignancy [1]. MCL comprises 6% of all non-Hodgkin’s lymphomas, with an incidence of approximately 5,000 cases per year in the United States [1]. The median age of diagnosis of MCL is the mid-60s, with a 3:1 male predominance. Extra-nodal involvement is frequent, and the general prognosis of MCL patients is poor. Although the disease frequently responds to initial treatments, therapy-resistant relapses eventually develop in almost all cases. As such, the median overall survival (OS) remains below three years [2].
The 2003 FDA approval of bortezomib (BTZ; Figure 1) for treatment of Multiple Myeloma (MM) validated the therapeutic potential of targeting the 20S proteasome and the ubiquitin proteasome system (UPS) in cancer and especially in hematologic malignancies [3] (Table 1). BTZ inhibits the 20S core proteasome, resulting in cancer cell death via multiple mechanisms; including, induction of reactive oxygen species (ROS), suppression of the unfolded protein response (UPR), accumulation of ubiquitinated proteins, inhibition of the cellular NFκB survival pathway via accumulation of IκBα, and stabilization of tumor suppressor proteins such as p21, p27, Bax and p53 [4–5].
Figure 1. Chemical Structure of BTZ.
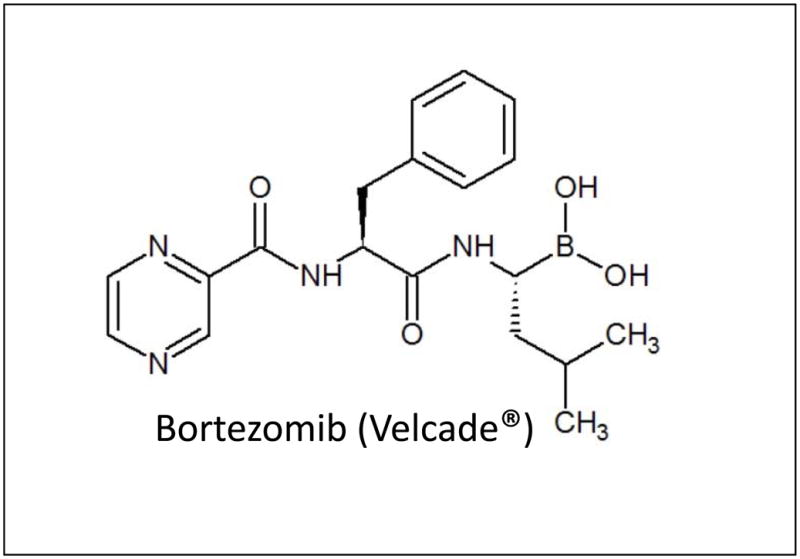
Bortezomib (BTZ, Trade name Velcade®) is a dipeptide boronic acid which forms non-covalent bonds with the N-terminal hydroxyl groups of threonine residues of the 20S proteasome via the Boron atom’ activity as an electron receiver, thus forming stable tetrahedral intermediates which inhibit the catalytic activity of specific β-subunits (β5 mainly, and β1) resulting in inhibition of chymotrypsin-like and PGPH-like activities.
Table 1.
Major events in the clinical development of Bortezomib
| Year | Events In Bortezomib Development | Source |
|---|---|---|
| 1995 | Synthesis by a team led by Julian Adams. | Adams, J. et al. Boronic ester and acid compounds. US Patent 5,780,554 (1995). |
| 2000 | Phase-I clinical trial shows BTZ is tolerable at 1.04 mg/m2. It also shows activity in multiple myeloma. | Orlowski, R. et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin. Oncol. 20, 4420–4427 (2002). |
| 2003 | Phase II clinical trial shows BTZ being active in refractory multiple myeloma. This prompted accelerated FDA approval for BTZ as treatment of refractory multiple myeloma. | Richardson, P. G. et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 348, 2609–2617 (2003). |
| 2005 | Phase III trial confirms BTZ activity in multiple myeloma. The FDA approved BTZ for the treatment of multiple myeloma with one prior therapy. | Kane RC et al. United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin Cancer Res. 2006 May 15;12(10):2955–60. |
| 2006 | Phase II trial shows BTZ activity in mantle cell lymphoma. The FDA approved BTZ for the treatment of mantle cell lymphoma with at least one prior therapy. | Kane RC et al. Bortezomib for the treatment of mantle cell lymphoma. Clinical cancer research, 13(18 pt 1): 5291–4, 2007. |
| 2008 | Phase III trial reports superior activity when combining BTZ with melphalan and prednisone as initial treatment for multiple myeloma. The FDA approved BTZ to be used in the initial treatment of multiple myeloma. | Miguel, J.F.S. et al. Bortezomib plus Melphalan and Prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 359, 906–917 (2008). |
| 2014 | Phase III trial reports at least 59% increased efficacy when BTZ replaces vincristine in a drug combination that includes rituximab, cyclophosphamide, doxorubicin, and prednisone for the initial treatment of mantle cell lymphoma. The FDA approved BTZ for the initial treatment of mantle cell lymphoma. | Robak T. et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N. Engl. J. Med. 372:944–953 (2015). |
Based on the findings and successes of preclinical model studies, BTZ was examined as a potential chemotherapeutic in the treatment of MCL. BTZ induced responses in approximately one-third of patients with previously-treated MCL in a single-agent Phase 2 trial [6]. A subsequent Phase 3 trial comparing a BTZ-containing combination to standard chemotherapeutic regimens demonstrated superior results with regard to quality and duration of patient responses in the BTZ-containing arm [7]. Consequently, BTZ was approved by the FDA for the treatment of relapsed Mantle Cell Lymphoma (MCL) on December 8th, 2006 for patients who have received at least one prior therapy, and on October 9th, 2014 as a frontline therapy for use in previously untreated MCL patients (Table 1) [8].
The intentions of this review are to provide a biological background on the role of the proteasome in cancer, and the molecular rationale of proteasome inhibition in MCL. In addition, the review will provide a historical and scientific accounting of the discovery, preclinical and early clinical development of BTZ as treatment of MCL. Finally, we will examine the potential limitations of BTZ therapy and discuss potential means of addressing these issues.
2. The Role of the Proteasome and the UPS in Cancer
The UPS is a complex, dynamic, and critical system in both normal and cancer cells, and disruption of the UPS, such as by a proteasome inhibitor, is associated with extensive modulation of signaling pathways, cellular activities and apoptosis/autophagy [9–12]. As a posttranslational modification system, the UPS modulates the fate of diverse proteins through the dynamic addition, branching and removal of ubiquitin (Ub) moieties. Ub is a highly conserved and small (76 amino acids, 8.5 kDa) regulatory protein moiety which, through a series of molecular processes, is linked to lysine residues of targeted proteins. The fate of the target proteins is dependent on the dynamic pattern of ubiquitin branching [13–15].
2.1 The Structure and characteristics of the 26S Proteasome
The 26S proteasome is the central molecular machinery of the UPS, consisting of a 20S catalytic core which enzymatically degrades targeted, ubiquitinated proteins, as well as two 19S regulatory caps which regulate the removal and recycling of the linked Ub chains and linearizing and inserting the targeted proteins into the 20S proteasome for catalytic degradation (Figure 2A). The 20S core is composed of two identical inner β-rings and two identical outer α-rings. Each β-ring contains 3 proteolytic sites on β1, β2 and β5 subunits, responsible for the post-glutamyl peptide hydrolase-like (PGPH) (or caspase-like), trypsin-like, or chymotrypsin-like (CT) activities, respectively (Figure 2A) [16–17].
Figure 2. The 26S Proteasome Structure and Proteolytic Activities.


A) The 26S proteasome consists of a 20S catalytic core and two 19S regulatory caps, which removes/recycles linked Ub, linearize and direct targeted proteins into the 20S Core. The 20S core is composed of 2-inner β-rings and 2-outer α-rings. As seen the β-rings contain 3 proteolytic sites (β1, β2 and β5 subunits), possessing post-glutamyl peptide hydrolase-like (PGPH) (or caspase-like), trypsin-like, or chymotrypsin-like (CT) activities, respectively. BTZ primarily binds to the β5 and β1 subunits blocking proteasome activities. B) The Ubiquitin Proteasome System (UPS) is composed of a series of ubiquitinating enzymes which work through the specific and targeted addition and branching of Ubiquitin to proteins which are differentially targeted by different E3-Ligases (which possess target specificity). Through the utilization of ATP, Ub is conjugated to the E1 Ligase, and then transferred to an E2 Ligases which subsequently transfers the Ub Moiety to a target protein or an existing Ub-chain. The complexity of the Ub branching pattern is determined by the interaction of numerous UPS components including the Deubiquitinating enzymes (DUBs) which serve to both recycle as well as modulate the branching patterns of ubiquitinated proteins. Most poly-ubiquitinated proteins then pass through the 19S cap which linearizes the peptide and passes it into the 20S Core proteasome for catalytic degradation.
2.2 Regulators of the UPS: Ubiquitin-Conjugating Enzymes E1, E2 and E3
Regulation of protein ubiquitination involves a multi-step process that transfers Ub-moieties to target proteins by the conjugation and ligation of different lengths and branching patterns of Ub. Ub conjugation occurs via an enzymatic cascade, involving three distinct enzymes: (i) Ub-activating (E1), (ii) Ub-conjugating (E2), and (iii) Ub-ligating enzymes (E3 Ligases) (Figure 2B). Protein ubiquitination is initiated by the ATP-dependent formation of a thioester linkage between the C-terminus of the Ub moiety and a cysteine residue of the E1-activating enzyme [13–14]. The Ub moiety is then transferred to an E2-Ubiquitin-conjugating enzyme through the formation of an E1-Ub-E2 complex via the formation of a thio-ester intermediate complex [15]. Finally, the Ub is transferred from the E2-conjugating enzyme to a target specific E3-ligase bound to a specific protein-substrate directly or via a third high-energy thio-ester intermediate, which results in the formation of an isopeptide bond between the C-terminus of the Ub and a specific lysine residue on the substrate protein, or expanding a preexisting Ub-chain of variable lengths and branching patterns [15–18].
A complex hierarchal pyramid of E1, E2, and E3 ubiquitin-conjugating enzymes allows for fine-tuned modulation of protein degradation via variable ubiquitination and branching patterns, intricately coordinated by differential expression and localization of the components of the UPS [18].
2.3 UPS-Mediated Regulation of Target Specificity and Cellular Fate
The extent and complexity of Ub-branching patterns significantly affects the behavior of the target protein and provides an extensive and diverse set of potential targets for future UPS-targeted drugs distinct from 20S-Core proteasome targeted by BTZ and other PIs [19–20]. The UPS regulates cellular activities and functions through protein binding specificity, and also differential expression and cellular localization of the individual UPS components (such as E1, E2, E3 and Deubiquitinases/DUBs) [15, 18]. For example, the UPS regulates cell-cycle progression and apoptosis through the turnover of key proteins, such as the cyclins, p21 and p53 [21–22]. UPS also plays an essential role in regulating one of the most important cell survival pathways, the NFκB pathway [23–24]. Cancer cells have been shown to utilize the UPS to maintain aberrant cell growth and resistance to apoptosis through enhanced degradation of the NFκB inhibitor, IκB. Inhibition of the proteasome by BTZ has been shown to induce cell-cycle arrest and apoptotic cell death selectively in human cancer cells such as multiple myeloma [25–26]. Proteasome inhibition causes these effects through modulation of a variety of cellular pathways in cancer cells, specifically, accumulation of p27, Bax, p53 and IκBα, inhibition of NFκB, and induction of the unfolded protein response (UPR) and DNA damage responses (DDR) (Figure 3) [12, 16, 18–22 24–235]. These UPS-mediated effects have been observed in pre-clinical studies across a wide spectrum of tumors, including MM and MCL.
Figure 3. Mechanisms of Action of BTZ and Cellular Effects and Pathways.

The 26S proteasome is responsible for the degradation of numerous critical cellular proteins, thus regulating multiple critical cellular pathways. Depicted, are a few key pathways associated with the proteasome activity and regulated by BTZ-mediated proteasome inhibition. Inhibition of the proteasome results in the stabilization and accumulation of multiple tumor suppressors (Blue) such as: p27, p53, Rb, IκBα, Bax and NOXA as well as ROS. Accumulation of p27 reverses the inhibition on RB, resulting in blockage of cell cycle progression through inhibition of E2F, a critical regulator of cell cycle progression (Oncogene/Oncogenic: Red). Accumulation and stabilization of Bax induces Cytochrome-C dependent apoptosis, and accumulation of IκBα inhibits NFκB, a crucial regulator of cell survival and anti-apoptotic pathways. Induction of NOXA and ROS by BTZ also contribute to its apoptosis-inducing and anti-MCL activities. Therefore, proteasome inhibition by BTZ obstructs three key cancer pathways: (i) Cell cycle progression, (ii) Cell survival pathways, and (iii) Cellular proliferation, the combination of which results in significant inhibition of cancer cell growth.
3. Discovery of Bortezomib
BTZ (Figure 1) is a reversible inhibitor of the 20S proteasome. The discovery and development of BTZ, then known as PS-341, a dipeptide boronic acid, was accomplished by Dr. Julian Adams and his team, who sought to inhibit the proteasome’s enzymatic functions in order to diminish the aberrant proteasome activity associated with cancer and inflammation [27–28]. Numerous peptidyl aldehydes have demonstrated the ability to inhibit the chymotrypsin (CT)-like activity of the proteasome complex [5]. Utilizing X-ray crystallography, it was discovered that the aldehyde inhibitors formed hemi-acetal adducts with the active site threonine nucleophile of β-subunits (Figure 2A) [28]. In order to further enhance the interaction with the β-subunits, Boron was incorporated into the compound which significantly enhanced the potency of the prototype proteasome inhibitors (PIs). BTZ is one such boron-containing PI.
In vitro, myeloma cells are differentially sensitive to BTZ-induced proteasome inhibition and apoptosis relative to normal cells. One reason for this is that non-malignant cells do not divide as rapidly as cancer cells therefore they are less dependent on the proteasome for protein turnover [4–5 19,]. As mentioned, a specific action of BTZ is the inhibition of NFκB pathway through the stabilization of its inhibitor protein IκB. Myeloma cells specifically depend on NFκB-mediated transcription of cytokine growth factor interleukin-6 (IL6), angiogenesis through vascular endothelial growth factor (VEGF), and the cell adhesion molecule VCAM-1 for adherence of the plasma cells to the stromal tissue in bone marrow [25–26, 30]. Dr. Adams and his collaborators found that even at low nanomolar concentrations, BTZ was highly effective in the abrogation of transcription of NFκβ-dependent genes. BTZ also increases p27Kip1 levels, and consequently reduces Cyclin-D1, thus inhibiting cell-cycle progression [31]. Proteasome inhibition by BTZ has also been shown to upregulate pro-apoptotic genes and downregulate anti-apoptotic genes [32–33]. For example, BTZ selectively increases levels of a pro-apoptotic protein NOXA in malignant cells [34–35]. Furthermore, it has been demonstrated that BTZ induces ER stress, ultimately leading to cytochrome C release and subsequent caspase-mediated apoptosis [33, 36]. The key molecular mechanisms of action of BTZ-dependent cancer cell death have been summarized in Figure 3.
4. Molecular Characteristics of MCL
Immuno-phenotyping of MCL typically reveals a CD5+, CD20+, CD10−, FMC7+, CD23−, CD43+, and cyclin D1+ profile [37–38]. Additionally, MCL is characterized by the specific chromosomal translocation t(11;14)(q13;q32) [39], which results in the overexpression of CCND1. MCL cells have increased expression of the transcription factor SOX11, which has been shown to promote tumor growth of MCL cells in vivo and is known to regulate a broad set of transcriptional programs that includes B-cell differentiation, cell proliferation, apoptosis, and angiogenesis [40–41]. MCL cells also exhibit inherent genetic instability with a tendency to accumulate alterations in cell cycle regulatory genes, DNA damage response pathway, and cell survival mechanisms which may contribute to the inevitable development of resistance to therapy seen in patients with MCL [42].
Recent genome-wide studies using next-generation sequencing (NGS) have expanded the understanding of genes and pathways involved in the development of MCL [43]. It has been shown that the most common secondary alteration in MCL is the mutation of the DNA damage sensor ATM, particularly in cells over-expressing SOX11 [40, 42]. Mutations in several chromatin modifiers such as WHSK1 (10%), MLL2 (14%), and MEF2B (3%) have also been detected almost exclusively in MCL cells expressing SOX11. Other aberrations identified include activating mutations in NOTCH1/2 in 10% of tumors associated with an aggressive evolution and somatic mutations in regulatory genes of NFκB pathway in 10% to 15% of MCL [44–45].
5. Preclinical studies of Bortezomib in Mantle Cell Lymphoma
5.1 Molecular mechanisms of Bortezomib activity and resistance in MCL
The cytotoxicity of bortezomib in MCL cells results from the effects of proteasome inhibition on several intracellular mechanisms [46–51]. An analysis of primary MCL cells by Chiarle et al showed that the protein levels of the p27 CDK inhibitor is decreased due to degradation by the proteasome [46]. The protein p27 is a negative regulator of the cell cycle, and its inhibition or down-regulation is associated with tumor progression. Although this research group used a different proteasome inhibitor (hemin), it nevertheless linked proteasome inhibition to the diminished tumorigenesis in MCL. BTZ was also shown to cause cell cycle arrest and apoptosis by inhibition of the NFκB pathway in MCL cell lines (Mino and DB-sp53) and primary tumor samples [31]. NFκB, an important signaling pathway regulating cell cycle progression and cell survival, is constitutively expressed in these cell lines and tissues. Inhibition of the proteasome by BTZ down-regulates the expression of NFκB, which can lead to G1 cell cycle arrest and apoptosis. The NFκB survival pathway may be sustained by protein kinase CK2, which phosphorylates and activates NFκB. Inhibition of CK2 enhances BTZ’s cytotoxic effect in MCL cell lines (Granta 519, Jeko-1, and Rec-1) [50–51]. Thus, high levels of CK2 may be involved in BTZ resistance. However, a study comparing BTZ-sensitive and BTZ-resistant MCL cell lines found no correlation between NF-kB activity and resistance status [35]. Furthermore, NF-kB activity varies widely among the sensitive and resistant cell lines [35]. As an example, NF-kB activity is ~2.5-fold higher in Granta cells than it is in UPN1 cells, but Granta is as sensitive to BTZ as UPN1 (35). Therefore, whether NF-kB pathway is involved in BTZ resistance remains unclear.
As further discussed in Section 6, a main problem observed through the use of BTZ is inherent and acquired resistance to the drug. Preclinical studies have suggested several potential mechanisms involved in development of bortezomib resistance, including: plasmacytic differentiation via up-regulation of IRF4 and CD38 and expression of CD138 (52); MCL-initiating or stem-like cell phenotype (lacking of prototypic B-cell marker CD19 (53); a defect in regulation or function of the pro-apoptotic BH3-only protein NOXA (35).
Consistently, targeting these involved pathways would overcome BTZ resistance. Indeed, harnessing NOXA demethylation was shown to be able to overcome BTZ resistance in MCL (54). Furthermore, resistance to BTZ could be overcome by the Hsp90 inhibitor IPI-504 (55), the dual PI3K and mTOR inhibitor NVP-BEZ235 (56), Sorafenib (57), calcium blockers (58), and inhibition of Lyn (59),
In addition, treatment of MCL cell lines (JVM-2, GRANTA-519, JEKO, and REC-1) with BTZ leads to depolarization of the mitochondria membrane, ROS production, and induction of the pro-apoptotic NOXA protein, resulting in apoptosis [35]. NOXA, in particular, appears to be important, as siRNA-induced downregulation of NOXA inhibits BTZ-induced apoptosis [35]. PRDM1 (PR domain zinc finger protein 1) is a transcription factor that represses the expression of proteins needed for B-cell identity and proliferation and helps to drive B-cells through their final differentiation stage to become antibody-secreting cells. PRDM1 is also a mediator of NOXA-induced apoptosis. Interestingly, PRDM1 was shown to be needed for BTZ-induced apoptosis in MCL cell lines (Mino and Jeko-1) and primary tumor samples [49]. This finding further strengthens the role of NOXA in BTZ-mediated anticancer activity in MM and MCL.
5.2 Rationale for combining BTZ with other chemotherapeutic agents in MCL
Given the fact that the majority of MCL patients do not have a clinical response to single-agent BTZ, and that it had been shown to be easily combined with other antineoplastic agents in the treatment of multiple myeloma, there was interest in developing BTZ-containing combination therapy in MCL.
Work involving several MCL cell lines (Granta-519, HBL-2, and Jeko-1) showed synergistic anti-MCL activity resulting from sequential use of the traditional chemotherapy drug cytarabine and BTZ. This approach was used with some success in two MCL patients treated by the same investigators [51]. A synergistic anti-tumor effect of the HDAC inhibitor vorinostat (SAHA) and BTZ, possibly due to vorinostat-mediated interference of NFκB transcription and signaling, as well as generation of ROS and also caspase activation, in human MCL cell lines (Jeko-1 and Granta-519) was also reported [60]. Treatment of MCL cell lines (Mino, Jeko, Granta, and DBsp53) and primary patient samples with the combination of the JAK/STAT pathway inhibitor Degrasyn and BTZ synergistically inhibits growth and induces apoptosis. In MCL xenograft studies using SCID mice, the drug combination treatment prevents tumor development and prolongs animal survival [61]. Arsenic Trioxide (ATO), which has been shown to upregulate pro-apoptotic proteins and induce apoptosis in MCL through downregulation of the NFκB pathway, also displays synergy with BTZ [62–63]. This has been demonstrated in multiple MCL cell lines (Jeko-1, SP-53, Mino, and REC-1) and primary patient samples.
More recently, a synergistic inhibitory effect of idelalisib (CAL-101) and BTZ on the growth of human MCL cell lines (Z138, HBL-2, and Jeko-1) was reported [64]. Treatment of MCL cell lines with both CAL-101 and BTZ resulted in inhibition of the Akt and NFκB signaling pathways and concurrent increased apoptosis [65]. Idelalisib is a PI3K inhibitor that is FDA-approved for chronic lymphocytic leukemia and follicular lymphoma [65].
Perhaps most importantly, the combination of BTZ and the anti-CD20 monoclonal antibody rituximab was shown to have increased response in MCL cell lines (SP-53, Mino, and Jeko) and patient samples [64]. Combination of BTZ and Rituximab leads to enhanced inhibition of NFκB and Akt signaling pathways and induction of apoptosis. These findings related to rituximab and BTZ in MCL are of particular significance given the central role the antibody plays in clinical management of B-cell lymphomas.
In summary, there is ample pre-clinical evidence that BTZ has synergistic anti-MCL activity with other drugs, including ones with central importance in the clinical management of MCL such as rituximab.
6. Clinical Development of BTZ in MCL treatment
Phase I studies found that the maximally tolerated dose (MTD) of BTZ in previously-treated NHL patients was similar to that for MM: 1.04 to 1.5 mg/m2 [6, 66–67]. The phase II PINNACLE trial in 2006, in which patients with relapsed/refractory MCL were treated with BTZ monotherapy, showed an overall response rate of 32%. This trial formed the basis for the initial FDA approval of BTZ for the treatment of MCL with at least one prior therapy (Table 1) (6, 68). Subsequent Phase II trials which combined BTZ and rituximab with either purine analogues (69–71) or alkylating agents (72–73) generally demonstrated overall response rates of over 50%. Impressively, the combination of BTZ with a standard alkylator-containing regimen (Rituxan + HyperCVAD) in newly-diagnosed MCL is associated with a response rate of 90–100% (74–75). BTZ-containing front-line combinations in MCL are generally tolerated, with myelosuppression, gastrointestinal side effects, and therapy-emergent peripheral sensory neuropathy seen (74–80). In 2015, the FDA approved a BTZ-based drug combination as the front-line treatment for MCL after a large phase III study demonstrated that compared to R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), the VcR-CAP regimen (BTZ, rituximab, cyclophosphamide, doxorubicin, and prednisone) is associated with more durable disease control and better clinical responses [7–8] (Table 1).
There have been multiple clinical trials utilizing BTZ in combination with other chemotherapeutics in the treatment of MCL, since the initial recognition of BTZ’s efficacy in MCL (Table 2). Of particular interest is a trial combining BTZ and rituximab with bendamustine, an alkylating agent, in patients with relapsed/refractory indolent NHL and MCL [73]. This combination was associated with an impressive 83% response rate, and expected gastrointestinal and neuropathic side effects. In contrast, the combination of BTZ, rituximab and lenalidomide was somewhat disappointing. This is a regimen widely used in the initial and subsequent treatment of multiple myeloma, and incorporates another FDA-approved therapy for MCL. Though the combination was tolerable, the overall efficacy (response rate 45%) was not sufficiently increased to justify further exploration of the combination [81]. Similarly, other recent Phase 1/2 trials combining BTZ with clinically available drugs such as everolimus [82] and temsirolimus [83] did not demonstrate clinical activity commensurate to reported pre-clinical synergy.
Table 2.
Ongoing clinical trials of Bortezomib in Mantle Cell Lymphoma
| Name of Study | Status | Condition | Intervention |
|---|---|---|---|
| Bortezomib (Velcade) Plus Rituximab-HyperCVAD in Patients With Mantle Cell Lymphoma | Active, Not recruiting | Mantle Cell Lymphoma | Bortezomib, Rituximab, Cyclophosphamide, Vincristine, Methotrexate, Cytarabine, Doxorubicin |
| EPOCH-R Chemotherapy Plus Bortezomib to Treat Mantle Cell Lymphoma | Active, Not recruiting | Mantle Cell Lymphoma | EPOCH, Rituximab, Bortezomib |
| Rituximab, Lenalidomide, and Bortezomib in Mantle Cell Lymphoma | Active, Not recruiting | Mantle Cell Lymphoma | Rituximab, Bortezomib, Lenalidomide |
| Combination of Ibrutinib and Bortezomib to Treat Patients With Mantle Cell Lymphoma | Recruiting | Mantle Cell Lymphoma | Ibrutinib, Bortezomib |
| Bortezomib (VELCADE), Cladribine and Rituximab (VCR) in Mantle Cell Lymphoma (PSHCI 10–011) | Recruiting | Mantle Cell Lymphoma | Cladribine, Rituximab, Bortezomib |
| Trial of Bortezomib, Cytarabine, and Dexamethasone in Mantle Cell Lymphoma | Not Yet Recruiting | Mantle Cell Lymphoma | Bortezomib, Cytarabine, Dexamethasone, Pegfilgrastim |
| Bortezomib and Lenalidomide in Treating Patients With Relapsed or Refractory Mantle Cell Lymphoma | Active, Not recruiting | Mantle Cell Lymphoma | Bortezomib, Lenalidomide |
| Bortezomib, Cladribine, and Rituximab in Treating Patients With Advanced Mantle Cell Lymphoma or Indolent Lymphoma | Active, Not recruiting | Lymphoma, mantle cell lymphoma, indolent lymphoma, SLL | Cladribine, Rituximab, Bortezomib |
| Alisertib, Bortezomib, and Rituximab in Treating Patients With Relapsed or Refractory Mantle Cell Lymphoma or B-cell Low Grade Non-Hodgkin Lymphoma | Recruiting | Recurrent and refractory B-cell non-Hodgkin Lymphoma, recurrent and refractory mantle cell lymphoma | Alisertib, Rituximab, Bortezomib |
| Bortezomib and Vorinostat in Treating Patients With Recurrent Mantle Cell Lymphoma or Recurrent and/or Refractory Diffuse Large B-Cell Lymphoma | Active, Not recruiting | Recurrent adult diffuse large cell lymphoma, recurrent mantle cell lymphoma | Vorinostat, Bortezomib |
| Efficacy and Safety of R-HAD Alone or in Combination With Bortezomib in Patients With Relapsed or Refractory MCL | Recruiting | Mantle cell lymphoma | Rituximab, high dose Ara-C, Dexamethasone, Bortezomib |
Data obtained from clinicaltrials.gov
There are two main problems encountered from the use of BTZ: (i) toxicities related to proteasomal inhibition and off-target actions, and (ii) inherent and acquired resistance to the drug. Common toxicities associated with BTZ include gastrointestinal side effects, myelosuppression, and neurotoxicity (usually peripheral sensory neuropathy, but occasionally motor or autonomic). A meta-analysis of over 30 myeloma and NHL trials utilizing intravenous BTZ demonstrated a 34% incidence of treatment-associated neuropathy (8% severe) [84]. Weekly, rather than twice-weekly, dosing has not been shown to reduce the frequency or severity of neuropathy in NHL patients [85], though this is held to be true in myeloma. Subcutaneous BTZ administration in myeloma has been shown to reduce neurotoxicity without affecting the anti-cancer effects of the drug [86], and has led to the general use of subcutaneous administration in NHL patients, as well. Several second-generation proteasome inhibitors have been developed, each with unique chemical structure, biochemical properties, binding affinity, binding reversibility, potency and/or selectivity. Carfilzomib (CFZ) has been approved by the FDA for treating MM in 2013. It has been shown that CFZ binds the 20S proteasome with more specificity than BTZ, with little or no off-target activity outside of the proteasome (87). Carfilzomib is somewhat less neurotoxic than BTZ, a finding postulated to be related to the differing on-and off-target binding of the drugs. The free α–amino group required for adduct formation with CFZ does not interact with serine and cysteine proteases that can be inhibited by BTZ (88). CFZ selectively targets the β5 over β1 and β2 subunits in the constitutive 20S proteasome.
Most patients treated with BTZ- and rituximab-containing induction regimens respond to initial therapy but relapses are invariable. Once chemotherapy for relapsed disease is required, response rates are dramatically lower, including general response rates of less than 50% with most BTZ-containing salvage regimens.
BTZ resistance remains to be a great challenge in the field. Although several potential mechanisms have been identified in preclinical studies (Section 5.1), they should be confirmed in clinical settings. Other pathways should also be examined as potential strategies to overcome BTZ resistance. It may be possible to target other components of the UPS pathway such as E3 ubiquitin ligase [89] and 19S proteasome-associated DUBs [90–91] to induce anti-tumor responses in BTZ-sensitive and resistant conditions.
7. Conclusions
Traditional therapies for MCL have limited efficacy, with almost all patients eventually relapsing. Recent work is rapidly elucidating the molecular basis of tumorigenesis and drug resistance, with an increasing number of targetable cellular proteins and pathways identified – including ones relevant to the use of BTZ and other proteasome inhibitors. Research involving MCL cell lines, primary tumor samples, and animal models has demonstrated that BTZ inhibits the pro-survival NFκB pathway via CK2, induces the pro-apoptotic protein NOXA via PRDM1, and causes mitochondrial membrane depolarization, associated with ROS production and apoptosis induction. As with other therapies, primary and acquired resistance to BTZ is a limitation to its use versus MCL. Preclinical work supports the use of BTZ-containing combinations to partially overcome the problem of resistance. Numerous clinical trials in MCL have confirmed that combining BTZ with other anti-lymphoma therapies is both feasible and effective. Future work should focus, at least on: better understanding the mechanisms of action of BTZ responsible for its efficacy and resistance; developing more potent, more specific and less toxic USP inhibitors that have activity in BTZ-resistant systems; deeper insight on the clonal heterogeneity and predictive/prognostic biomarkers of MCL; further identification of specific, critical, essential cellular pathways for this disease. All these could lead to development of novel strategies for selectively targeting MCL and improving its treatment outcomes.
8. Expert Opinion
The key findings described above relate to the biological effects of BTZ in pre-clinical models and the promising results from subsequent clinical trials. The mechanism of BTZ activity is likely due to its effect on multiple intracellular processes. It is not clear which UPS substrate(s) is most important as a mediator of therapeutic response. Further, there may be multiple resistance mechanisms relevant to BTZ therapy in MCL, and these have not been fully elucidated.
Optimization of MCL therapy not only entails identifying the best antineoplastic agents, but also the most effective schedule and duration of therapy. Maintenance therapy with other drugs, namely rituximab, has been shown to prolong disease control in some types of NHL, raising the question as to whether a “continuous therapy” strategy might be helpful in MCL management. Long-term maintenance therapy with BTZ has been shown to be feasible and beneficial in another hematologic malignancy, multiple myeloma. This, as well as the recent development of a more convenient, well-tolerated oral proteasome inhibitor (Ixazomib ®, also called Ninlaro®) may make PI maintenance therapy more feasible in the future. Ultimately, even if combinations containing BTZ or other PIs fail to cure MCL, it is reasonable to hope they may make prolonged disease control possible for more patients.
Continued research into resistance pathways is going to be essential in achieving maximal benefit from BTZ and proteasome inhibitors in general in MCL. It may be possible to circumvent resistance to existing PIs by targeting other components of the UPS. For example, inhibition of some specific E3-ligases would stabilize expression of tumor suppressors IκBα, p27, etc., without generally inhibiting all proteasome activity globally, which is critical to normal cellular activities. Alternatively, new classes of drugs with completely different targets may potentiate the effects of BTZ. As one example, selinexor is a drug which selectively inhibits CRM-1 (also called exportin-1 or XPO-1) thereby blocking shuttling of tumor suppressor proteins out of the cell nucleus [91]. This oral agent has been shown to have preclinical activity in a variety of tumors, including solid tumors, NHL, and multiple myeloma. Preclinical work suggests treatment of PI-resistant myeloma cells with selinexor may partially restore PI-sensitivity [92]. This is being explored in clinical trials in relapsed/refractory myeloma presently [93]. Identifying key pathways and selectively targeting them in MCL cells (or the surrounding microenvironment, perhaps) should lead to an improved therapeutic index for these drugs. Clonal evolution and MCL cell heterogeneity may continue to pose an obstacle to completely overcoming drug resistance, as it has in other tumors. As we are now in an era in which molecular and genetic profiling of cancer cells is less time- and cost-prohibitive than ever before, it is likely that advances in cancer biology gleaned from other tumor types may be brought to bear against MCL. Targeted therapies approved for other cancers (or even non-malignant disease) may be able to be used versus MCL if patient-specific (even clone-specific) susceptibility factors could be identified. These factors are likely to change over the course of a single case of MCL as different therapies are used sequentially.
Article highlights.
The discovery and development of Bortezomib (BTZ), the first FDA approved 20S proteasome inhibitor drug for treating relapsed refractory multiple myeloma, has validated the therapeutic potential of targeting the proteasome and the ubiquitin-proteasome system (UPS) to selectively treat human cancer.
Mantle cell lymphoma (MCL) is an aggressive B-cell malignancy. Traditional therapies for MCL have met with limited efficacy, with almost all patients eventually experiencing relapse. Therefore, it is important to understand the molecular characteristics of MCL clonal heterogeneity, discover predictive/prognostic biomarkers, and develop new targeted therapies for treating this disease.
Preclinical MCL models demonstrated potent apoptotic effects of BTZ alone, as well as enhanced synergistic anti-MCL activity between BTZ and other chemotherapeutics.
BTZ alone induces clinical responses in approximately one-third of relapsed MCL patients, and BTZ-based combination therapies have further improved the quality and duration of clinical responses and enhanced survival rates, compared to standard front-line treatments in previously untreated MCL patients.
The cytotoxicity of BTZ in MCL cells results from its proteasome inhibitory activity,., through several potential mechanisms, including: accumulation of tumor suppressor proteins (p27, p21, p53 and NOXA), inhibition of the pro-survival NFκB pathway, mitochondrial membrane depolarization, ROS production, cell cycle arrest and apoptosis induction.
Inherent and acquired BTZ resistance remains the major challenge in MCL therapies. Preclinical studies of BTZ resistance have identified multiple possible mechanisms, such as plasmacytic differentiation, stem-like cell phenotypes, and defects in regulation or functions of NOXA. However, which of these is responsible for clinical BTZ resistance requires further investigation.
Next-generation proteasome inhibitors with different pharmacodynamic properties from BTZ may partially address the issue of resistance.
Targeting upstream components of the UPS pathway, such as E3 ubiquitin ligases and deubiquitinating enzymes may be a promising strategy for overcoming proteasome inhibitor resistance and improving the specificity and efficacy of BTZ-based combination therapies for treating patients with MCL.
Acknowledgments
We thank Dr. Gang Liu for his assistance with this manuscript.
FUNDING: This work was partially supported by National Cancer Institute grant R21CA184788 (to Q. Ping Dou) and National Institutes of Health grant P30 CA022453 (to the Karmanos Cancer Institute at Wayne State University).
Footnotes
DECLARATION OF INTEREST
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1.Vose JM. Mantle cell lymphoma: 2015 update on diagnosis, risk-stratification, and clinical management. American journal of hematology. 2015;90( 8):739–45. doi: 10.1002/ajh.24094. [DOI] [PubMed] [Google Scholar]
- 2.Ruan J, Martin P. Which Patients With Mantle Cell Lymphoma Do Not Need Aggressive Therapy. Current hematologic malignancy reports. 2016;11( 3):234–40. doi: 10.1007/s11899-016-0324-3. [DOI] [PubMed] [Google Scholar]
- 3.US Food and Drug Administration. [Accessed July 20, 2016];FDA approves bortezomib (Velcade) for the treatment of patients with mantle cell lymphoma who have received at least one prior therapy. 2009 www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm094929.htm.
- 4.Chen D, Frezza M, Schmitt S, et al. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011 Mar;11(3):239–53. doi: 10.2174/156800911794519752. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dou QP, Goldfarb RH. Bortezomib (millennium pharmaceuticals) IDrugs. 2002 Aug;5(8):828–34. [PubMed] [Google Scholar]
- 6*.Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24:4867–4874. doi: 10.1200/JCO.2006.07.9665. Clinical Trial demonstrating efficacy of BTZ in patients with Relapsed refractory MCL. [DOI] [PubMed] [Google Scholar]
- 7*.Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015 Mar 5;372(10):944–53. doi: 10.1056/NEJMoa1412096. Proved Enhanced Efficacy and statistically significant results for approval of BTZ in newly diagnosed MCL, not just in MCL patients receiving a previous treatment. [DOI] [PubMed] [Google Scholar]
- 8.Raedler L. Velcade (Bortezomib) Receives 2 New FDA Indications: For Retreatment of Patients with Multiple Myeloma and for First-Line Treatment of Patients with Mantle-Cell Lymphoma. Am Health Drug Benefits. 2015 Mar;8(Spec Feature):135–140. [PMC free article] [PubMed] [Google Scholar]
- 9.Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol. 1999;17:739–79. doi: 10.1146/annurev.immunol.17.1.739. Review. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003 Dec 18;426(6968):895–9. doi: 10.1038/nature02263. Review. [DOI] [PubMed] [Google Scholar]
- 11.Glickman MH, Ciechanover A. The ubiquitin–proteasome proteolytic pathway: Destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 12.Shen M, Schmitt S, Buac D, Dou QP. Targeting the ubiquitin-proteasome system for cancer therapy. Expert opinion on therapeutic targets. 2013;17(9):1091–1108. doi: 10.1517/14728222.2013.815728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thirunavukarasou A, Govindarajalu G, Singh, et al. Cullin 4A and 4B ubiquitin ligases interact with gamma-tubulin and induce its polyubiquitination. Molecular and cellular biochemistry. 2015;401( 1–2):219–28. doi: 10.1007/s11010-014-2309-7. [DOI] [PubMed] [Google Scholar]
- 14.Cole AJ, Clifton-Bligh R, Marsh DJ. Histone H2B monoubiquitination: roles to play in human malignancy. Endocrine-related cancer. 2015;22( 1):T19–33. doi: 10.1530/ERC-14-0185. [DOI] [PubMed] [Google Scholar]
- 15.Eletr ZM, Wilkinson KD. Regulation of proteolysis by human deubiquitinating enzymes. Biochimica et biophysica acta. 2014;1843( 1):114–28. doi: 10.1016/j.bbamcr.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams J. The proteasome: structure, function, and role in the cell. Cancer treatment reviews. 2003;29( Suppl 1):3–9. doi: 10.1016/s0305-7372(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 17.Peth A, Besche HC, Goldberg AL. Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol Cell. 2009;36(5):794–804. doi: 10.1016/j.molcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Wijk SJ, Timmers HT. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J. 2010 Apr;24(4):981–93. doi: 10.1096/fj.09-136259. Epub 2009 Nov 25. [DOI] [PubMed] [Google Scholar]
- 19.Pal A, Young MA, Donato NJ. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer research. 2014;74( 18):4955–66. doi: 10.1158/0008-5472.CAN-14-1211. [DOI] [PubMed] [Google Scholar]
- 20.Lilienbaum A. Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol. 2013;4(1):1–26. [PMC free article] [PubMed] [Google Scholar]
- 21.Chua JS, Liew HP, Guo L, et al. Tumor-specific signaling to p53 is mimicked by Mdm2 inactivation in zebrafish: insights from mdm2 and mdm4 mutant zebrafish. Oncogene. 2015 doi: 10.1038/onc.2015.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Nie Z, Chen W, et al. RNF115/BCA2 E3 ubiquitin ligase promotes breast cancer cell proliferation through targeting p21Waf1/Cip1 for ubiquitin-mediated degradation. Neoplasia. 2013;15( 9):1028–35. doi: 10.1593/neo.13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins PE, Colleran A, Carmody RJ. Control of NF-kappaB Subunits by Ubiquitination. Methods in molecular biology. 2015;1280:355–70. doi: 10.1007/978-1-4939-2422-6_21. [DOI] [PubMed] [Google Scholar]
- 24.Morelli M, Dennis AF, Patton JT. Putative E3 ubiquitin ligase of human rotavirus inhibits NF-kappaB activation by using molecular mimicry to target beta-TrCP. mBio. 2015;6(1) doi: 10.1128/mBio.02490-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001 Apr 1;61(7):3071–6. [PubMed] [Google Scholar]
- 26.Hideshima T, Chauhan D, Richardson P, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002 May 10;277(19):16639–47. doi: 10.1074/jbc.M200360200. Epub 2002 Feb 28. [DOI] [PubMed] [Google Scholar]
- 27.Adams J. Development of the proteasome inhibitor PS-341. The oncologist. 2002;7( 1):9–16. doi: 10.1634/theoncologist.7-1-9. [DOI] [PubMed] [Google Scholar]
- 28.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer cell. 2004;5( 5):417–21. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 29.Pellom ST, Jr, Shanker Anil. Development of Proteasome Inhibitors as Therapeutic Drugs. J Clin Cell Immunol. 2012 doi: 10.4172/2155-9899.S5-005. S5:005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-kB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20(1):87–92. doi: 10.1038/nm.3435. [DOI] [PubMed] [Google Scholar]
- 31.Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappy B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol. 2003 Jul 1;171(1):88–95. doi: 10.4049/jimmunol.171.1.88. [DOI] [PubMed] [Google Scholar]
- 32.Jia L, Gopinathan G, Sukumar JT, et al. Blocking Autophagy Prevents Bortezomib-Induced NF-κB Activation by Reducing I-κBα Degradation in Lymphoma Cells. PLoS ONE. 2012 doi: 10.1371/journal.pone.0032584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Periyasamy-Thandavan Sudharsan, Jackson William H, Samaddar Julia S, et al. Bortezomib blocks the catabolic process of autophagy via a cathepsin dependent mechanism, affects endoplasmic reticulum stress, and induces caspase dependent cell death in antiestrogen–sensitive and resistant ER+ breast cancer cells. Autophagy. 2010;6(1) doi: 10.4161/auto.6.1.10323. [DOI] [PubMed] [Google Scholar]
- 34**.Pérez-Galán P, Roué G, Villamor N, Montserrat E, et al. The proteasome inhibitor brotezomib induces apoptosis in mantle cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006 Jan 1;107(1):257–64. doi: 10.1182/blood-2005-05-2091. First to show ROS production and NOXA activation, important pathways mediating BTZ activity in MCL. [DOI] [PubMed] [Google Scholar]
- 35*.Rizzatti EG, Mora-Jensen H, Weniger MA, et al. Noxa mediates bortezomib-induced apoptosis in both sensitive and intrinsically resistant mantle cell lymphoma cells and this effect is independent of constitutive activity of the AKT and NF-kappaB pathways. Leuk Lymphoma. 2008 Apr;49(4):798–808. doi: 10.1080/10428190801910912. The pro-apoptotic protein NOXA is an important pathway in the anti-cancer effect of proteasome inhibition. This paper confirms its role controlling mantle cell lymphoma by BTZ. [DOI] [PubMed] [Google Scholar]
- 36.Wang CY, et al. NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 37.Akhter A, Mahe E, Street L, et al. CD10-positive mantle cell lymphoma: biologically distinct entity or an aberrant immunophenotype? Insight, through gene expression profile in a unique case series. Journal of clinical pathology. 2015;68( 10):844–8. doi: 10.1136/jclinpath-2015-202955. [DOI] [PubMed] [Google Scholar]
- 38.Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest. 2012;122(10):3416–3423. doi: 10.1172/JCI61272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin CC, Luthra R. Molecular detection of t(11;14)(q13;q32) in mantle cell lymphoma. Methods in molecular biology. 2013;999:211–6. doi: 10.1007/978-1-62703-357-2_14. [DOI] [PubMed] [Google Scholar]
- 40.Navarro A, Clot G, Royo C, et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 2012;72(20):5307–5316. doi: 10.1158/0008-5472.CAN-12-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Jima D, Moffitt AB, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123(19):2988–2996. doi: 10.1182/blood-2013-07-517177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmitt S, Deshmukh RR, Dou PQ. Resistance to Proteasome Inhibitors in Cancer. Springer; 2014. [Google Scholar]
- 43.Iqbal J, Liu Z, Deffenbacher K, et al. Gene expression profiling in lymphoma diagnosis and management. Best practice & research. Clinical haematology. 2009;22( 2):191–210. doi: 10.1016/j.beha.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Beà S, Valdés-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kridel R, Meissner B, Rogic S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119(9):1963–1971. doi: 10.1182/blood-2011-11-391474. [DOI] [PubMed] [Google Scholar]
- 46*.Chiarle R, Budel LM, Skolnik J, et al. Increased proteasome degradation of cyclin-dependent kinase inhibitor p27 is associated with a decreased overall survival in mantle cell lymphoma. Blood. 2000 Jan 15;95(2):619–26. First paper to show that inhibiting the proteasome can have anti-cancer activity in MCL. [PubMed] [Google Scholar]
- 47.Smolewski P, Witkowska M, Robak T. Treatment options for mantle cell lymphoma. Expert Opin Pharmacother. 2015;16(16):2497–507. doi: 10.1517/14656566.2015.1087507. Epub 2015 Sep 11. [DOI] [PubMed] [Google Scholar]
- 48.Wang CY, et al. NF-κB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy induced apoptosis. Mol Cell Biol. 1999;19:5923–5929. doi: 10.1128/mcb.19.9.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Desai S, Maurin M, Smith MA, et al. PRDM1 is required for mantle cell lymphoma response to Bortezomib. Mol Cancer Res. 2010 Jun;8(6):907–18. doi: 10.1158/1541-7786.MCR-10-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manni S, Brancalion A, Mandato E, et al. Protein kinase CK2 inhibition down modulates the NF-kB and STAT3 survival pathways, enhances the cellular proteotoxic stress and synergistically boosts the cytotoxic effect of bortezomib in multiple myeloma and mantle cell lymphoma cells. PLoS One. 2013 Sep 27;8(9):e75280. doi: 10.1371/journal.pone.0075280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51*.Weigert O, Pastore A, Rieken M, et al. Sequence-dependent synergy of the proteasome inhibitor bortezomib and cytarabine in mantle cell lymphoma. Leukemia. 2007 Mar;21(3):524–8. doi: 10.1038/sj.leu.2404511. One of the firsts to investigate the combination of Bortezomib with a cytotoxic drug. [DOI] [PubMed] [Google Scholar]
- 52.Pérez-Galán P, Mora-Jensen H, Weniger MA, et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood. 2011 Jan 13;117(2):542–52. doi: 10.1182/blood-2010-02-269514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jung HJ, Chen Z, Fayad L, et al. Bortezomib-resistant nuclear factor κB expression in stem-like cells in mantle cell lymphoma. Exp Hematol. 2012 Feb;40(2):107–18. e2. doi: 10.1016/j.exphem.2011.10.004. Epub 2011 Oct 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leshchenko VV, Kuo PY, Jiang Z, et al. Harnessing Noxa demethylation to overcome Bortezomib resistance in mantle cell lymphoma. Oncotarget. 2015 Sep 29;6(29):27332–42. doi: 10.18632/oncotarget.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roué G, Pérez-Galán P, Mozos A, et al. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival ER chaperone BiP/Grp78. Blood. 2011 Jan 27;117(4):1270–9. doi: 10.1182/blood-2010-04-278853. Epub 2010 Nov 24. [DOI] [PubMed] [Google Scholar]
- 56.Kim A, Park S, Lee JE, et al. The dual PI3K and mTOR inhibitor NVP-BEZ235 exhibits anti-proliferative activity and overcomes bortezomib resistance in mantle cell lymphoma cells. Leuk Res. 2012 Jul;36(7):912–20. doi: 10.1016/j.leukres.2012.02.010. Epub 2012 May 5. [DOI] [PubMed] [Google Scholar]
- 57.Xargay-Torrent S, López-Guerra M, Montraveta A, et al. Sorafenib inhibits cell migration and stroma-mediated bortezomib resistance by interfering B-cell receptor signaling and protein translation in mantle cell lymphoma. Clin Cancer Res. 2013 Feb 1;19(3):586–97. doi: 10.1158/1078-0432.CCR-12-1935. Epub 2012 Dec 11. [DOI] [PubMed] [Google Scholar]
- 58.Jung HJ, Chen Z, Wang M, et al. Calcium blockers decrease the bortezomib resistance in mantle cell lymphoma via manipulation of tissue transglutaminase activities. Blood. 2012 Mar 15;119(11):2568–78. doi: 10.1182/blood-2011-09-377598. Epub 2012 Jan 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim A, Seong KM, Kang HJ, et al. Inhibition of Lyn is a promising treatment for mantle cell lymphoma with bortezomib resistance. Oncotarget. 2015 Nov 10;6(35):38225–38. doi: 10.18632/oncotarget.5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heider U, von Metzler I, Kaiser M, et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in mantle cell lymphoma. Eur J Haematol. 2008 Feb;80(2):133–42. doi: 10.1111/j.1600-0609.2007.00995.x. [DOI] [PubMed] [Google Scholar]
- 61.Pham LV, Tamayo AT, Li C, Bornmann W, et al. Degrasyn potentiates the antitumor effects of bortezomib in mantle cell lymphoma cells in vitro and in vivo: therapeutic implications. Mol Cancer Ther. 2010 Jul;9(7):2026–36. doi: 10.1158/1535-7163.MCT-10-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jung HJ, Chen Z, McCarty N. Synergistic anticancer effects of arsenic trioxide with bortezomib in mantle cell lymphoma. Am J Hematol. 2012;87:1057–1064. doi: 10.1002/ajh.23317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao LL, Liu YF, Peng LJ, et al. Arsenic trioxide rewires mantle cell lymphoma response to bortezomib. Cancer Med. 2015 Nov;4(11):1754–66. doi: 10.1002/cam4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64*.Alinari L, White VL, Earl CT, et al. Combination bortezomib and rituximab treatment affects multiple survival and death pathways to promote apoptosis in mantle cell lymphoma. MAbs. 2009 Jan-Feb;1(1):31–40. doi: 10.4161/mabs.1.1.7472. First to show the Bortezomib-Rituximab combination, which is now part of the front-line treatment for MCL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qu FL, Xia B, Li SX, et al. Synergistic suppression of the PI3K inhibitor CAL-101 with bortezomib on mantle cell lymphoma growth. Cancer Biol Med. 2015 Dec;12(4):401–8. doi: 10.7497/j.issn.2095-3941.2015.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orlowski RZ, et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol. 2002 Nov 15;20(22):4420–7. doi: 10.1200/JCO.2002.01.133. [DOI] [PubMed] [Google Scholar]
- 67.O’Connor OA, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2005 Feb 1;23(4):676–84. doi: 10.1200/JCO.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 68.Goy A, et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Annals of Oncology. 2009;20(3):520–5. doi: 10.1093/annonc/mdn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weigert O, et al. A novel regimen combining high dose cytarabine and bortezomib has activity in multiply relapsed and refractory mantle cell lymphoma - long-term results of a multicenter observation study. Leuk Lymphoma. 2009 May;50(5):716–22. doi: 10.1080/10428190902856790. [DOI] [PubMed] [Google Scholar]
- 70.Kouroukis CT, et al. A phase II study of bortezomib and gemcitabine in relapsed mantle cell lymphoma from the National Cancer Institute of Canada Clinical Trials Group (IND 172) Leuk Lymphoma. 2011;52(3):394–9. doi: 10.3109/10428194.2010.546015. [DOI] [PubMed] [Google Scholar]
- 71.Orciuolo E, Buda G, Pelosini M, et al. Fludarabine, Bortezomib, Myocet and rituximab chemotherapy in relapsed and refractory mantle cell lymphoma. Br J Haematol. 2010 Mar;148(5):810–2. doi: 10.1111/j.1365-2141.2009.07998.x. [DOI] [PubMed] [Google Scholar]
- 72*.Musto PM, et al. A pilot study with rituximab, bortezomib and hyper-fractionated cyclophosphamide (rbc regimen) for the treatment of advanced mantle cell lymphoma in elderly patients. Haematologica. 2008;93(suppl 1) abstr 0273. Of particular interest is a trial combining BTZ and rituximab with bendamustine, an alkylating agent, in patients with relapsed/refractory indolent NHL and MCL. [Google Scholar]
- 73*.Friedberg JW, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood. 2011 Mar 10;117(10):2807–12. doi: 10.1182/blood-2010-11-314708. Study demonstrating efficiacy of incorporating BTZ into MCL treatment regimens that led to its FDA approval for clinical trials and approval as front line treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74*.Romaguera JE, et al. Phase I trial of bortezomib in combination with rituximab-HyperCVAD alternating with rituximab, methotrexate and cytarabine for untreated aggressive mantle cell lymphoma. Br J Haematol. 2010 Oct;151(1):47–53. doi: 10.1111/j.1365-2141.2010.08315.x. Study demonstrating efficiacy of incorporating BTZ into MCL treatment regimens that led to its FDA approval for clinical trials and approval as front line treatment. [DOI] [PubMed] [Google Scholar]
- 75.Kahl B, et al. VcR-CVAD Produces a High Complete Response Rate in Untreated Mantle Cell Lymphoma: A Phase II Study from the Wisconsin Oncology Network. Blood. 2008;112 abstr 265. [Google Scholar]
- 76.Ribrag V, et al. Final Results of a Randomized Phase 2 Multicenter Study of Two Bortezomib Schedules In Patients with Recurrent or Refractory Follicular Lymphoma. Groupe d’Etude Des Lymphomes De l’Adulte (GELA) Study FL-05. Blood. 2010;116 abstr 768. [Google Scholar]
- 77.Di Bella N, et al. Results of a phase 2 study of bortezomib in patients with relapsed or refractory indolent lymphoma. Blood. 2010 Jan 21;115(3):475–80. doi: 10.1182/blood-2009-08-233155. [DOI] [PubMed] [Google Scholar]
- 78.O’Connor OA, et al. Time to treatment response in patients with follicular lymphoma treated with bortezomib is longer compared with other histologic subtypes. Clin Cancer Res. 2010 Jan 15;16(2):719–26. doi: 10.1158/1078-0432.CCR-08-2647. [DOI] [PubMed] [Google Scholar]
- 79.Sehn LH, et al. Bortezomib ADDED to R-CVP is safe and effective for previously untreated advanced-stage follicular lymphoma: a phase II study by the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2011 Sep 1;29(25):3396–401. doi: 10.1200/JCO.2010.33.6594. [DOI] [PubMed] [Google Scholar]
- 80.Nabhan C, Dalal N, Tolzien K, et al. Bortezomib (VELCADE), Rituximab, Cyclophosphamide, Dexamethasone (VRCD) Combination Therapy In Front-Line Low-Grade Non-Hodgkin Lymphoma (LG-NHL) Is Active In Elderly Patient Population. Blood. 2010;116 abstr 1769. [Google Scholar]
- 81.Morrison VA, Jung SH, Johnson J, et al. Therapy with bortezomib plus lenalidomide for relapsed/refractory mantle cell lymphoma: final results of a phase II trial (CALGB 50501) Leuk Lymphoma. 2015 Apr;56(4):958–64. doi: 10.3109/10428194.2014.938333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang M, Popplewell LL, Collins RH, Jr, et al. Everolimus for patients with mantle cell lymphoma refractory to or intolerant of bortezomib: multicentre, single-arm, phase 2 study. Br J Haematol. 2014 May;165(4):510–8. doi: 10.1111/bjh.12780. [DOI] [PubMed] [Google Scholar]
- 83.Fenske TS, Shah NM, Kim KM, et al. A phase 2 study of weekly temsirolimus and bortezomib for relapsed or refractory B-cell non-Hodgkin lymphoma: A Wisconsin Oncology Network study. Cancer. 2015 Oct 1;121(19):3465–71. doi: 10.1002/cncr.29502. [DOI] [PubMed] [Google Scholar]
- 84.Agathocleous A, Rohatiner A, Rule S, et al. Weekly versus twice weekly bortezomib given in conjunction with rituximab, in patients with recurrent follicular lymphoma, mantle cell lymphoma and Waldenström macroglobulinaemia. Br J Haematol. 2010 Nov;151(4):346–53. doi: 10.1111/j.1365-2141.2010.08340.x. [DOI] [PubMed] [Google Scholar]
- 85*.Peng L, Ye X, Zhou Y, et al. Meta-analysis of incidence and risk of peripheral neuropathy associated with intravenous bortezomib. Support Care Cancer. 2015 Sep;23(9):2813–24. doi: 10.1007/s00520-015-2648-2. Study demonstrating the risk and types of side effects associated with BTZ. [DOI] [PubMed] [Google Scholar]
- 86.Moreau P, Pylypenko H, Grosicki S, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011 May;12(5):431–40. doi: 10.1016/S1470-2045(11)70081-X. [DOI] [PubMed] [Google Scholar]
- 87*.Ruschak AM, Slassi M, Kay LE, et al. Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst. 2011;103:1007–17. doi: 10.1093/jnci/djr160. Studies that introduce novel (Irreversible) PIs as potential treatments to counteract BTZ resistance in refractory MCL. [DOI] [PubMed] [Google Scholar]
- 88.Arastu-Kapur S, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res. 2011 May 1;17(9):2734–43. doi: 10.1158/1078-0432.CCR-10-1950. Epub Mar 1 (2011)’. [DOI] [PubMed] [Google Scholar]
- 89.Jones RJ, Bjorklund CC, Baladandayuthapani V, et al. Drug resistance to inhibitors of the human double minute-2 E3 ligase is mediated by point mutations of p53, but can be overcome with the p53 targeting agent RITA. Mol Cancer Ther. 2012 Oct;11(10):2243–53. doi: 10.1158/1535-7163.MCT-12-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.D’Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther. 2015 Mar;147:32–54. doi: 10.1016/j.pharmthera.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 91.Farshi P, Deshmukh RR, Arkwright RT, et al. Deubiquitinases (DUBs) and DUB inhibitors: a patent review. Expert Opin Ther Pat. 2015;25(10):1191–208. doi: 10.1517/13543776.2015.1056737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Das A, Wei G, Parikh K, et al. Selective inhibitors of nuclear export (SINE) in hematological malignancies. Exp Hematol Oncol. 2015 Mar 1;4:7. doi: 10.1186/s40164-015-0002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosebeck S, Alonge MM, Kandarpa M, et al. Synergistic Myeloma Cell Death via Novel Intracellular Activation of Caspase-10-Dependent Apoptosis by Carfilzomib and Selinexor. Mol Cancer Ther. 2016 Jan;15(1):60–71. doi: 10.1158/1535-7163.MCT-15-0488. [DOI] [PubMed] [Google Scholar]