

Summary
The G protein-coupled receptor Smoothened (Smo) is the signal transducer of the Sonic Hedgehog (Shh) pathway. Smo signals through G protein-dependent and independent routes, with G protein-independent canonical signaling to Gli effectors requiring Smo accumulation in the primary cilium. Mechanisms controlling Smo activation and trafficking are not yet clear, but likely entail small-molecule binding to pockets in its extracellular cysteine-rich domain (CRD) and/or transmembrane bundle. Herein we demonstrate cytosolic phospholipase cPLA2α is activated through Gβγ downstream of Smo to release arachidonic acid. Arachidonic acid binds Smo and synergizes with CRD-binding agonist, promoting Smo ciliary trafficking and high-level signaling. Chemical or genetic cPLA2α inhibition dampens Smo signaling to Gli, revealing an unexpected contribution of G protein-dependent signaling to canonical pathway activity. Arachidonic acid displaces the Smo transmembrane domain inhibitor cyclopamine to rescue CRD agonist-induced signaling, suggesting arachidonic acid may target the transmembrane bundle to allosterically enhance signaling by CRD agonist-bound Smo.
eTOC Blurb
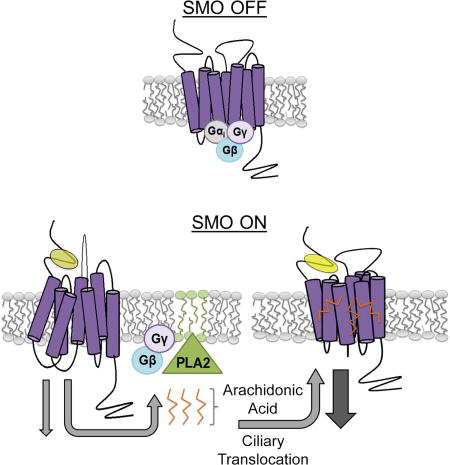
Arensdorf et al report phospholipase A2 is activated downstream of the G protein-coupled receptor Smoothened to produce arachidonic acid. Arachidonic acid binds the Smoothened transmembrane domain to promote Smoothened ciliary trafficking and high-level signaling. These results identify arachidonic acid as a candidate allosteric regulator of Smoothened.
Introduction
Hedgehog (Hh) morphogens activate a signaling cascade that provides instructional information for developmental tissue patterning and contributes to post-developmental tissue homeostasis (Jiang and Hui, 2008). Vertebrates encode three Hh ligands, Sonic (Shh), Indian and Desert, with Shh being most broadly expressed (Ingham and McMahon, 2001). Signaling is initiated by Shh binding to a receptor complex comprised of the sterol-sensing domain (SSD) containing transmembrane protein Patched (Ptch1 or Ptch2) and the adhesion molecule CDO or BOC (Allen et al., 2011; Marigo et al., 1996; Motoyama et al., 1998; Yao et al., 2006). In the absence of Hh, Ptch maintains the pathway in an off state by preventing the G protein-coupled receptor (GPCR) Smoothened (Smo) from signaling to intracellular effectors (Arensdorf et al., 2016). Shh binding quenches Ptch-mediated repression, thereby allowing Smo to signal to downstream effectors. The precise mechanism by which Ptch inhibits Smo is not yet clear, but is thought to involve modulation of sterol or phospholipid availability to affect Smo membrane trafficking (Huang et al., 2016; Jiang et al., 2016; Luchetti et al., 2016; Nachtergaele et al., 2012; Yavari et al., 2010). For maximal signaling, Smo must traffic into the primary cilium (PC). The PC is gated by membrane microdomains including the ciliary pocket, a zone of membrane curvature between plasma and ciliary membranes, and the transition zone, which works with the ciliary pocket to govern phospholipid and protein entry (Benmerah, 2013) (Milenkovic et al., 2015; Rohatgi et al., 2007). Membrane phospholipid compositions of the ciliary base and body differ, and must be maintained for proper Smo regulation (Chavez et al., 2015; Garcia-Gonzalo et al., 2015).
Phospholipids are substrates for lipid kinases, phosphatases and phospholipases. Select lipid kinases and phosphatases can modulate Smo signaling, but evidence for phospholipase-mediated regulation is lacking (Garcia-Gonzalo et al., 2015; Jiang et al., 2016; Yavari et al., 2010). Phospholipase enzymes are divided into four classes based upon where they cleave phospholipids: PLA, PLB, PLC and PLD. PLA is further divided into PLA1 or PLA2 subclasses (Fig. 1A). PLA1 and PLA2 cleave at the SN-1 or SN-2 acyl chains, respectively, releasing a lysophospholipid and a free fatty acid, most commonly the eicosanoid precursor arachidonic acid. The PLA2 group includes secretory and cytosolic enzymes, the latter of which is classified into Ca2+ dependent (cPLA2) and independent enzymes (iPLA2). PLB enzymes have both PLA1 and PLA2 activity. PLC cleaves between the phosphate and the acyl chains, leading to production of diacylglycerol and inositol triphosphate. PLD clips between the phosphate and the phospholipid head group to release phosphatidic acid and alcohol. PLA, PLC and PLD participate in a number of signal transduction cascades by producing intracellular second messengers or remodeling membrane lipids in response to wide-ranging extracellular cues (Murakami et al., 2011; Richmond and Smith, 2011). Although PLC is activated downstream of Smo to influence Ca release in developing neurons (Belgacem and Borodinsky, 2011), a role for phospholipases upstream of Smo signaling has not been reported.
Figure 1.

cPLA2 inhibitors modulate Shh signaling. For all panels, *** indicates p ≤ 0.0001; ns, p > 0.05. Error bars indicate standard error of the mean (SEM). A. Schematic of a phospholipid showing phospholipase hydrolysis sites. B. Light2 reporter cells were pretreated with PLA2 inhibitors (MAFP (5 µM), GIRI (5 µM) and BEL (5 µM); orange), PLC inhibitor NEO (200 µM; pink) or PLD inhibitors (FIPI (40 nM), PLD1i (40 nM) and PLD2i (40 nM); purple) prior to stimulation with ShhN conditioned media. Statistical significance was determined using a one-way ANOVA. C. Light2 cells were pretreated with cPLA2 inhibitor (GIRI, diamond) or iPLA2 inhibitor (BEL, square) prior to ShhN conditioned media treatment. Statistical significance was determined using a two-way ANOVA. D. NIH3T3 cells were pretreated with GIRI (4 µM) prior to stimulation with ShhN conditioned media. Gli1 and Gli3 protein levels were analyzed in nuclear fractions by western blot. The experiment was performed twice. A representative blot is shown. Arrowheads indicate Gli3 activator and repressor species. Lamin C is the nuclear marker. E. Diagram of Smo ligand binding pockets. F–G. Light2 cells were pretreated with GIRI or MAFP prior to SAG (100 nM, square), 20-OHC (10 µM, triangle) or ShhN conditioned media (circle). Statistical significance was determined using a two-way ANOVA. For all experiments involving Light2 cells, Gli-luciferase reporter activity was normalized to tk-renilla control and expressed relative to the agonist-stimulated control. PL inhibitors were added 2 hours prior to agonist for pretreatment. Assays were repeated a minimum of three times in triplicate and all data pooled.
Herein, we test for phospholipase regulation of Shh signaling, and provide evidence cPLA2α contributes to pathway activity by enhancing Smo ciliary entry. In response to Shh or direct Smo agonist, cPLA2α is activated in a Smo-and Gβγ-dependent manner to release arachidonic acid. Arachidonic acid binds Smo and synergizes with the cysteine rich domain (CRD)-binding agonist 20(S)-OHC to enhance Smo activity, suggesting PLA2 activation drives a positive feed-forward regulatory loop. Accordingly, chemical inhibition or genetic targeting of cPLA2α blocks arachidonic acid release and attenuates Shh- and 20(S)-OHC-induced Smo activation. Strikingly, Smo activated by the 7-transmembrane (7TM) agonist SAG is largely insensitive to cPLA2 inhibition, and is competent to reach ciliary membranes and induce high-level signaling despite Gβγ or cPLA2 inhibition. This is likely due to arachidonic acid and SAG targeting overlapping 7TM pockets, as suggested by the ability of arachidonic acid to displace the 7TM-binding inverse agonist cyclopamine. We provide a testable model whereby cPLA2α is activated through Gβγ to release arachidonic acid, which targets the 7TM core of active Smo to potentiate its ciliary trafficking and signaling.
Results
To screen for phospholipase effects on Shh signaling, Shh-responsive Light2 reporter cells (Taipale et al., 2000) were treated with phospholipase inhibitors, and ShhN-stimulated gli-luciferase reporter activity was monitored (Fig. 1A–B). For initial screening, inhibitors were tested at their IC50. Targeting PLC by neomycin sulfate (NEO) or PLD by 5-Fluoro-2-indolyl des-chlorohalopemide hydrochloride hydrate (FIPI), VU0359595 (PLD1i) or VU0364739 (PLD2i) did not significantly alter signaling (Fig. 1B, pink and purple). Conversely, reduced reporter activity was observed following treatment with cytosolic PLA2 inhibitors methyl arachidonyl fluorophosphate (MAFP), which inhibits both cPLA2 and iPLA2, and Giripladib (GIRI, also referred to as Wyeth-2 and PLA-659), a specific inhibitor of cPLA2 (Duvernay et al., 2015; Thotala et al., 2013). These inhibitors reduced signaling by ~60%, narrowing focus to this phospholipase class. The iPLA2 inhibitor bromoenol lactone (BEL) failed to significantly alter reporter activity, suggesting suppressive effects were specific to inhibition of cPLA2 enzymes. To test this, dose curves of GIRI and BEL were performed. GIRI triggered pronounced, dose-dependent inhibition, reducing maximal signal by ~50–80% (Fig. 1C, F). iPLA2-specific BEL failed to inhibit at any dose tested (Fig. 1C).
To confirm reporter results were indicative of effects on endogenous pathway components, ShhN effects on nuclear pools of transcriptional effectors Gli1 and Gli3 were tested in the presence of GIRI (Fig. 1D). ShhN stimulation reduced Gli3R, shifting the Gli3A:Gli3R ratio to favor the full-length activator species (arrowheads, lanes 1 compared to 3). Accordingly, Gli1 protein levels increased in nuclear fractions, indicating ShhN-induced transcription of the Gli1 target gene (lane 1 vs. 3). GIRI treatment reduced nuclear Gli1 and attenuated Gli3R reduction (lane 4 compared to 3), consistent with cPLA2 impacting the Shh pathway at a level upstream of Gli transcriptional effectors.
In the absence of Shh, Ptch-mediated Smo inhibition can be bypassed by direct Smo agonists such as 7TM-binding SAG and CRD-binding 20(S)-OHC (Fig. 1E and Chen et al., 2002b; Nachtergaele et al., 2013). These compounds stabilize Smo in active conformations insensitive to Ptch, allowing for signal induction in the absence of Shh. To determine whether cPLA2 impacted signaling upstream or downstream of Smo, GIRI and MAFP dose curves were performed on Light2 cells stimulated with ShhN conditioned media, SAG or 20(S)-OHC (Fig. 1F–G). Both GIRI and MAFP reduced reporter activity in cells stimulated with ShhN or 20(S)-OHC (Fig. F–G). The ability of inhibitors to blunt signaling by the direct Smo agonist 20(S)-OHC indicated cPLA2 function in the signal cascade at or downstream of Smo. Despite this, SAG-induced pathway activity was partially resistant to GIRI, and almost fully resistant to MAFP, showing only a ~10% reduction at the highest concentration of MAFP tested (Fig. 1F–G, squares).
High-level signaling correlates with Smo having increased binding durations in the primary cilium, evidenced in fixed cells by intensified Smo signal throughout the ciliary body and at ciliary tips (Milenkovic et al., 2015). Regulatory processes controlling Smo ciliary trafficking are not yet clear, but activating ligands, and paradoxically, the inverse agonist cyclopamine, promote its ciliary accumulation (Rohatgi et al., 2009; Wilson et al., 2009). To determine whether differential MAFP and GIRI sensitivity of ShhN- and SAG-induced reporter activity correlated with Smo ciliary localization, vehicle control or cPLA2 inhibitor-treated NIH3T3 cells were stimulated with ShhN or SAG, and ciliary localization of endogenous Smo was quantified (Fig. 2). Stimulation with ShhN conditioned media or SAG lead to increased Smo signal intensity (Fig. 2A, G, gray and C, E, J, L, green; arrowheads mark ciliary tips). cPLA2 inhibition altered this localization; MAFP pretreatment concentrated Smo at the ciliary base and GIRI nearly ablated Smo ciliary entry in response to ShhN (Fig. 2A, G, orange and D, K, green). Gli3, which accumulates at ciliary tips in response to Smo signaling, showed reduced tip intensity in GIRI-pretreated ShhN-stimulated cells (Fig. 2H and J–K, magenta). Remarkably, ciliary localization of Smo and Gli3 in SAG-treated cells was unaffected by MAFP or GIRI (Fig. 2 A, F, G, H, M), further suggesting SAG-stimulated Smo does not require cPLA2 for ciliary translocation or signaling. Ciliary trafficking of Smo induced by the 7TM-binding inverse agonist cyclopamine was also unaffected by MAFP (Fig. 2N–Q), consistent with 7TM-binding compounds not requiring cPLA2 for Smo ciliary entry.
Figure 2.

cPLA2 influences ShhN-, but not SAG-induced Smo ciliary translocation. For all panels *** indicates p ≤ 0.0001; * indicates p ≤ 0.05; ns indicates p > 0.05; nt, not tested. Error bars indicate SEM. A–M. NIH3T3 cells pretreated with MAFP (5 µM) or GIRI (5 µM) were stimulated with ShhN conditioned media or SAG (100 nM) and imaged by immunofluorescence confocal microscopy. Ciliary tips are indicated by arrowheads. Smo and/or Gli3 signal in primary cilia was quantified by counting ≥100 cells over a minimum of 2 experiments. Significance indicated in A, G and H was calculated based upon total cell number analyzed using a two-way ANOVA. N–Q. Smo localization was quantified as above in NIH3T3 cells treated with cyclopamine (10 µM) +/− MAFP (5 µM). Significance was determined using a two-tailed Student’s t-test. For all cilia shown, Smo is green, Gli3 is magenta, the ciliary marker acetylated α-tubulin is blue and the ciliary base marker Cep290-GFP is magenta.
Because most cPLA2 enzymes are not constitutively active (Murakami et al., 2011), we hypothesized the cPLA2 enzyme influencing Smo might be under Shh control. To test this, PLA2 activity assays were performed. Cellular lysates prepared from NIH3T3 cells or Smo−/− mouse embryo fibroblasts (MEFs) treated with ShhN, SAG and MAFP were tested using a phospholipid reporter that fluoresces upon PLA2 cleavage (Fig. 3A). Lysates from NIH3T3 cells treated with ShhN or SAG showed statistically significant increases in PLA2 activity, supporting PLA2 activation downstream of Smo (Fig. 3B, gray). Because GIRI and BEL results (Fig. 1) indicated Shh effects were cPLA2-specific, we did not expect large changes in total PLA2 activity following Smo activation, but did expect a significant decrease by cytosolic PLA2 inhibitor MAFP. Accordingly, MAFP reduced total PLA2 activity in ShhN-stimulated cells to below baseline (Fig. 3B, orange). ShhN conditioned media failed to raise activity over baseline in Smo−/− cells (Fig. 3C, SmoKO), consistent with cPLA2 activation occurring downstream of Smo. This failure was not due to general inactivity of PLA2 enzymes or ability of the assay to detect activity in this cell type because MAFP treatment reduced basal PLA2 activity similarly in both Smo−/− and NIH3T3 cells (Fig. 3C, orange).
Figure 3.

PLA2 is activated in a Smoothened-dependent manner. For all panels *** indicates p ≤ 0.0001; ** indicates p ≤ 0.001; ns indicates p > 0.05. Error bars indicate SEM. A. PLA2 activity assay diagram. B–C. Lysates were prepared from NIH3T3 or Smo−/− MEFs treated with ShhN conditioned media or SAG (100nM) +/− MAFP (5 µM). Results are represented as fold activity change relative to vehicle. The experiment was repeated four times in duplicate and all data pooled. Significance relative to vehicle controls were determined using a one-way ANOVA (B) or two-way ANOVA (C). D–E. Total fatty acid pool in treated and untreated cells was calculated from the sum of individual fatty acids quantified using the d8-20:4 internal standard. E. Absolute levels of 20:4 were determined using mass spectrometry with d8-20:4 internal standard. The experiment was conducted four times and all data pooled. Significance was determined using a one-way ANOVA.
Activation of cPLA2 enzymes triggers release of the eicosanoid precursor arachidonic acid (Balsinde et al., 2002). To directly test whether Smo induction increased arachidonic acid levels, cells were treated with vehicle, 20(S)-OHC or SAG for two hours, and 20:4 arachidonic acid levels were determined by mass spectrometry using deuterated d8–20:4 as an internal standard. Levels of total fatty acids were not significantly altered by 20(S)-OHC or SAG (Fig. 3D). However, both triggered statistically significant increases in arachidonic acid (Fig. 3E). In untreated cells, 20:4 comprised 2.8 ± 0.1% of the total fatty acid pool, and rose to 4.8 ± 0.2% and 4.6 ± 0.1% of total fatty acids in cells treated with 20(S)-OHC and SAG, consistent with a selective increase in cellular arachidonic acid following Smo activation.
Multiple cPLA2 genes encoding numerous PLA2 isozymes are present in mammals. In mouse, these include Pla2g4a, Pla2g4b, Pla2g4d, Pla2g4e, Pla2g4f and Plb1 (Murakami et al., 2011). Given compensatory activity by these gene products in vivo (Bonventre, 1999), the specific cPLA2 affecting Smo signaling in fibroblasts could not be predicted based upon knockout animal phenotypes. To empirically determine which Pla2 genes were expressed in NIH3T3 cells, RNA was harvested and qRT-PCR analysis was performed (Fig. 4A). Pla2g4d and Pla2g4e transcripts fell below the level of detection. Pla2g4a, which encodes cPLA2α was most abundantly expressed. Pla2g4b, Pla2g4f and Plb1 were also detectable, but showed exponentially lower expression levels, making cPLA2α the most likely candidate affecting Smo ciliary trafficking. Accordingly, antibody specific to active phospho-cPLA2α revealed cPLA2α signal at the ciliary base in 100% of cells examined in both stimulated and unstimulated conditions (Fig. 4B). Consistent with previous reports, significant phospho-cPLA2α signal was also evident in the nucleus (Grewal et al., 2002; Zhu et al., 2009). Although cPLA2α did not directly overlap with ciliary Smo signal (Fig. 4B’), its localization to the PC base placed it in an appropriate subcellular localization to influence Smo ciliary translocation.
Figure 4.

cPLA2α contributes to Smo signaling. For all panels *** indicates p ≤ 0.0001; ns indicates p > 0.05; nd, not detected. Error bars indicate SEM. A. RNA was harvested from NIH3T3 cells and analyzed for expression of cPLA2 genes by qRT-PCR. Analysis was performed three times in triplicate and all results pooled. B–B’. Cells were immunostained using antibody against active phospho-cPLA2α. Images are representative of ~200 cells analyzed over two independent experiments. B’ shows Smo and cPLA2α localization overlay with α-Tub c (top) and without (bottom). C. Western blot of cPLA2α in Pla2g4a wild type and knockout MEFs using total and phospho-specific antibodies. Tubulin is the loading control. D. Western blot of cytoplasmic and nuclear extracts from control and cPLA2α knockout MEFs. Loading controls are Tubulin (cytoplasmic) and Lamin C (nuclear). The experiment was repeated 3 times, a representative blot is shown. E–F. Fatty acid quantification in Pla2g4a wild type and knockout MEFs. The total fatty acid pool 20:4 absolute level was calculated as in Fig. 3D–E. The experiment was performed four times and all data pooled. Significance was determined using a two-way ANOVA. G. NIH3T3 cells were transfected with pCDNA-cPLA2αHA in the absence and presence of ShhN conditioned media. Gli1 protein was analyzed by immunoblot. Tubulin and Lamin C are the cytoplasmic and nuclear loading controls. The experiment was performed twice. A representative blot is shown. H. NIH3T3 cells were transfected with pCDNA-cPLA2αHA (yellow) and stimulated with control (VEH) or ShhN conditioned media. Cilia were examined for Smo (green) and Gli3 (magenta) ciliary localization. Acetylated α tubulin is blue. Arrowhead marks the ciliary tip. Due to excessive signal, HA was split from overlay and is shown below. Approximately 200 cells were analyzed over two independent experiments. Representative cells are shown.
To genetically test for involvement of cPLA2α in Shh signaling to Gli transcriptional effectors, MEFs derived from Pla2g4a knockout animals or littermate controls were examined for Gli1 nuclear localization in the absence and presence of ShhN. (Fig. 4C–D). Control MEFs showed nuclear Gli1 in the absence of ligand, and increased Gli1 in its presence. Conversely, nuclear Gli1 was nearly undetectable in knockout MEFs, and only increased to wild type baseline levels in response to ligand, consistent with genetic cPLA2α loss compromising Shh signaling to Gli. To confirm cPLA2α was the gene product responsible for Shh pathway-induced arachidonic acid release, we tested modulation of arachidonic acid levels in control and knockout MEFs following pathway activation (Fig. 4E–F). cPLA2α knockout and wild type littermate control MEFs were stimulated with 20(S)-OHC for 2 hours, and cell lysates were analyzed for 20:4 levels as above. The total non-esterified fatty acid pool was slightly lower in cPLA2α knockout cells than in control MEFs, and these levels did not change in response to 20(S)-OHC stimulation (Fig. 4E). Arachidonic acid content increased significantly in control MEFs in response to 20(S)-OHC stimulation, but failed to change in cPLA2α knockout MEFs (Fig. 4F). Taken together, these results suggest the Pla2g4a gene product cPLA2α is the primary PLA2 enzyme functioning in the Shh pathway in mouse fibroblasts.
To determine whether cPLA2α overexpression was sufficient to induce Smo signaling in the absence of pathway activation, cPLA2αHA was overexpressed in NIH3T3 cells, and effects on Smo, Gli1 and Gli3 were examined in ShhN-stimulated or unstimulated cells (Figs. 4G–H). Despite its high expression, cPLA2αHA failed to alter Gli1 protein levels or Smo and Gli3 ciliary tip enrichment in either condition. As such, increased expression of cPLA2α is not sufficient to induce Shh pathway activity in the absence of agonist, and does not augment signaling in its presence.
cPLA2α can be activated by recruitment to membranes, Ca2+ flux, activation of Gα i or Gαo-coupled GPCRs, direct Gβγ binding, or through phosphorylation by cAMP-dependent protein kinase (PKA), PKC or Erk1/2 (Bechler and Brown, 2014; Murakami et al., 2011). Smo is a GPCR that couples to Gα iβγ heterotrimeric G proteins (Riobo et al, 2006), leading us to hypothesize Smo-dependent cPLA2α activation involved Smo-activated Gβγ. NIH3T3 cells were treated with the Gβγ inhibitor Gallein (GALL, Lehmann et al., 2008), and Shh-induced PLA2 activity was quantified (Fig. 5A). GALL blocked PLA2 induction by ShhN, but did not reduce activity below baseline like MAFP (Fig. 3B, orange and Fig. 5A, green), consistent with GALL silencing the specific ShhN-induced signal elevating PLA2 activity. Loss of this signal compromised downstream signaling to Gli, evidenced by potent dose-dependent decreases in Gli reporter activity in GALL-treated cells (Fig. 5B circle). SAG-mediated activation was resistant to GALL (Fig. 5B, square). Similar effects were observed for Smo ciliary localization. Like GIRI and MAFP, GALL significantly reduced ShhN-stimulated Smo ciliary signal intensity (Fig. 2 and Fig. 5C, green; E compared to H), but did not affect SAG-stimulated Smo trafficking (Fig. 5C and I).
Figure 5.

Smoothened activates cPLA2α through Gβγ. For all experiments * indicates p ≤ 0.05; *** indicates p ≤ 0.0001; ns, p > 0.05. Error bars indicate SEM. A. PLA2 activity assays were performed on lysates from NIH3T3 cells stimulated with ShhN conditioned media in the presence or absence of Gβγ inhibitor GALL (25 µM). Results are shown as fold change in activity relative to vehicle control. The experiment was performed three times in triplicate and all data pooled. Significance was determined using a one-way ANOVA. B. Light2 cells were treated with ShhN conditioned media or SAG (100 nM) +/− GALL as indicated. Normalized percent reporter activity is expressed relative to the agonist-stimulated condition set to 100%. The experiment was performed three times in triplicate and all data pooled. Significance was determined using a two-way ANOVA. C. Cells were treated with ShhN conditioned media or SAG (100 nM) +/− GALL (25 µM) as indicated. Smo localization was analyzed in ≥100 cells across two experiments by immunofluorescence confocal microscopy and ciliary signal intensity was quantified. Significance was calculated based upon number of cells using a two-way ANOVA. D–I. Representative images for each condition are shown. Primary cilia are marked by acetylated α-tubulin (α-Tub c, blue) and the base indicated by Cep290-GFP (magenta). Smo is green.
To clarify the curious results suggesting differential sensitivity of ShhN, 20(S)-OHC and SAG-stimulated Smo to cPLA2 inhibition, we interrogated the mechanism by which cPLA2α impacted Smo signaling activity. To test whether its product arachidonic acid affected Smo signaling, Light2 cells were treated with increasing concentrations of the fatty acid. Exogenous arachidonic acid induced modest reporter activity in the absence of pathway agonist, failed to significantly affect ShhN-induced signaling, and increased SAG activity by ~10% (Fig. 6A–A’). Conversely, arachidonic acid showed pronounced synergy with CRD-binding agonist 20(S)-OHC (Fig. 6A’), enhancing reporter induction by ~80%. Moreover, exogenous arachidonic acid rescued MAFP-mediated inhibition of reporter activity back to 20(S)-OHC-stimulated levels (Fig. 6B), supporting cooperation between the two compounds in modulating Smo signaling. To determine whether this occurred through direct arachidonic acid-Smo binding, bead capture assays were performed. Arachidonic acid agarose beads were generated using click chemistry, and incubated with lysate from SmoYFP-expressing cells alone or in the presence of 20(S)-OHC (Fig. 6C). Arachidonic acid-clicked beads captured Smo-YFP in both the absence and presence of 20(S)-OHC (lanes 2–3). Binding was attenuated, in a dose dependent manner, by addition of free arachidonic acid to cell lysates (lanes 4–7). Addition of the endocannabinoid yV-stearoyldopamine (SD) failed to impact Smo bead capture (lanes 8–9 compared to 3), supporting specificity of the Smo-arachidonic acid interaction in this assay.
Figure 6.

Arachidonic acid binds Smo and synergizes with 20(S)-OHC. For all panels, * indicates p ≤ 0.05; *** indicates p ≤ 0.0001; ns indicates p > 0.05. Error bars indicate SEM. A–A’. Light2 cells were treated with increasing concentrations of arachidonic acid. Normalized baseline reporter activity is shown relative to the vehicle-treated ShhN-stimulated level of reporter activity, set to 100%, shown in A’. A’. Light2 reporter cells were stimulated with ShhN conditioned media, 20(S)-OHC (10 µM) or SAG (100 nM) plus increasing arachidonic acid. Enhancement is shown relative to the vehicle-treated, Smo agonist-stimulated level of reporter activity set to 100% for each agonist. Significance was determined using a two-way ANOVA. B. Light2 cells were treated with 20(S)-OHC (10 µM) +/−MAFP (5 µM). MAFP-treated cells were treated with arachidonic acid, as indicated. The line indicates the baseline 20(S)-OHC stimulated level, set to 100%. Significance was determined relative to this control using a one-way ANOVA. C. Arachidonic acid was coupled to agarose beads using click chemistry and beads were incubated with lysate from SmoYFP-expressing cells in the presence of 20(S)-OHC or vehicle. Binding was competed with cold arachidonic acid. The endocannabinoid N-stearoyldopamine (SD) failed to compete Smo from arachidonic acid beads. The experiment was performed two times. A representative blot is shown. D–D’. Light2 cells were stimulated with 20(S)-OHC (10 µM) and treated with increasing concentrations of cyclopamine and arachidonic acid. D’. X marks show the calculated IC50 for cyclopamine at each arachidonic acid concentration tested. Each X represents one independent experiment done in triplicate. For all reporter assays, experiments were repeated two or three times in triplicate and all data pooled. E. Binding of fluorescent BODIPY-cyclopamine to Smo was monitored by flow cytometry. The experiment was performed three times. A representative experiment is shown. F. A model for cPLA2α modulation of Smo signaling. Initiation of Smo signaling by a CRD oxysterol agonist activates cPLA2α, resulting in lysophospholipid (green) and arachidonic acid (orange) production. Arachidonic acid is proposed to target the TM domain of active Smo to enhance ciliary translocation and bolster signal output. Smo activated by the 7TM agonist SAG induces PLA2, but does not require PLA2-generated lipids for optimal ciliary translocation and high-level signaling.
Enhanced synergy between arachidonic acid and 20(S)-OHC compared to synergy observed for SAG suggested SAG and arachidonic acid might target overlapping binding sites within the 7TM core. The ability of arachidonic acid to reverse reporter gene inhibition by the 7TM-binding inverse agonist cyclopamine was tested as a readout for 7TM occupancy (Fig. 6D). Light2 cells were treated with increasing concentrations of cyclopamine in the presence of 20(S)-OHC, and reduction in reporter activity was monitored. The IC50 of cyclopamine was determined to be ~4.4 nM (Fig. 6D–D’). Introduction of increasing concentrations of arachidonic acid in the presence of cyclopamine shifted the IC50 to ~8.4 nM (Fig. 6D’). Moreover, arachidonic acid reversed inhibition by 4 nM cyclopamine when provided at higher concentrations (Fig. 6D, black vs. green and yellow). To directly test whether arachidonic acid could displace cyclopamine from Smo, binding of fluorescent BODIPY-cyclopamine was monitored by flow cytometry (Khaliullina et al., 2015). Treatment of NIH3T3 cells with BODIPY-cyclopamine labeled approximately 90% of the cell population (Fig. 6E, center). Addition of arachidonic acid competed BODIPY-cyclopamine binding, evidenced by a ~25% decrease the labeled population (right), further supporting arachidonic acid binds the Smo 7TM domain.
Discussion
Smo trafficking and signaling are tightly linked, making regulation of its ciliary trafficking an effective control point for modulation of Shh pathway activity. Movement of proteins through the ciliary membrane is a regulated process, with the primary cilium being partitioned into checkpoints including the ciliary pocket, the transition zone and the Elis van Creveld (EvC2) zone (Benmerah, 2013; Dorn et al., 2012). Distinct phospholipids are associated with specific ciliary membrane domains; the ciliary membrane proper is enriched in PI4P while PI(4,5)P2 enriches at the ciliary base (Chavez et al., 2015; Garcia-Gonzalo et al., 2015). Inactivation of inositol polyphosphate 5-phosphatase E (Inpp5e) at the transition zone allows spreading of PI(4,5)P2 along the ciliary membrane, which promotes ciliary retention of negative pathway regulators Tulp3 and GPR161 (Garcia-Gonzalo et al., 2015). However, inactivation of Inpp5e does not disrupt Shh-mediated Smo ciliary translocation (Chavez et al., 2015; Garcia-Gonzalo et al., 2015), suggesting other lipid remodeling enzymes may be recruited to the ciliary base to provide this control. Herein, we propose cPLA2α, which binds PI(4,5)P2 phospholipids with high affinity (Murakami et al., 2011) is one such enzyme because it localizes to the ciliary base and promotes Smo ciliary accumulation.
Precedent exists for membrane recruitment of lipid remodeling enzymes by active GPCRs, the most relevant being the closely related Wnt receptor Frizzled (Fz) (Qin et al., 2009). Phosphatidylinositol kinases are induced by Fz/Dishevelled complexes to recruit essential components of the active Wnt signalosome to the membrane (Kim et al., 2013; Qin et al., 2009). We propose that like Wnt, Shh influences lipid metabolism to promote pathway activity. This could occur through arachidonic acid influencing an active Smo structure to enhance its lateral transport from the ciliary pocket into the ciliary membrane, disrupting confinement interactions with factors anchoring it at the ciliary base or EvC2 zone, and/or enhancing its interaction with ciliary motors (Milenkovic et al., 2009; Milenkovic et al., 2015). Pitchfork and G Protein Coupled Receptor Associated Sorting Protein 2 (Gprasp2) are potential candidates for the latter scenario because Shh stabilizes association of these proteins with Smo for its ciliary translocation (Jung et al., 2016).
For most GPCRs, activation occurs in response to agonist binding and stabilizing the receptor in a specific active conformation. More than one ligand binding pocket can exist on a given receptor, with differential small molecule binding stabilizing the target receptor in different active or inactive conformations (Strachan et al., 2014). Smo has two known ligand binding pockets, one in the amino-terminal CRD which binds sterols and one in the 7TM that binds SAG and cyclopamine (Byrne et al, 2016; Chen et al, 2002b; Huang et al., 2016; Luchetti et al., 2016; Nachtergaele et al., 2013). The ability of arachidonic acid to enhance 20(S)-OHC-induced Smo signaling is most consistent with arachidonic acid acting through the 7TM core. We hypothesize that upon binding of 20(S)-OHC to the CRD, the 7TM remains accessible for arachidonic acid to bind and enhance signal output. Conversely, binding of SAG to the 7TM domain likely occludes arachidonic acid from effectively working through this interface. Based upon studies interrogating CRD function (Byrne et al., 2016; Luchetti et al, 2016; Myers et al., 2013; Nachtergaele et al, 2012; Nachtergaele et al, 2013; Nedelcu et al, 2013; Rana et al., 2013), we speculate Shh activates signaling by impacting availability or binding of a CRD-directed agonist, and that the ligand binding pocket in the 7TM behaves as a modulatory site for allosteric control. We propose a testable model in which cPLA2α is activated by CRD agonist-occupied Smo at the ciliary base or transition zone to produce arachidonic acid derivatives, which in turn target the 7TM of active Smo to enhance its ciliary trafficking and activity (Fig. 6F). In this model, Shh stimulation is predicted to trigger both CRD and 7TM occupancy, providing a potential explanation for the observed failure of exogenous arachidonic acid to synergize with ShhN.
Once activated, Smo is thought to signal through two distinct pathways, the canonical pathway to Gli transcriptional effectors, believed to be Gα i-independent, and the Gα i-dependent noncanonical pathway (Arensdorf et al, 2016). Gβγ activation being required for optimal Smo ciliary trafficking and target gene activation is seemingly inconsistent with separation of canonical and noncanonical effector routes. However, our results predict cPLAα functions in a modulatory capacity. Because of this, effects of uncoupling Smo from Gα iβγ on canonical signaling would be less pronounced than what would be observed following loss of an essential canonical pathway component. It should also be considered the studies presented here analyzed activity of endogenous Smo and cPLA2α in response to three different activators, ShhN, SAG and 20(S)-OHC. Studies performed using over-expressed Smo, or the 7TM-directed activator SAG, may mask loss of modulatory activity by effectors such as Gβγ and cPLA2α that control molecules acting through the 7TM core.
A number of endocannabinoids, which are in the arachidonic acid metabolic network, have been reported to modulate Smo activity. Of the endocannabinoids examined, cannabinol, cannabidiol and N-acylethanolamide 20:4 were found to compete with the 7TM-docking inverse agonist cyclopamine for binding (Khaliullina et al., 2015). Although these compounds were identified as negative modulators of Smo signaling, their ability to displace cyclopamine further supports lipids in the arachidonic acid family can act through the 7TM ligand binding pocket to affect Smo. The ability of arachidonic acid network molecules to either promote or inhibit Smo activity through this pocket is likely explained by binding of distinct metabolites stabilizing Smo in different conformations. Consistent with this notion, crystallographic evidence suggests the difference between a small molecule functioning as a Smo agonist or antagonist can occur based upon shifts resulting from alteration of a single hydrogen bond (Wang et al., 2014).
Although our data are most consistent with arachidonic acid being the cPLA2α product responsible for affecting Smo, we cannot rule out contributions by cPLA2α-produced lysophospholipids. Lysophospholipids, when intercalated into the phospholipid bilayer, promote membrane curvature (Murakami et al., 2011). The ciliary pocket is an area of membrane curvature near the ciliary base that contributes to gating of ciliary proteins (Benmerah, 2013). As such, it is possible cPLA2α may impact pocket architecture, via lysophospholipid membrane insertion, to assist in Smo ciliary entry or docking. The importance of precise control of ciliary protein trafficking is underscored by human ciliopathies including Joubert syndrome, Bardet-Biedl syndrome, Ellis van Creveld syndrome, and polycystic kidney disease (Quinlan et al., 2008). Although such syndromes do not eliminate SHH signaling, pathway activity is commonly altered to affect disease etiology. As such, loss of a positive modulator of SMO ciliary translocation resulting in a ~40–80% reduction of in vitro pathway activity may result in overt disease in vivo. Future studies will be needed to determine whether cPLA2α is one such modulator.
Experimental Procedures
Chemicals
SAG (Enzo) and 20(S)-OHC (Steraloid, Inc.), were reconstituted in DMSO and stored at −20°C. Cyclopamine (LC laboratories) was reconstituted in ethanol and stored at −80°C. MAFP (IC50 = 0.6 µM, Cayman Chemical Company) was purchased, solvent exchanged to DMSO and stored at −80°C. BEL (IC50 = 60 nM, Cayman Chemical Company), FIPI (IC50 = 10 nM, EMD Millipore), BODIPY-cyclopamine (BioVision) and GIRI (Duvernay et al., 2015) were reconstituted in DMSO and stored at −20°C. GALL (IC50 = 422 nM, (Lehmann et al., 2008), SCBT), PLD1i (IC50 = 3.7 nM) and PLD2i (IC50 = 20 nM, (Lewis et al., 2009), were dissolved in DMSO and stored at 4°C. NEO (IC50 = 10 µM, SCBT) was reconstituted in H2O and stored at −20°C. Arachidonic acid (Cayman Chemical Company) and N-stearoyldopamine (SD, Tocris Bioscience) were stored in ethanol at −20°C.
Statistical Methods
Statistical analyses were performed using Graphpad Prism and a one- or two-way unpaired ANOVA with Bonferroni correction as appropriate. Statistics for reporter assays were calculated based upon at least two independent experiments performed in triplicate. For each sample luciferase reporter activity was normalized to an internal renilla luciferase control, then indicated relative to the vehicle-treated, pathway-activated response. For cyclopamine IC50 determination, data were plotted as nonlinear regression curves using Graphpad Prism log(inhibitor) vs. response variable slope function. Statistics for ciliary quantification were calculated from two independent experiments with at least 60 cilia per condition per experiment. The average signal intensity of each experimental cilia was normalized to signal of vehicle-treated ShhN- or SAG-stimulated conditions set to 100%. Statistics for PLA2 activity and HPLC/MS/MS experiments were based upon 2–3 experiments as indicated with three samples per condition. For all statistical analyses, graphs show the average of all data points pooled with error bars indicating standard error of the mean (s.e.m.).
Reporter Assays
Light2 cells were pretreated with drug for 2 hours at 37°C in serum-free media (DMEM supplemented with 0.1mM nonessential amino acids, 2mM L-Glutamine, 1mM sodium pyruvate and 1% Pen-Strep). After incubation, media was replaced with low serum (0.5%) media containing drug plus Smo modulator (SAG (100 nM), ShhN conditioned media (100–300 µl /mL), 20(S)-OHC (10 µM), cyclopamine or arachidonic acid (as indicated)) and incubated for 36 hours. ShhN conditioned media was generated as described (Maity et al., 2005; Marada et al., 2015). Luciferase activity was measured using Dual-Luciferase Reporter assay system (Promega).
Nuclear Extraction and Immunoblot Analysis
Subcellular fractions were isolated using the Active Motif kit. Fractions were normalized for protein content, and analyzed by immunoblot. Blots were incubated in blocking buffer (5% non-fat dry milk, 1X TBS, 0.1% Tween) for 30 minutes, then incubated overnight at 4°C with anti-Gli3 (R&D), anti-Gli1 (Cell Signaling), anti-Lamin AC (Santa Cruz) and anti-tubulin (Cell Signaling) antibodies in blocking buffer. Blots were washed 3x in TBST buffer (1X TBS, 0.1% Tween) then incubated for 1 hour with donkey anti-mouse HRP (Jackson Immuno), donkey anti-goat HRP (Jackson Immuno), or IR 800 mouse (Li-COR). Blots were developed using an Odyssey Fc imaging system (Li-COR) and either ECL prime chemiluminescent substrate (GE Amersham) or IR800 dyes (Li-COR).
Immunofluorescence Analysis
NIH3T3 cells were plated onto coverslips (Corning) and pretreated with drug in serum-free media. After 2 hours, media was changed to low serum media containing drug plus SAG (100 nM), ShhN conditioned media (100–300 µL/mL) or cyclopamine (10 µM) and cells were incubated for 20 hours. For transition zone staining, cells were transfected with pEGFP-mCep290 (Valente et al., 2006) (Addgene) 24 hours prior to drug treatment using Lipofectamine 3000. Immunofluorescence analysis was performed as described (Garcia-Gonzalo et al., 2011; Marada et al., 2015). Cells were incubated at 4°C overnight with anti-Smo (SCBT), anti-acetylated-alpha tubulin (Cell Signaling), anti-Gli3 (R & D systems), anti-HA (Roche) and/or anti-p-cPLA2 (1:1000, Cell Signaling) in blocking buffer. Cells were washed 3x prior to addition of secondary antibodies and incubation at room-temperature for 1 hour. AlexaFluor 488, 555 and 647 conjugated secondary antibodies (Life Technologies) and anti-GFP-Dylight488 (Rockland) were used. Cells were washed 3x in wash buffer then 3x in PBS prior to mounting with Prolong Diamond without DAPI (Life Technologies). Images were collected using a Zeiss LSM 710 or Leica TCS SP8 STED 3x confocal microscope with a 63x oil-immersion objective and processed using ZEN 2012 (blue edition, Zeiss), LAS X (Leica) and Photoshop CS6. For all immunofluorescence experiments, multiple cells (≥100) were examined over a minimum of two experiments and representative images are shown.
Quantification of Ciliary Signal Intensity
At least three randomly chosen fields of view were selected and all cilia within the field quantified using ZEN 2012/LAS X. Each cilium was traced from the base to the tip using spline profile function. Line scan intensity values along the cilium were exported and analyzed in Excel or Igor Pro (Wavemetrics) with custom written macro in automated manner. Cilia were divided into three equal regions and Smo or Gli3 intensity within all regions measured.
PLA2 Activity Assay
NIH3T3 or Smo−/− MEFs were pretreated with drug in serum-free media. After 2 hours, media was changed to low serum media containing drug and pathway activator as above. Cells were harvested in 500 µL/well chilled hypotonic lysis buffer (20mM HEPES, 10mM KCl, pH 7.4), transferred to a pre-chilled 1 mL dounce homogenizer, and incubated on ice for 30 minutes. Cells were lysed with 20 strokes of the tight pestle twice, transferred to 1.5 mL microfuge tubes and centrifuged at 2,000 × g for 10 minutes at 4°C. PLA2 activity in the supernatant was tested using EnzChek Phospholipase A2 Assay Kit (ThemoFisher). PLA2 activity was normalized to total protein concentration of the lysate determined by BCA assay (Bio-Rad).
Fatty Acid Extraction and Quantification by HPLC/MS/MS
NIH3T3 or Pla2g4a cells were incubated in serum-free media for 2 hours then stimulated with vehicle, 20-OHC (10 µM) or SAG (100nM). After 2 hours, cells were harvested in ice-cold PBS. Non-esterified (free) fatty acids and lipids were extracted from cells as described (Bligh and Dyer, 1959). Samples were spiked with 400 pmol of d8-arachidonic acid (d8–20:4) (Sigma-Aldrich) and analyzed using workflow as described (Nzoughet et al., 2015). Free fatty acids were separated from lipids and analyzed using a Shimadzu Prominence UFLC attached to a Sciex QTrap 4500 equipped with a Turbo V ion source (Sciex). Samples were injected onto an Acquity UPLC® BEH HILIC, 1.7 µm, 2.1 × 150 mm column (Waters) using a flow rate of 0.2 ml/min. Solvent A was 100% acetonitrile, and Solvent B was 15 mM ammonium acetate, pH 3. The HPLC program was: 0 to 2 min isocratic at 4% B; 2 to 20 min linear gradient to 80% B; 20 to 23 min isocratic at 80% B; 23 to 25 min linear gradient to 4% B; 25 to 30 min isocratic with 4% B. The QTrap 4500 was operated in the negative mode, and the ion source parameters were: ion spray voltage, −4500 V; curtain gas, 25 psi; temperature, 350 °C; collision gas, medium; ion source gas 1, 40 psi; ion source gas 2, 60 psi; declustering potential, −45 V and collision energy, −15 V. The MRM transitions (Q1 / Q3) for the fatty acids analyzed in this study were: 16:0 (255.2 / 211), 16:1 (253.2 / 209), 18:0 (283.2 / 239), 18:1 (281.2 / 237), 18:2 (279.2 / 235), 20:3 (305.2 / 261), 20:4 (303.2 / 259), 20:5 (301.2 / 257), 22:4 (331.2 / 287), 22:5 (329.2 / 285), 22:6 (327.2 / 283), and d8–20:4 (311.2 / 267). The system was controlled by the Analyst® software (Sciex) and quantitatively analyzed with MultiQuant™ 3.0.2 software (Sciex).
Quantitative RT-PCR
RNA was isolated with Trizol (Invitrogen). cDNA was synthesized with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR reactions were performed on a QuantStudio 7 Flex PCR machine using PowerUp Sybr Green Master Mix (Applied Biosystems). Pre-designed KiCqStart SYBR green primers against Pla2g4b, Pla2g4d, Pla2g4e, Pla2g4f and Plb1 were purchased (Sigma-Aldrich). Primers against Ppia (5’-AGCACTGGAGAGAAAGGATT; 5’-ATTATGGCGTGTAAAGTCACCA), Btf3 (5’-GAACAACATCTCTGGTATTGAAGA; 5’-AATGGTGAAGGTGTTTGCTG) and Pla2g4a (5’-TGGTGTGATGAAGGCACTG; 5’-CTGGAAAATCGGGGTGAG) were purchased (Invitrogen). Primer sets were tested and optimized such that PCR efficiencies were ~100%. mRNA expression level of each cPla2 gene was calculated using standard comparative CT method with minor adjustments. The DCT for each gene was calculated using the average CT of two housekeeping genes (Ppia and Btf3). The 2^(−DCT) value for each gene was then normalized to the 2^(−DCT) of Ppia. Assays were conducted at least three times in triplicate then averaged together.
Arachidonic Acid Bead Capture
Arachidonic acid beads were freshly prepared by click chemistry using the Click-iT Cell Reaction Buffer Kit (Invitrogen). Azide-coated agarose beads (Click Chemistry Tools) were washed 3x in H2O, supernatant removed, and beads resuspended in Click-iT reaction mix containing arachidonic acid alkyne (Caymen Chemicals). Reactions were incubated at room-temperature for 30 minutes with inversion, then beads pelleted at 2,000 × g for 2 minutes at 4°C and supernatant removed. Beads were washed 3 times in washing/binding buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 0.25% NP40).
Membrane fractions were prepared as described (Nachtergaele et al., 2013) with minor modifications. NIH3T3 cells were transfected with pCS2+-mSmo-YFP (Addgene) using Lipofectamine 3000. After 36 hours, cells were harvested and swelled in 1 mL hypotonic SEAT buffer (250 mM sucrose, 1 mM EDTA, 10 mM acetic acid, 10 mM triethanolamine and SigmaFast EDTA-free protease inhibitor cocktail) for 30 mins on ice. Cells were lysed with 40 strokes using the tight dounce pestle twice, transferred to 1.5 mL microfuge tube and centrifuged at 900 × g for 5 minutes at 4°C. Membranes were isolated by ultracentrifugation (95,000 × g, 30 mins at 4°C) and solubilized in n-dodecyl-β-D-maltopyranoside (DDM) buffer (50 mM Tris pH 7.4, 150 mM NaCl, 10% glycerol, 0.5% DDM and SigmaFast EDTA-free protease inhibitor cocktail) for 2 hours at 4°C with inversion. Supernatant was precleared with unclicked agarose beads for 2 hours at 4°C with inversion. Arachidonic acid-agarose beads in washing/binding buffer (30% slurry, preparation described above) were added and vehicle, 20-OHC (10 µM), free arachidonic acid or SD added with Smo-containing membranes. The mixture was incubated at 4°C for 2.5 hours with inversion. Beads were collected at 2,000 × g for 1 minute, washed 3x, then resuspended in protein loading buffer (10% SDS, 10 mM DTT, 20% glycerol, 0.2 M Tris-HCl pH 6.8, and 0.05% Bromophenol blue) and incubated at 4°C with inversion for 1 hour.
BODIPY-Cyclopamine Competition Analysis
This assay was performed as described (Chen et al., 2002a). NIH3T3 were incubated in serum-free media for two hours at 37°C, then treated with BODIPY-Cyclopamine (5nM) and arachidonic acid (12.5µM) in phenol-red free DMEM supplemented with 10% FBS for 5 hours at 37°C. Cells were collected and binding was stopped by adding an equal volume of fresh phenol red free DMEM supplemented with 0.5% BCS. Cells were pelleted as above and resuspended in 300 µL phenol red free DMEM supplemented with 0.5% BCS. GFP positive cells were detected by flow cytometry on a BD Canto FACS machine and analyzed using FlowJo software.
Highlights.
Sonic Hedgehog activates cytosolic phospholipase A2 to release arachidonic acid.
Phospholipase A2 is activated by Gβγ downstream of Smoothened.
Arachidonic acid binds Smoothened to promote its ciliary accumulation and signaling.
Arachidonic acid is a candidate allosteric regulator of Smoothened
Acknowledgments
Studies were supported by NIH NIGMS grants GM101087 (SKO), GM034496 (COR), Cancer Center Support Grant CA021765 and by ALSAC of St. Jude Children’s Research Hospital. Confocal microscopy was performed at the SJCRH Cell and Tissue Imaging Center which is supported by SJCRH and NCI P30CA021765. We thank P. Beachy, J. Bonventre, A. Brown, V. Frohlich, R. Rohatgi, M. Turnis and members of the Ogden lab for reagents, advice, assistance and discussion, and J. Temirov for assistance with ciliary signal quantification.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, SKO, AMA; Investigation, AMA, MED, JMM, MWF; Validation, AMA; Formal Analysis, AMA, MWF, COR; Resources, AMA, SKO, COR; Data Curation; AMA, SKO; Writing, SKO, AMA, COR; Visualization, AMA, MWF, COR; Supervision, SKO, COR; Project Administration, SKO, AMA; Funding Acquisition, SKO, COR.
The authors declare no conflicts of interest.
References
- Allen BL, Song JY, Izzi L, Althaus IW, Kang JS, Charron F, Krauss RS, McMahon AP. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell. 2011;20:775–787. doi: 10.1016/j.devcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arensdorf AM, Marada S, Ogden SK. Smoothened Regulation: A Tale of Two Signals. Trends Pharmacol Sci. 2016;37:62–72. doi: 10.1016/j.tips.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsinde J, Winstead MV, Dennis EA. Phospholipase A(2) regulation of arachidonic acid mobilization. FEBS Lett. 2002;531:2–6. doi: 10.1016/s0014-5793(02)03413-0. [DOI] [PubMed] [Google Scholar]
- Bechler ME, Brown WJ. Gbeta1gamma2 activates phospholipase A-dependent Golgi membrane tubule formation. Front Cell Dev Biol. 2014;2:0004. doi: 10.3389/fcell.2014.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgacem YH, Borodinsky LN. Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc Natl Acad Sci U S A. 2011;108:4482–4487. doi: 10.1073/pnas.1018217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmerah A. The ciliary pocket. Curr Opin Cell Biol. 2013;25:78–84. doi: 10.1016/j.ceb.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Bonventre JV. The 85-kD cytosolic phospholipase A2 knockout mouse: a new tool for physiology and cell biology. J Am Soc Nephrol. 1999;10:404–412. doi: 10.1681/ASN.V102404. [DOI] [PubMed] [Google Scholar]
- Byrne EF, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, Tully MD, Mydock-McGrane L, Covey DF, Rambo RP, et al. Structural basis of Smoothened regulation by its extracellular domains. Nature. 2016;535:517–522. doi: 10.1038/nature18934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, Schiffmann SN. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Dev Cell. 2015;34:338–350. doi: 10.1016/j.devcel.2015.06.016. [DOI] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002a;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A. 2002b;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn KV, Hughes CE, Rohatgi R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev Cell. 2012;23:823–835. doi: 10.1016/j.devcel.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernay MT, Matafonov A, Lindsley CW, Hamm HE. Platelet Lipidomic Profiling: Novel Insight into Cytosolic Phospholipase A2alpha Activity and Its Role in Human Platelet Activation. Biochemistry. 2015;54:5578–5588. doi: 10.1021/acs.biochem.5b00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Phua SC, Roberson EC, Garcia G, 3rd, Abedin M, Schurmans S, Inoue T, Reiter JF. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev Cell. 2015;34:400–409. doi: 10.1016/j.devcel.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal S, Morrison EE, Ponnambalam S, Walker JH. Nuclear localisation of cytosolic phospholipase A2-alpha in the EA.hy.926 human endothelial cell line is proliferation dependent and modulated by phosphorylation. J Cell Sci. 2002;115:4533–4543. doi: 10.1242/jcs.00146. [DOI] [PubMed] [Google Scholar]
- Huang P, Nedelcu D, Watanabe M, Jao C, Kim Y, Liu J, Salic A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell. 2016;166:1176–1187. doi: 10.1016/j.cell.2016.08.003. e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–812. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Liu Y, Fan J, Zhang J, Li XA, Evers BM, Zhu H, Jia J. PI(4)P Promotes Phosphorylation and Conformational Change of Smoothened through Interaction with Its C-terminal Tail. PLoS Biol. 2016;14:e1002375. doi: 10.1371/journal.pbio.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung B, Padula D, Burtscher I, Landerer C, Lutter D, Theis F, Messias AC, Geerlof A, Sattler M, Kremmer E, et al. Pitchfork and Gprasp2 Target Smoothened to the Primary Cilium for Hedgehog Pathway Activation. PLoS One. 2016;11:e0149477. doi: 10.1371/journal.pone.0149477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliullina H, Bilgin M, Sampaio JL, Shevchenko A, Eaton S. Endocannabinoids are conserved inhibitors of the Hedgehog pathway. Proc Natl Acad Sci U S A. 2015;112:3415–3420. doi: 10.1073/pnas.1416463112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Pan W, Jones SA, Zhang Y, Zhuang X, Wu D. Clathrin and AP2 are required for PtdIns(4,5)P2-mediated formation of LRP6 signalosomes. J Cell Biol. 2013;200:419–428. doi: 10.1083/jcb.201206096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann DM, Seneviratne AM, Smrcka AV. Small molecule disruption of G protein beta gamma subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol. 2008;73:410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Scott SA, Lavieri R, Buck JR, Selvy PE, Stoops SL, Armstrong MD, Brown HA, Lindsley CW. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg Med Chem Lett. 2009;19:1916–1920. doi: 10.1016/j.bmcl.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, Covey DF, Siebold C, Rohatgi R. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife. 2016;5 doi: 10.7554/eLife.20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity T, Fuse N, Beachy PA. Molecular mechanisms of Sonic hedgehog mutant effects in holoprosencephaly. Proc Natl Acad Sci U S A. 2005;102:17026–17031. doi: 10.1073/pnas.0507848102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marada S, Navarro G, Truong A, Stewart DP, Arensdorf AM, Nachtergaele S, Angelats E, Opferman JT, Rohatgi R, McCormick PJ, et al. Functional Divergence in the Role of N-Linked Glycosylation in Smoothened Signaling. PLoS genetics. 2015;11:e1005473. doi: 10.1371/journal.pgen.1005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- Milenkovic L, Scott MP, Rohatgi R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J Cell Biol. 2009;187:365–374. doi: 10.1083/jcb.200907126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic L, Weiss LE, Yoon J, Roth TL, Su YS, Sahl SJ, Scott MP, Moerner WE. Single-molecule imaging of Hedgehog pathway protein Smoothened in primary cilia reveals binding events regulated by Patched1. Proc Natl Acad Sci U S A. 2015;112:8320–8325. doi: 10.1073/pnas.1510094112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama J, Takabatake T, Takeshima K, Hui C. Ptch2, a second mouse Patched gene is co-expressed with Sonic hedgehog. Nat Genet. 1998;18:104–106. doi: 10.1038/ng0298-104. [DOI] [PubMed] [Google Scholar]
- Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog Lipid Res. 2011;50:152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Myers BR, Sever N, Chong YC, Kim J, Belani JD, Rychnovsky S, Bazan JF, Beachy PA. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell. 2013;26:346–357. doi: 10.1016/j.devcel.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF, Rohatgi R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat Chem Biol. 2012;8:211–220. doi: 10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, Ingham PW, Covey DF, Siebold C, Rohatgi R. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife. 2013;2:e01340. doi: 10.7554/eLife.01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelcu D, Liu J, Xu Y, Jao C, Salic A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat Chem Biol. 2013;9:557–564. doi: 10.1038/nchembio.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nzoughet JK, Gallart-Ayala H, Biancotto G, Hennig K, Dervilly-Pinel G, LeBizec B. Hydrophilic interaction (HILIC) and reverse phase liquid chromatography (RPLC)-high resolution MS for characterizing lipids profile disruption in serum of anabolic implanted bovines. Metabolomics. 2015;11:1884–1895. [Google Scholar]
- Qin Y, Li L, Pan W, Wu D. Regulation of phosphatidylinositol kinases and metabolism by Wnt3a and Dvl. J Biol Chem. 2009;284:22544–22548. doi: 10.1074/jbc.M109.014399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan RJ, Tobin JL, Beales PL. Modeling ciliopathies: Primary cilia in development and disease. Curr Top Dev Biol. 2008;84:249–310. doi: 10.1016/S0070-2153(08)00605-4. [DOI] [PubMed] [Google Scholar]
- Rana R, Carroll CE, Lee HJ, Bao J, Marada S, Grace CR, Guibao CD, Ogden SK, Zheng JJ. Structural insights into the role of the Smoothened cysteine-rich domain in Hedgehog signalling. Nat Commun. 2013;4:2965. doi: 10.1038/ncomms3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond GS, Smith TK. Phospholipases A(1) International journal of molecular sciences. 2011;12:588–612. doi: 10.3390/ijms12010588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riobo NA, Saucy B, Dilizio C, Manning DR. Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci U S A. 2006;103:12607–12612. doi: 10.1073/pnas.0600880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Corcoran RB, Scott MP. Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc Natl Acad Sci U S A. 2009;106:3196–3201. doi: 10.1073/pnas.0813373106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Strachan RT, Sun JP, Rominger DH, Violin JD, Ahn S, Thomsen AR, Zhu X, Kleist A, Costa T, Lefkowitz RJ. Divergent Transducer-Specific Molecular Efficacies Generate Biased Agonism At A G Protein-Coupled Receptor (GPCR) J Biol Chem. 2014 doi: 10.1074/jbc.M114.548131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- Thotala D, Craft JM, Ferraro DJ, Kotipatruni RP, Bhave SR, Jaboin JJ, Hallahan DE. Cytosolic phospholipaseA2 inhibition with PLA-695 radiosensitizes tumors in lung cancer animal models. PLoS One. 2013;8:e69688. doi: 10.1371/journal.pone.0069688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- Wang C, Wu H, Evron T, Vardy E, Han GW, Huang XP, Hufeisen SJ, Mangano TJ, Urban DJ, Katritch V, et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat Commun. 2014;5:4355. doi: 10.1038/ncomms5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CW, Chen MH, Chuang PT. Smoothened adopts multiple active and inactive conformations capable of trafficking to the primary cilium. PLoS One. 2009;4:e5182. doi: 10.1371/journal.pone.0005182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S, Lum L, Beachy P. The ihog cell-surface proteins bind hedgehog and mediate pathway activation. Cell. 2006;125:343–357. doi: 10.1016/j.cell.2006.02.040. [DOI] [PubMed] [Google Scholar]
- Yavari A, Nagaraj R, Owusu-Ansah E, Folick A, Ngo K, Hillman T, Call G, Rohatgi R, Scott MP, Banerjee U. Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev Cell. 2010;19:54–65. doi: 10.1016/j.devcel.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Hu C, Sheng W, Tan KS, Haidekker MA, Sun AY, Sun GY, Lee JC. NAD(P)H oxidase-mediated reactive oxygen species production alters astrocyte membrane molecular order via phospholipase A2. Biochem J. 2009;421:201–210. doi: 10.1042/BJ20090356. [DOI] [PubMed] [Google Scholar]