

Abstract
The accumulation of reactive oxygen species (ROS) have implicated the pathogenesis of several human diseases including neurodegenerative disorders, stroke, and traumatic brain injury, hence protecting neurons against ROS is very important. In this study, we focused on sigma-1 receptor (Sig-1R), a chaperone at endoplasmic reticulum, and investigated its protective functions. Using hydrogen peroxide (H2O2)-induced ROS accumulation model, we verified that apoptosis-signaling pathways were elicited by H2O2 treatment. However, the Sig-1R agonists, dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS), reduced the activation of apoptotic pathways significantly. By performing protein-protein interaction assays and shRNA knockdown of Sig-1R, we identified the brain Zinc finger protein 179 (Znf179) as a downstream target of Sig-1R regulation. The neuroprotective effect of Znf179 overexpression was similar to that of DHEAS treatment, and likely mediated by affecting the levels of antioxidant enzymes. We also quantified the levels of peroxiredoxin 3 (Prx3) and superoxide dismutase 2 (SOD2) in the hippocampi of wild-type and Znf179 knockout mice, and found both enzymes to be reduced in the knockout versus the wild-type mice. In summary, these results reveal that Znf179 plays a novel role in neuroprotection, and Sig-1R agonists may be therapeutic candidates to prevent ROS-induced damage in neurodegenerative and neurotraumatic diseases.
Keywords: Sig-1R, Znf179, DHEA/DHEAS, reactive oxygen species, hydrogen peroxide
Introduction
Reactive oxygen species (ROS) including free radicals, such as superoxide anion (·O2−) and hydroxyl radical (OH), as well as non-radical oxidants, such as singlet oxygen (1O2) and hydrogen peroxide (H2O2), are produced as by-products of cellular metabolism, majorly in the mitochondria [1]. In normal cellular processes, ROS are regulated at relatively low steady-state levels (background levels) by endogenous antioxidant enzymes. However, many neurodegenerative diseases, including Alzheimer’s (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and neurotraumatic diseases, including stoke, traumatic brain injury (TBI), show a reduction in antioxidant capacity and/or an increase in ROS production [2–5]. They may result from impaired mitochondrial function or environmental stressors including sugar/oxygen deprivation in ischemia, cytokine release in inflammation, excitatory neurotransmitter release, and metabolic depression [4–6]. Left unchecked, the accumulated ROS like the hydroxyl free radical will damage DNA and induce apoptosis. Therefore, preventing ROS accumulation or enzymatically eliminating them is an important subject in neurological disorders therapy.
The sigma-1 receptor (Sig-1R) is widely expressed in the endoplasmic reticulum (ER) and mitochondrial and plasma membranes of neurons throughout the central nervous system [7]. It binds certain neuroleptic drugs, psychotropic drugs, and steroid hormones [7, 8]. These drugs regulate the ability of Sig-1R to associate with other ER protein partners, including an inositol 1-4-5-triphosphate receptor (IP3R), the potassium voltage-gated channel Kv1.2, and Binding immunoglobulin protein (BiP) [9, 10]. Activation of Sig-1R appears to provide neuroprotection and neurorestoration in cellular and animal models of brain ischemia, and in neurodegenerative diseases such as AD, PD, and ALS [11, 12].
Dehydroepiandrosterone (DHEA) and its sulfated analog (DHEAS) are steroid hormone agonists of Sig-1R [13–15]. DHEA/DHEAS are secreted by the adrenal gland and brain, and can prevent excitatory amino acid-induced neurotoxic actions in primary hippocampal neurons and retinal cells, reduce apoptosis in a model of serum-free PC12 cell death, and attenuate the lesion area of focal brain injury [16, 17]. Additionally, some studies indicate that the two steroid hormones perform antioxidant functions in age-related neurodegenerative disorders and cardiovascular disease by increasing the expression of antioxidant proteins such as superoxide dismutases (SODs) [18, 19]. However, the relationship between DHEA/DHEAS-mediated antioxidant protein expression and Sig-1R signaling, and whether either activity is applicable in neurological disorder therapy, is still unknown.
The zinc finger protein 179 (Znf179), also known as RING finger protein 112 (Rnf112), is predominantly expressed in the central nervous system [20], and is known to be active in nervous system development during embryogenesis [21]. Although the roles of Znf179 post-embryogenesis still remain unclear, it is a downstream target of Sig-1R, and could theoretically mediate the neuroprotective effects of DHEA/DHEAS. This study evaluates the functional effects of both Sig-1R and Znf179 in relation to ROS-induced damage.
Materials and methods
Cells
Mouse neuroblastoma Neuro-2a (N2a) cells (ATCC) were cultured in minimum essential medium Eagle (MEM, Invitrogen) containing 10% fetal bovine serum (FBS), 100 μg/ml streptomycin sulfate, and 100 U/ml penicillin-G sodium at 37°C and 5% CO2. N2a cells were differentiated after serum withdrawal by incubation in MEM/BSA medium (MEM supplemented with 0.1% bovine serum albumin and without FBS) [22]. The 90% confluent cells were treated with DHEA (1 μM or 10 μM, Sigma-Aldrich), DHEAS (1 μM or 10 μM, Sigma-Aldrich), or BD1063 (1 μM, Tocris Bioscience) for 30 min before H2O2 treatment. Transfection of cells with protein-expressing vectors or shRNA plasmids was performed by using Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen). Each transfection experiment was performed three times and each sample in each experiment was prepared in duplicate.
Western blot analysis
Protein samples were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto a PVDF membrane (Bio-Rad Laboratories). Subsequently, the membrane was blocked with 5% nonfat milk in TBST buffer (10 mM Tris. pH 8.0, 150 mM NaCl, and 0.5% Tween 20) for 1 h and incubated with primary antibodies: Anti-Sig-1R (1:500), anti-emerin (1:2000), anti-Nesprin1 (1:500), anti-Sun2 (1:500), anti-(activating transcription factor6) ATF6 (1:500) antibodies from Santa Cruz Biotechnology, anti-p53 (1:3000), anti-phospho-p53 (Ser15) (1:3000), anti-p38 (1:2000), anti-phospho-p38 (Thr180/Tyr182) (1:1000), anti-JNK (1:2000), anti-phospho-JNK (Thr183/Tyr185) (1:500), anti-ERK (1:3000), anti-phospho-ERK (Thr202/Tyr204) (1:3000), anti-(poly (ADP-ribose) polymerase) PARP (1:2000), anti-TNF-alpha (1:1000) antibodies from Cell Signaling Technology, anti-SOD1 (1:3000), anti-SOD2 (1:3000), anti-Prx3 (1:1000), anti-caspase 3 (1:500) antibodies from GeneTex, anti-actin (1:3000), anti-Sp3 (1:1000), HDAC1 (1:2000), anti-HDAC2 (1:2000) antibodies from Millipore, anti-MeCP2 (1:1000) antibody from Abcam, anti-(binding immunoglobulin protein) BiP (1:3000) from BD biosciences, and anti-Znf179 (1:1000) antibody [21, 23], for 2 h at room temperature. After incubation with primary antibodies, the membranes were washed three times (5 min each) with TBST buffer, and then incubated with a 1:3000 dilution of horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. Finally, the membranes were washed again and the peroxidase was developed by chemiluminescence using Amersham Hyperfilm ECL (GE Healthcare).
His-pull-down assay
His-Sig-1R expressing cells were lysed by using modified RIPA buffer (50 mM Tris, pH 7.8, 150 mM NaCl, 5 mM EDTA, 0.5% Triton-X100, 0.1% Nonidet P-40, and protease inhibitors) contained 1 mM phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 1 mM imidazole. The cell lysates were then added with 30 μl of HisPur Ni-NTA Superflow Agarose (Thermo Scientific) and incubated at 4°C with shaking for 2 h. The beads were subsequently washed three times in modified RIPA buffer containing 10 mM imidazole. Bound proteins were eluted by using electrophoresis sample buffer and analyzed by western blot using specific antibodies, such as ER proteins (binding immunoglobulin protein (BiP), activating transcription factor (ATF) 6, etc.), nuclear envelope proteins (emerin, laminB, lamin-B receptor, lamin A/C, Nesprin1, Sun2, etc.), transcription factors (Sp3, MeCP2, barrier-to-autointegration factor (BAF), etc.), and cytoplasmic- nuclear proteins (Histone deacetylases (HDACs), Znf179, extracellular signal-regulated kinases (ERK) 1 and 2, etc.).
Immunoprecipitation
Cells (1 × 107) were washed with PBS, and the cellular lysates were prepared by using modified RIPA buffer. Rabbit polyclonal anti-Znf179 antibodies or control rabbit IgG were then added (1: 500) to the lysates and incubated at 4°C with rotation. After 2 h, protein-A/G agarose beads (30 μl, Santa Cruz Biotechnology) were also added to the mixture and further incubated for 1 h. The protein beads were washed three times with modified RIPA buffer. Bound proteins were eluted by using electrophoresis sample buffer.
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay
N2a cells were plated onto 24-well culture plates at an initial density of 1 × 105 cells/well. After the induction of neuronal differentiation, cells were treated with different doses of H2O2 for 24 h or were treated with 50 μM H2O2 for different time intervals. Subsequently, fresh medium containing MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent (final concentration of 0.5 mg/ml, Sigma Aldrich) was added to each well, and cells were incubated for 1 h at 37°C, after which the MTT solution was removed. The resultant formazan crystals were dissolved in 200 μl DMSO, and the absorbance readings of DMSO extracts measured at 570 nm using the iMark Microplate Absorbance Reader (Bio-Rad).
Experimental animals
We used male wild-type mice (C57BL/6, National Laboratory Animal Center, Taipei, Taiwan) and Znf179 knockout mice (129/Sv. μ C57BL/6) weighing 30–40 g, housed five per cage in an air-conditioned vivarium with free access to food and water. Throughout the study, a 12-h light/dark cycle was maintained with lights on at 8 AM. All procedures adhered to the Guidelines for Care and Use of Experimental Animals of the Taipei Medical University (Taipei, Taiwan).
Measurement of cellular ROS
Intracellular ROS levels (superoxide anion, ·O2−) were measured by using dihydroethidium (DHE), which can be oxidized by ·O2− into 2-hydroxyethidium and exhibits red fluorescence (Ex/Em=518/605 nm; Invitrogen). Differentiated N2a cells (1.5 × 104 in each well of a 96-well plate) were treated with 50 μM H2O2 (1h up to 3h). Then, the cells were washed once with pre-warmed PBS supplemented with 10 μM DHE. After incubation at 37 °C for 30 min, cells were washed again and DHE fluorescence was detected by the FL2 laser of a flow cytometer (BD FACSCalibur), and analyzed by BD CellQuest software.
Statistical Analysis
The statistical analyses for data from the western blots, MTT assays, and cellular ROS quantification were calculated using the unpaired, two-tailed Student’s t-test, with a significance level set at p < 0.05.
Results
The activation of Sig-1R by DHEA/DHEAS attenuates H2O2-induced cell death
H2O2 treatment were used in a neuron-like cell model (differentiated N2a cells) to induce intracellular ROS production. By examining the cell death-related signaling pathways, we showed that the phosphorylated levels of p53, p38, and JNK were rapidly increased as early as 10 min after H2O2 treatment, and were clearly visible for up to 3 h in H2O2-treated cells compared to control cells (Fig. 1A and 1B). Since previous studies have identified neuroprotective effects of Sig-1R activity [11, 12], agonists of Sig-1R (DHEA/DHEAS) were used to study whether activated Sig-1R protects cells against H2O2-induced cytotoxicity (Fig. 1C to 1F). The data in Fig. 1C and 1D demonstrates that DHEA and DHEAS treatment significantly attenuated the phosphorylation of p53, p38, and JNK in H2O2-treated N2a cells. In addition, the levels of PARP 1 cleavage fragments, remnants of an apoptotic marker following cleavage by caspases 3 and 7, and of phospho-p53, revealed that DHEAS significantly reduced not only p53 signaling in an early stage of apoptosis, but also downstream components (the late-stage nuclear events) of the apoptotic pathway (Fig. 1E and 1F). Notably, the neuroprotective effects of DHEAS were inhibited by the selective Sig-1R antagonist, BD1063 (Fig. 1G). Quantification of the levels of phospho-p53 and phospho-JNK in H2O2−, DHEAS-, and/or BD1063-treated cells confirmed that DHEAS, via Sig-1R activation, suppressed H2O2-induced cell death signaling (Fig. 1H). Taken together, these data suggest Sig-1R is capable of reducing ROS-induced cytotoxic damage.
Figure 1.
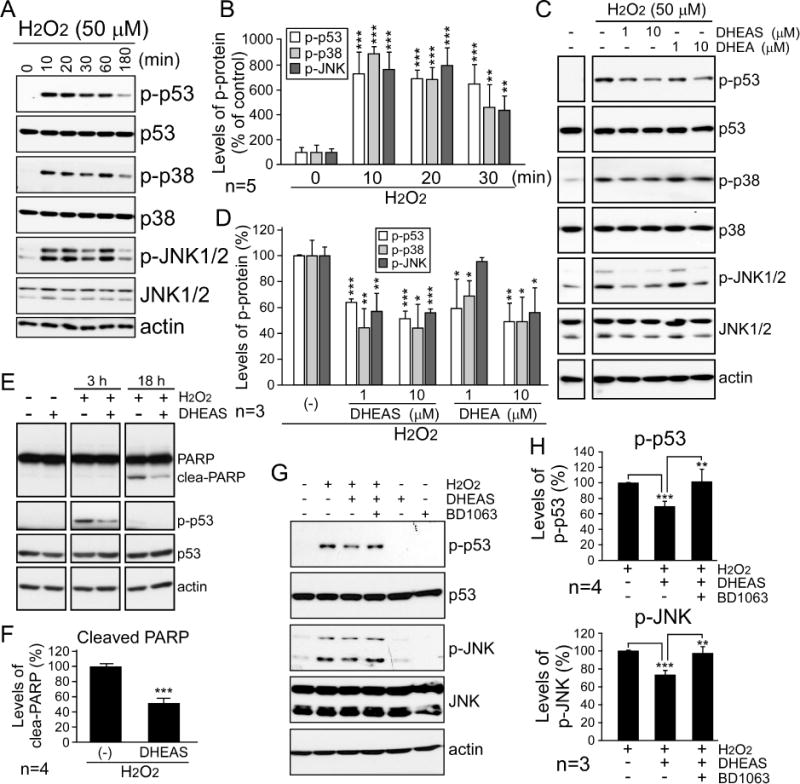
Activated sigma-1 receptor (Sig-1R) by dehydroepiandrosterone sulfate (DHEA/S) attenuates the activation of cell death signaling pathways significantly. (A) N2a cells, which were differentiated into neuron-like cells, were treated with 50 μM H2O2 and collected after different time intervals (0, 10, 20, 30, 60 and 180 min). Proteins were analyzed via western blot using the indicated antibodies. (B) Results from five independent experiments in A (t-test: **p < 0.01, ***p < 0.001). (C to F) The cells were treated with 50 μM H2O2 or co-treated with different doses of DHEAS and DHEA as indicated for 1 hour (C), or 3 and 18 hours (E). The cells were then analyzed by western blot using the indicated antibodies. The phosphorylated levels of p53 (Ser15), p38 (Thr180/Tyr182), and JNK (Thr183/Tyr185) normalized to their total protein expression (D) and the cleaved (clea-) PARP normalized to total PARP (F) were quantified. Bars represent means ± s.e.m. from three to four independent experiments (t-test: *p < 0.05, **p < 0.01, ***p < 0.001). (G) The cells were pre-treated with (+) or without (−) 1 μM BD1063, a Sig-1R antagonist, for 30 min as indicated before H2O2 treatment and/or DHEAS co-treatment, and were then analyzed by immunoblotting. (H) Results from three to four independent experiments in G (t-test: **p < 0.01, ***p < 0.001).
Sig-1R associates with Znf179 and affects Znf179 protein expression
We next examined the downstream targets of Sig-1R. By using an in vitro His-tag protein pull-down assay, we found that Sig-1R associated with several proteins, including ER proteins (BiP), nuclear envelope proteins (emerin, lamin A/C, Nesprinl, Sun2), transcription factors (Sp3, BAF), and cytoplasmic-nuclear proteins (HDAC1, HDAC2, HDAC3, Znf179), but not with ATF6, MeCP2, lamin B, lamin-B receptor, ERK, etc., which are shown in Fig. 2A and our more recent study [24]. Immunoprecipitation assay was subsequently performed to confirm the in vivo protein interactions. A result showed here that the interaction between Sig-1R and Znf179 was significantly increased after H2O2 treatment (Fig. 2B and 2C). In analyzing the functional effect of Sig-1R on Znf179, DHEA and DHEAS were found to significantly elevate Znf179 protein levels (Fig. 2D and 2E). By contrast, a significant decrease in Znf179 was detected in BD1063 treatment (Fig. 2D and 2E) and Sig-1R knockdown (Fig. 2F and 2G) relative to control treatment. In summary, Sig-1R stimulation plays a critical role in the regulation of Znf179 expression levels.
Figure 2.
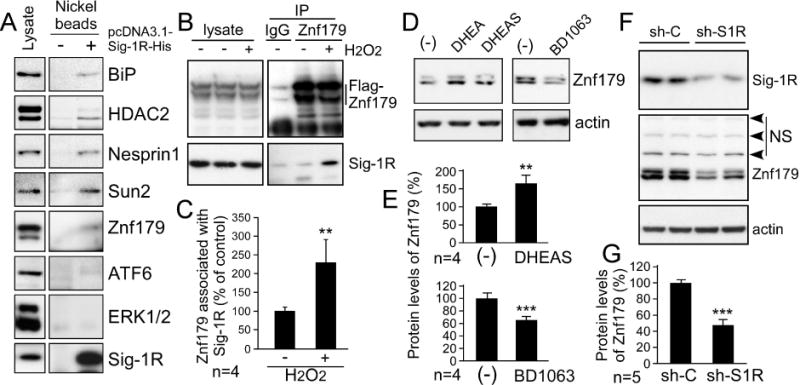
Oxidative stress induces the interaction of sigma-1 receptor (Sig-1R) and Znf179, and Sig-1R affects the expression level of Znf179. (A) Differentiated N2a cells were transiently transfected with (+) or without (−) pcDNA3.1-Sig-1R-His to overexpress Sig-1R-His. The cell lysates were then incubated with Ni-NTA agarose to precipitate Sig-1R-His and its associating proteins. The precipitated samples were then analyzed by immunoblotting analysis with antibodies as indicated. (B) Differentiated N2a cells were treated with (+) or without (−) 50 μM H2O2 for 30 min. After treatment, these cells were used for the immunoprecipitation assay with anti-Znf179 antibodies and rabbit IgG, and analyzed via immunoblotting. (C) H2O2 increased Sig-1R association with Znf179. Bars represent means ± s.e.m. from four independent experiments. (t-test: **p < 0.01) (D) Cells were treated with 10 μM dehydroepiandrosterone (DHEA), 10 μM DHEA sulfate (DHEAS), or 1 μM BD1063 as indicated for 18 h, and protein was then analyzed via immunoblotting. (E) Results from four independent experiments in D (t-test: **p < 0.01, ***p < 0.001). (F) Differentiated N2a cells were transfected with control shRNA (sh-C) or Sig-1R shRNA (sh-S1R). After 48 h incubation, cells were analyzed via western blot with the indicated antibodies. NS is nonspecific bands at 80 kDa, 100 kDa, and 130 kDa recognized by the anti-ZNF179 antibody. (G) Sig-1R knockdown reduced Znf179 protein levels. Bars represent means ± s.e.m. from five independent experiments (t-test: ***p < 0.001).
Znf179 protects cell survival against cell death signaling pathway
Since H2O2 increased the interaction of Sig-1R and Znf179, and Sig-1R affected Znf179 expression, we studied the expression of Znf179 after peroxide intoxication to correlate Znf179 expression with oxidative stress. Differentiated N2a cells were treated with H2O2 for different time intervals (0–24 h, Fig 3A and 3B) or with different doses of H2O2, for 18 h and 24 h (Fig. 3C and 3D). Western blotting results demonstrated that Znf179 was a statistically significant increase in response to H2O2 exposure in a time- and dose-dependent manner (Fig. 3).
Figure 3.
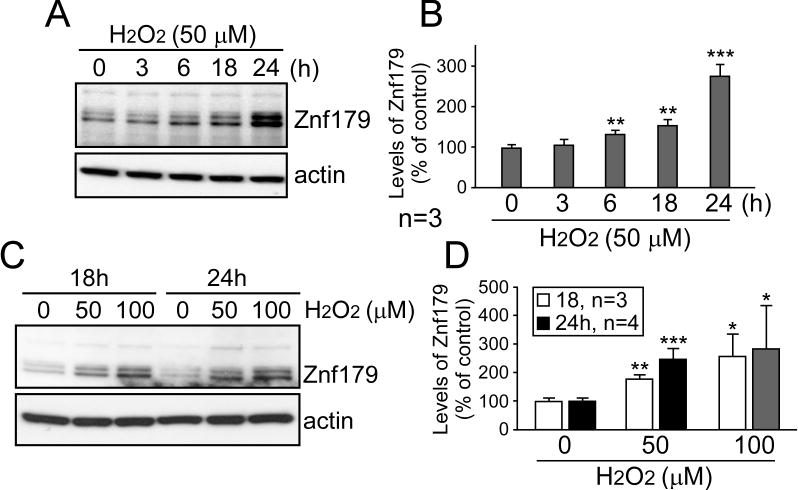
The protein level of Znf179 is upregulated in H2O2 injury cell model. Differentiated N2a cells were treated with 50 μM H2O2 and collected after different time intervals (0, 3, 6, 18, and 24 hours, A and B) or were treated with different doses of H2O2 as indicated for 18 and 24 hours (C and D). Proteins from these cells were then analyzed via western blot with the indicated antibodies. (B and D) Znf179 levels normalized to the loading control were quantified in A and C. Bars represent means ± s.e.m. from three to four independent experiments (t-test: *p < 0.05, **p < 0.01, ***p < 0.001).
Znf179 overexpression was subsequently used to study the role of Znf179 in oxidative stress. Increasing Znf179 levels attenuated H2O2-induceded phosphorylation of p53, p38, and JNK, but did not affect ERK (Fig. 4A to 4C). In addition, the apoptotic markers cleaved-caspase 3 and non-cleaved-PARP 1, and the inflammation marker tumor necrosis factor alpha (TNF-alpha), were also investigated in Znf179-overexpressing N2a cells (Fig. 4D). We found that Znf179 reduced apoptosis and TNF-alpha production after H2O2 exposure. However, a significant increase in p53 activation was detected when Znf179 was knocked down using a short hairpin RNA (Fig. 4E and 4F). Taken together, these results provide the first demonstration of Znf179 expression as being neuroprotective against oxidative stress.
Figure 4.
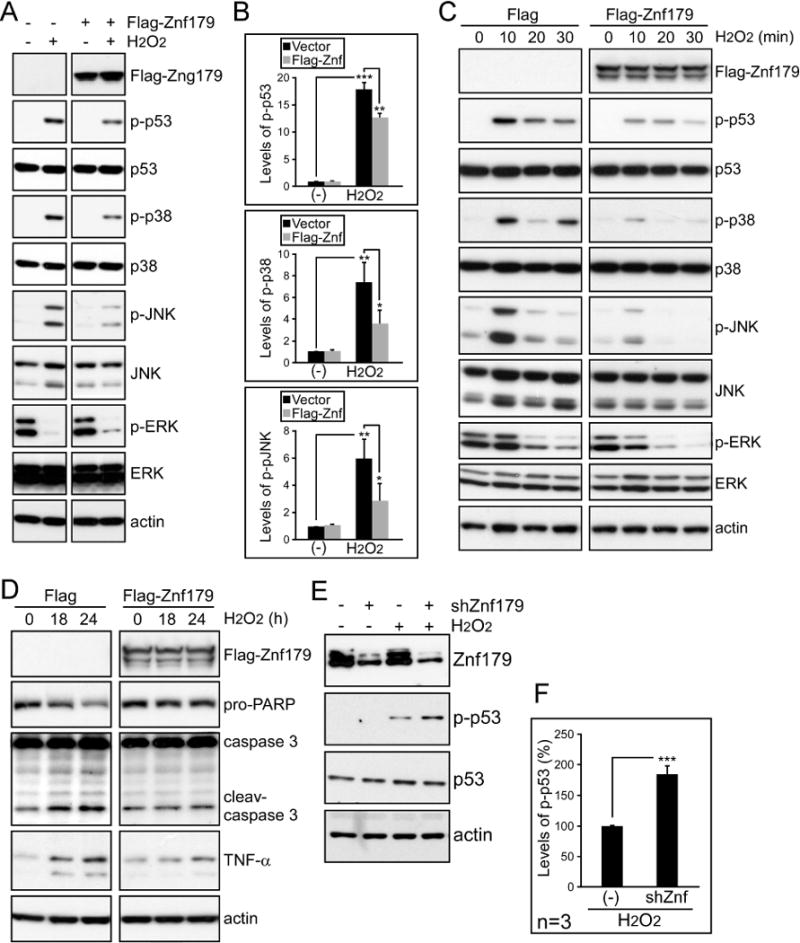
Znf179 expression suppresses H2O2-induced activation of cell death pathways. (A) Differentiated N2a cells were transfected with pCMV-Tag 2A control vector or Flag-Znf179 expressing vector. After 1 day transfection, cells were treated with or without 50 μM H2O2 for 1 hour and the lysates from these cells were then analyzed via western blot with the indicated antibodies. (B) The phosphorylated levels of p53, p38, and JNK normalized to their total protein expression were quantified (t-test: *p < 0.05, **p < 0.01, ***p < 0.001). (C and D) Transfected cells with Flag- or Flag-Znf179-expressed vector were treated with 50 μM H2O2 and collected after different time intervals (0, 10, 20, and 30 min in C, and 18 and 24 hours in D). The cells were then analyzed by using immunoblotting with indicated antibodies. (E) Differentiated N2a cells were transfected with or without Znf179 shRNA. After 24 hours incubation, cells were treated with 50 μM H2O2 for 1 hour and analyzed by immunoblotting. (F) Bars represent means ± s.e.m. of phosphorylated p53 that were normalized to total p53 from three independent experiments (t-test: ***p < 0.001).
Zn179 expression reduces oxidative injury via increasing antioxidant enzyme levels
In order to clarify the protective effects of Znf179, a stable cell line for constitutive Znf179 expression was established (Fig. 5A). This cell line and control cells were then treated with different doses of H2O2 for 24 h (Fig. 5B), or with 50 μM H2O2 for different time intervals (Fig. 5C), and cell viability was then measured using the MTT assay. Znf179 was able to prevent cell death induced by oxidative stress. Based on this result, antioxidant-related enzyme levels were analyzed in the hippocampus of control and Znf179 knockout mice (Fig. 5D and 5E). Znf179 knockout was correlated with decreased levels of peroxiredoxin 3 (Prx3) and superoxide dismutase 2 (SOD2), but not SOD1. We also quantified cellular ROS and found that elevating Znf179 protein reduced ROS levels at 3 h after H2O2 treatment (Fig. 5F). Taken together, Znf179 modulates antioxidant enzyme levels in a manner suggestive of a neuroprotective effort.
Figure 5.
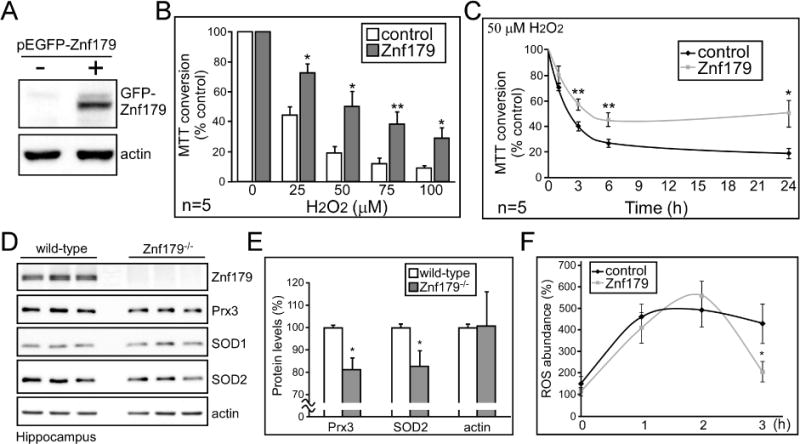
Znf179 protects cell survival and affects expression of antioxidant enzymes including peroxiredoxin 3 (Prx3) and superoxide dismutase 2 (SOD2). (A) Green fluorescent protein (GFP-) and GFP-Znf179 expressed N2a stable cells were analyzed by immunoblotting with anti-GFP antibodies to confirm Znf179 protein levels. (B and C) Differentiated N2a cells were treated with different doses of H2O2 as indicated for 24 hours (B), or were treated with 50 μM H2O2 and collected after different time intervals (C). Cell viability was then assessed by colorimetric MTT assay. (D and E) Hippocampus from wild-type or Znf179 knockout (−/−) mice were analyzed by western blot with the indicated antibodies. (E) Bar graphs represent means ± s.e.m. from three independent experiments (t-test: *p < 0.05). (F) GFP- and GFP-Znf179 expressed N2a stable cells were treated with 100 μM H2O2 and collected after different time intervals. Intracellular reactive oxygen species (ROS) levels were measured by using dihydroethidium (DHE). (Results from three independent experiments, t-test: *p < 0.05)
The Sig-1R-Znf179 pathway is critical for the protective effect of DHEAS against cell apoptosis
Since Sig-1R activity and Znf179 expression were regulated by DHEAS, we studied whether the Sig-1R-Znf179 pathway mediated the ability of DHEAS to protect against oxidative stress using a short hairpin RNA-mediated Znf179 knockdown. Figure 6 demonstrates that reduced Znf179 levels attenuated the protective capability of DHEAS similarly to the Sig-1R antagonist BD1063 (Fig. 1G and 1H). Taken together, the data suggest that DHEAS utilizes the Sig-1R-Znf179 pathway to prevent cell death from oxidative injury.
Figure 6.
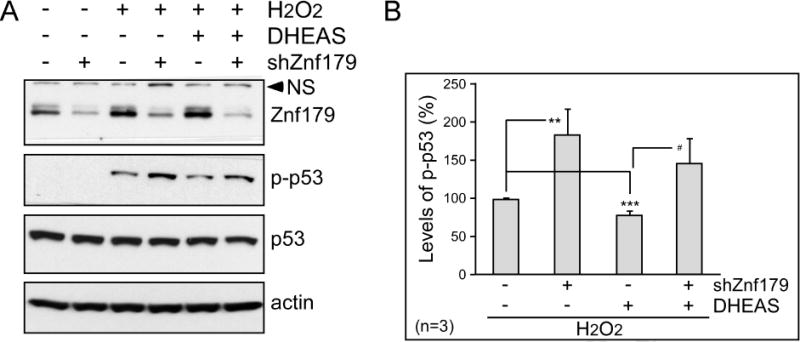
Knocking down Znf179 blocks protection by dehydroepiandrosterone sulfate (DHEAS). (A) Differentiated N2a cells were transfected with or without Znf179 shRNA as indicated. After 24 h incubation, cells were treated with 50 μM H2O2 or co-treated with 10 μM DHEAS for 1 h, and analyzed by immunoblotting. NS is a nonspecific band at 80 kDa recognized by the anti-ZNF179 antibody. (B) Bars represent means ± s.e.m. of phosphorylated p53 that were normalized to total p53 from three independent experiments (The effects of Znf179 knockdown and DHEAS in H2O2-treated N2a cells, t-test: **p < 0.01 and ***p < 0.001; the effects of Znf179 knockdown in H2O2- and DHEAS-treated N2a cells, t-test: #p < 0.05).
Discussion
Overall, these data demonstrate that stimulation of the Sig1-R-Znf179 pathway plays a role in mitigating cell death following cellular injury and oxidative stress. Specifically, we demonstrated here that: 1) Znf179 is a downstream effector of Sig-1R; 2) Znf179 is a novel neuroprotective factor and its accumulation correlates with H2O2 exposure time; and 3) agonist stimulation of Sig-1R is necessary for Sig-1R to provide maximal protection. Oxidative stress may partially induce endogenous Sig-1R-Znf179 pathway activity, but ROS accumulation is more potent at inducing cell death than this partial induction is at protecting the cells. In this study, the neurosteroids DHEA/DHEAS, known agonists of Sig-1R [13–15], served to activate the Sig-1R-Znf179 pathway, and in doing so significantly decreased cell death. Indeed, our current data demonstrated that the protective effect of DHEAS was blocked by the Sig-1R antagonist, BD1063. Notably, Znf179 was upregulated by DHEA/DHEAS, and downregulated by BD1063 as well as by Sig-1R knockdown. When Znf179 was overexpressed in cells, its protective ability after H2O2 exposure was similar to that of DHEAS treatment. We confirmed this ability of Znf179 by demonstrating that ROS accumulation in oxidative stress was decreased by Znf179 overexpression. Additionally, Znf179 knockdown blocked the protective effect of DHEAS similarly to Sig-1R antagonist treatment. Overall, our findings indicate that the Sig-1R-Znf179 neuroprotective pathway is enhanced by DHEA/DHEAS, and could provide new therapeutic approaches to mitigate or prevent the extensive damage in neurodegenerative and neurotraumatic diseases (Fig. 7).
Figure 7.
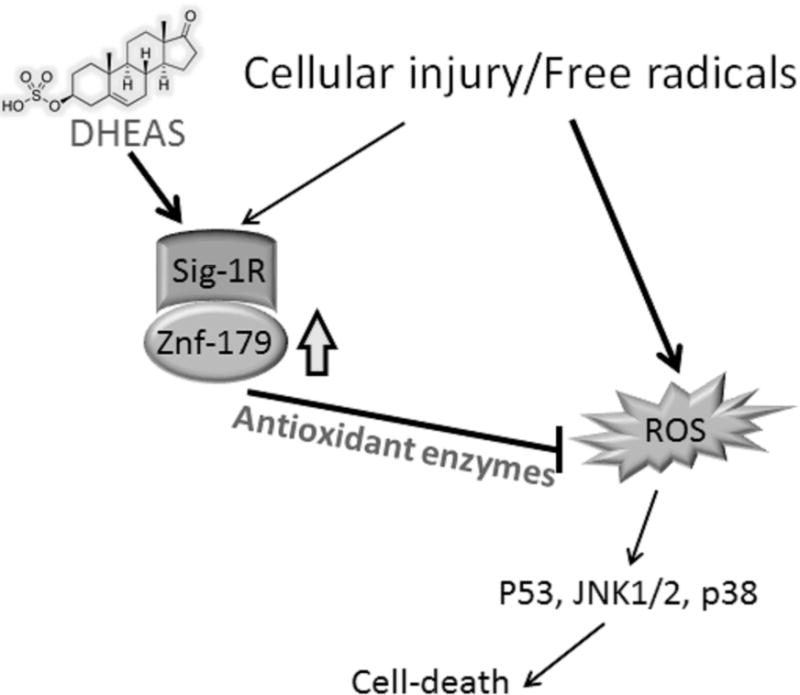
Simplified diagram illustrating the neuroprotective mechanism of dehydroepiandrosterone sulfate (DHEAS), which regulates Sig-1R-Znf179 pathway to affect antioxidant enzyme expression and mitigate reactive oxygen species (ROS)-mediated cytotoxic effects.
ROS/free radicals are accumulated, and contribute to cell death, during traumatic injury. This is a complex process involving oxidative stress, glutamate excitotoxicity, inflammatory damage, and other mechanisms [5, 25, 26], though they all support the idea that free radicals and ROS play a decisive role in the pathology of neurotraumatic diseases. In addition, a key role of ROS for neuronal damage is further indicated by studies on the pathology of neurodegenerative diseases, such as AD, PD, or ALS, where increased ROS levels were detected in degenerating neurons [27]. Therefore, it is noteworthy that inhibition of ROS-induced cell death is important for both neurotraumatic and neurodegenerative diseases. Although DHEA/DHEAS and Sig-1R protect neurons against oxidative stress [28–31], the mechanism of ROS damage mitigation by DHEA/DHEAS and Sig-1R was never examined. Our current data indicate Znf179, a downstream target of Sig-1R, regulates antioxidant enzymes, and that both DHEA and DHEAS increase Znf179 expression. Even though the mechanism of how Znf179 alters enzyme expression is still unclear, the Sig-1R-Znf179 pathway appears to be a possible way to regulate ROS levels after DHEA/DHEAS treatment. Further study of the mechanism by which Sig-1R and Znf179 regulate gene expression will be performed in future.
In nervous system development, Znf179 is critically important during embryogenesis [21]. When P19 cells are treated with retinoic acid to induce neuronal differentiation, Znf179 is upregulated and increases p35 gene expression and p27 protein accumulation, which causes cell cycle arrest at the G0/G1 phase and initiates cell differentiation. By using RNA interference to inhibit Znf179 expression, neuronal differentiation is significantly suppressed [21]. A further study indicated that Znf179 may affect gene regulation via altering the levels of transcription repressors, such as Plzf [23]. When Znf179 is co-overexpressed with Plzf together in cells, Znf179 associated physically with Plzf. The interaction then results in a translocation of Znf179 from cytoplasm to nucleus, and an increase in Plzf protein abundance [23].
A recent study indicated that Znf179 in astrocytes can be upregulated by CCAAT/enhancer binding protein delta (CEBPD) upon pro-inflammatory cytokine (interleukin-1β) treatment. Znf179 subsequently associates with Plzf to play an anti-apoptotic role in astrocytes. The protein complex suppresses several pro-apoptotic genes, including insulin-like growth factor binding protein 3 (IGFBP3) and BCL2-interacting killer (BIK) [32]. Here, we demonstrated that Znf179 is also upregulated in cortical brain tissues (data not shown) and N2a neuron-like cells after environmental stress. In addition, we find the protective functions of Znf179 not only to reduce apoptotic actions, but also to increase antioxidant enzymes. The previous study detected 339 downregulated genes and 98 upregulated genes in Znf179-overexpressing glioblastoma cells [32]. We assume that Znf179 downregulates gene expression through enhancing transcriptional repressors such as Plzf, and upregulates gene expression through enhancing a transcriptional activator. However, the mechanisms of Znf179-altered downstream gene expression still need to be further examined.
Sig-1R interacts with antipsychotic drugs (such as haloperidol), psychotropic drugs (such as pentazocine, SKF10047, and fluvoxamine), and steroid hormones (such as DHEA/DHEAS) [7–9, 17]. These drugs regulate the ability of Sig-1R to bind with other protein partners, improve learning and memory, and reduce depression and anxiety [33]. As mentioned above, agonist stimulation of Sig-1R also has neuroprotective functions [34–36]. For example, 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) prevents neuronal death in an experimental stroke model [34], and Pre084 protects motor neuron survival in a mouse ALS model [35]. Although previous studies indicated that those agonists act through Sig-1R to promote neuronal survival [37], the molecular mechanisms were not discerned. Our current results indicated that Znf179 is a previously unidentified downstream target of Sig-1R, and the Sig-1R-Znf179 pathway contributes to the neuroprotection of DHEA/DHEAS against oxidative stress. It is possible that this pathway can be also regulated by other Sig-1R agonists. Notably, in our results, the activity of Sig-1R may be critical for Znf179 protein expression. If the Sig-1R-Znf179 pathway is indeed activated by multiple Sig-1R agonists, it implies that better and more stable compounds than DHEA/DHEAS could stimulate the neuroprotective ability of the Sig-1R-Znf179 pathway. Finally, we hope that the finding can be applied to animal models and clinical treatment of neurological disorders.
Highlights.
Znf179 is a downstream effector of Sig-1R
Znf179 is a novel neuroprotective factor
DHEA/DHEAS treatment increase the expression of Znf179
DHEA/DHEAS treatment enhance Sig-1R-Znf179 neuroprotective pathway
Agonist stimulation of Sig-1R is necessary for Sig-1R to provide maximal protection
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology (MOST 103-2320-B-038-046-MY3, MOST 103-2321-B-038-001, and MOST 104-2923-B-038-002-MY3) and Taipei Medical University (TMU102-AE1-B25).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 2.Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology. 2001;40:959–975. doi: 10.1016/s0028-3908(01)00019-3. [DOI] [PubMed] [Google Scholar]
- 3.Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–190. doi: 10.1016/j.cell.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 5.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- 7.Kourrich S, Su TP, Fujimoto M, Bonci A. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012;35:762–771. doi: 10.1016/j.tins.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi T, Su TP. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc Natl Acad Sci U S A. 2001;98:491–496. doi: 10.1073/pnas.98.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishikawa MHK. The role of sigma-1 receptors in the pathophysiology of neuropsychiatric diseases. J Receptor Ligand Channel Res. 2010;2010:25–36. [Google Scholar]
- 10.Kourrich S, Hayashi T, Chuang JY, Tsai SY, Su TP, Bonci A. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell. 2013;152:236–247. doi: 10.1016/j.cell.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen L, Lucke-Wold BP, Mookerjee SA, Cavendish JZ, Robson MJ, Scandinaro AL, Matsumoto RR. Role of sigma-1 receptors in neurodegenerative diseases. J Pharmacol Sci. 2015;127:17–29. doi: 10.1016/j.jphs.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Ruscher K, Wieloch T. The involvement of the sigma-1 receptor in neurodegeneration and neurorestoration. J Pharmacol Sci. 2015;127:30–35. doi: 10.1016/j.jphs.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Su TP, London ED, Jaffe JH. Steroid binding at sigma receptors suggests a link between endocrine, nervous, and immune systems. Science. 1988;240:219–221. doi: 10.1126/science.2832949. [DOI] [PubMed] [Google Scholar]
- 14.Monnet FP, Mahe V, Robel P, Baulieu EE. Neurosteroids, via sigma receptors, modulate the [3H]norepinephrine release evoked by N-methyl-D-aspartate in the rat hippocampus. Proc Natl Acad Sci U S A. 1995;92:3774–3778. doi: 10.1073/pnas.92.9.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurice T, Roman FJ, Privat A. Modulation by neurosteroids of the in vivo (+)-[3H]SKF-10,047 binding to sigma 1 receptors in the mouse forebrain. J Neurosci Res. 1996;46:734–743. doi: 10.1002/(SICI)1097-4547(19961215)46:6<734::AID-JNR10>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 16.Charalampopoulos I, Tsatsanis C, Dermitzaki E, Alexaki VI, Castanas E, Margioris AN, Gravanis A. Dehydroepiandrosterone and allopregnanolone protect sympathoadrenal medulla cells against apoptosis via antiapoptotic Bcl-2 proteins. Proc Natl Acad Sci U S A. 2004;101:8209–8214. doi: 10.1073/pnas.0306631101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kokona D, Charalampopoulos I, Pediaditakis I, Gravanis A, Thermos K. The neurosteroid dehydroepiandrosterone (DHEA) protects the retina from AMPA-induced excitotoxicity: NGF TrkA receptor involvement. Neuropharmacology. 2012;62:2106–2117. doi: 10.1016/j.neuropharm.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 18.Grimm A, Schmitt K, Lang UE, Mensah-Nyagan AG, Eckert A. Improvement of neuronal bioenergetics by neurosteroids: implications for age-related neurodegenerative disorders. Biochim Biophys Acta. 2014;1842:2427–2438. doi: 10.1016/j.bbadis.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Camporez JP, Akamine EH, Davel AP, Franci CR, Rossoni LV, Carvalho CR. Dehydroepiandrosterone protects against oxidative stress-induced endothelial dysfunction in ovariectomized rats. J Physiol. 2011;589:2585–2596. doi: 10.1113/jphysiol.2011.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimura T, Arakawa Y, Inoue S, Fukushima Y, Kondo I, Koyama K, Hosoi T, Orimo A, Muramatsu M, Nakamura Y, et al. The brain finger protein gene (ZNF179), a member of the RING finger family, maps within the Smith-Magenis syndrome region at 17p11.2. Am J Med Genet A. 1997;69:320–324. [PubMed] [Google Scholar]
- 21.Pao PC, Huang NK, Liu YW, Yeh SH, Lin ST, Hsieh CP, Huang AM, Huang HS, Tseng JT, Chang WC, et al. A novel RING finger protein, Znf179, modulates cell cycle exit and neuronal differentiation of P19 embryonal carcinoma cells. Cell Death Differ. 2011;18:1791–1804. doi: 10.1038/cdd.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evangelopoulos ME, Weis J, Kruttgen A. Signalling pathways leading to neuroblastoma differentiation after serum withdrawal: HDL blocks neuroblastoma differentiation by inhibition of EGFR. Oncogene. 2005;24:3309–3318. doi: 10.1038/sj.onc.1208494. [DOI] [PubMed] [Google Scholar]
- 23.Lin DY, Huang CC, Hsieh YT, Lin HC, Pao PC, Tsou JH, Lai CY, Hung LY, Wang JM, Chang WC, et al. Analysis of the interaction between Zinc finger protein 179 (Znf179) and promyelocytic leukemia zinc finger (Plzf) J Biomed Sci. 2013;20:98. doi: 10.1186/1423-0127-20-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai SY, Chuang JY, Tsai MS, Wang XF, Xi ZX, Hung JJ, Chang WC, Bonci A, Su TP. Sigma-1 receptor mediates cocaine-induced transcriptional regulation by recruiting chromatin-remodeling factors at the nuclear envelope. Proc Natl Acad Sci U S A. 2015;112:E6562–6570. doi: 10.1073/pnas.1518894112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendes Arent A, de Souza LF, Walz R, Dafre AL. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. Biomed Res Int. 2014;2014:723060. doi: 10.1155/2014/723060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez-Rodriguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A. Oxidative stress in traumatic brain injury. Curr Med Chem. 2014;21:1201–1211. doi: 10.2174/0929867321666131217153310. [DOI] [PubMed] [Google Scholar]
- 27.Di Matteo V, Esposito E. Biochemical and therapeutic effects of antioxidants in the treatment of Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Curr Drug Targets CNS Neurol Disord. 2003;2:95–107. doi: 10.2174/1568007033482959. [DOI] [PubMed] [Google Scholar]
- 28.Aly HF, Metwally FM, Ahmed HH. Neuroprotective effects of dehydroepiandrosterone (DHEA) in rat model of Alzheimer’s disease. Acta Biochim Pol. 2011;58:513–520. [PubMed] [Google Scholar]
- 29.Hanna DM, Tadros MG, Khalifa AE. ADIOL protects against 3-NP-induced neurotoxicity in rats: Possible impact of its anti-oxidant, anti-inflammatory and anti-apoptotic actions. Prog Neuropsychopharmacol Biol Psychiatry. 2015;60:36–51. doi: 10.1016/j.pnpbp.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Hirata Y, Yamamoto H, Atta MS, Mahmoud S, Oh-hashi K, Kiuchi K. Chloroquine inhibits glutamate-induced death of a neuronal cell line by reducing reactive oxygen species through sigma-1 receptor. J Neurochem. 2011;119:839–847. doi: 10.1111/j.1471-4159.2011.07464.x. [DOI] [PubMed] [Google Scholar]
- 31.Tuerxun T, Numakawa T, Adachi N, Kumamaru E, Kitazawa H, Kudo M, Kunugi H. SA4503, a sigma-1 receptor agonist, prevents cultured cortical neurons from oxidative stress-induced cell death via suppression of MAPK pathway activation and glutamate receptor expression. Neurosci Lett. 2010;469:303–308. doi: 10.1016/j.neulet.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Wang SM, Lee YC, Ko CY, Lai MD, Lin DY, Pao PC, Chi JY, Hsiao YW, Liu TL, Wang JM. Increase of zinc finger protein 179 in response to CCAAT/enhancer binding protein delta conferring an antiapoptotic effect in astrocytes of Alzheimer’s disease. Mol Neurobiol. 2015;51:370–382. doi: 10.1007/s12035-014-8714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urani A, Roman FJ, Phan VL, Su TP, Maurice T. The antidepressant-like effect induced by sigma(1)-receptor agonists and neuroactive steroids in mice submitted to the forced swimming test. J Pharmacol Exp Ther. 2001;298:1269–1279. [PubMed] [Google Scholar]
- 34.Goyagi T, Goto S, Bhardwaj A, Dawson VL, Hurn PD, Kirsch JR. Neuroprotective effect of sigma(1)-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) is linked to reduced neuronal nitric oxide production. Stroke. 2001;32:1613–1620. doi: 10.1161/01.str.32.7.1613. [DOI] [PubMed] [Google Scholar]
- 35.Peviani M, Salvaneschi E, Bontempi L, Petese A, Manzo A, Rossi D, Salmona M, Collina S, Bigini P, Curti D. Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol Dis. 2014;62:218–232. doi: 10.1016/j.nbd.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 36.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi T, Su T. The sigma receptor: evolution of the concept in neuropsychopharmacology. Curr Neuropharmacol. 2005;3:267–280. doi: 10.2174/157015905774322516. [DOI] [PMC free article] [PubMed] [Google Scholar]