

Abstract
The advent of second generation CARs and the CD19 paradigm have ushered a new therapeutic modality in oncology. In contrast to earlier forms of adoptive cell therapy, which were based on the isolation and expansion of naturally occurring T cells, CAR therapy is based on the design and manufacture of engineered T cells with optimized properties. A new armamentarium, comprising not only CARs but also chimeric costimulatory receptors, chimeric cytokine receptors, inhibitory receptors and synthetic Notch receptors, expressed in naïve, central memory or stem cell-like memory T cells, is being developed for clinical use in a wide range of cancers. Immunological principles are thus finding a new purpose thanks to advances in genetic engineering, synthetic biology and cell manufacturing sciences.
This article provides an account of why and how adoptive T cell therapies are breaking out of the mold of classic immunology and bringing on a new dimension to immunotherapy, one that is predicated on cell engineering and synthetic biology. The first embodiments of adoptive T cell therapies made use of naturally occurring T cells (Phase I). Chimeric antigen receptors (CARs), bolstered by the success of the CD19 paradigm, have introduced synthetic biology into clinical practice. Thus, current CAR therapy approaches utilize autologous T cells that are retargeted to a specified antigen and metabolically reprogrammed through synthetic receptors known as second generation CARs (Phase 2). This paradigm shift is paving the way for further T cell engineering that will not only substitute for insufficient T cell responses but provide “off-the-shelf” therapeutic T cells and possibly circumvent normal T cell ontogeny by generating T cells in vitro (Phase 3).
Phase I: Natural cell therapies
The technique of adoptive cell transfer was used to study tumor immunity well over half a century ago [1–3]. By the 1980’s, a large body of work had established that T lymphocytes harvested from immune mice could protect syngeneic recipients from a subsequent tumor challenge and sometimes mediate rejection of established tumors, reviewed in [4]. This common laboratory practice eventually inspired the use of T cell transfers for therapeutic purposes [5]. Also dating back to the 50’s, the clinical use of allogeneic bone marrow grafts would eventually lead to the recognition that donor T cells present in bone marrow grafts could mediate potent effects, some beneficial through the graft-versus-leukemia (GVL) effect and some deleterious resulting in graft-versus-host disease (GVHD). [6–9].
The clinical results obtained with early adoptive T cell transfers pointed to the need to devise better approaches to select and expand T cells with increased tumor specificity, and, especially in the allogeneic setting, decreased toxicity [10–12].
In the 80’s and 90’s, a succession of cell therapy approaches were developed with the intent to increase efficacy and minimize toxicity, reviewed in [13]. Therapies utilizing autologous lymphokine-activated killer (LAK) cells and donor leukocyte infusion (DLI) made use of bulk circulating mononuclear cells, non-specifically expanded in the presence of interleukin-2 in the case of LAK cells, or left unmanipulated in the case of DLI [14–17]. The shortcomings of these approaches pointed to the need to further enrich the infusion product with antigen-specific T cells. Starting from surgical explants rather than blood, a higher frequency of tumor-reactive cells could be retrieved and expanded from tumor-infiltrating lymphocytes (TILs), providing superior outcomes to LAK cell therapy [18]. TIL therapy remains in use to this date and is being applied to some other cancers, although its use cannot be generalized [19]. In the allogeneic setting, the establishment of a number of methodologies to isolate virus-specific T lymphocytes (VSTs) from peripheral blood proved to be fruitful against Epstein-Barr virus, cytomegalovirus and adenovirus, enabling the use of donor T cells with a markedly reduced risk of GVHD [20–22].
A common feature of these various cell therapies is their reliance on naturally occurring T cells, isolated from the patient or a donor (Figure 1, left). These varied technologies all depend on the existence of potentially therapeutic cells in the collected blood or tissue samples, and fall short when such cells cannot be retrieved [13]. The advent of T cell engineering singularly altered this overarching limitation. Armed with genetic technologies and further empowered by the design of supra-physiological, synthetic receptors, it would no longer be a cell harvested from the patient or a donor that would be adoptively transferred, but an engineered cell product that was designed and generated through ex vivo cell manufacturing (Figure 1, right). The founding example for this concept is CD19 CAR therapy.
Figure 1.
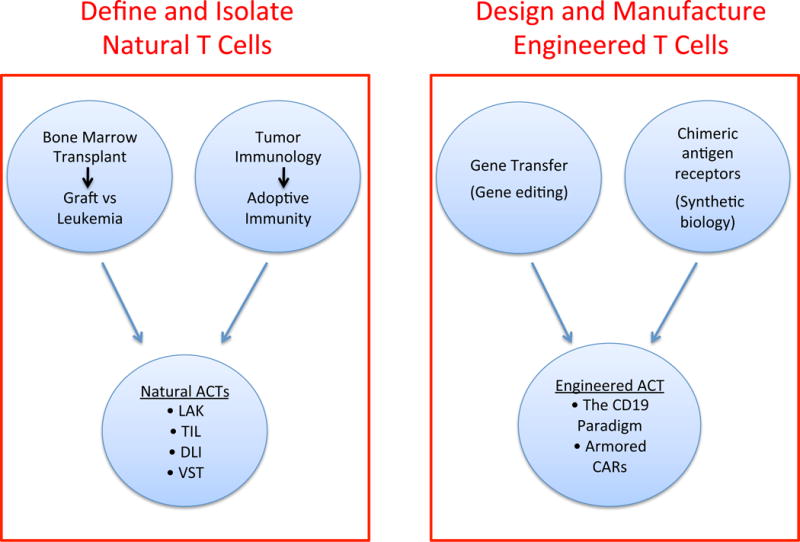
A paradigm switch in T cell therapy. The quest to isolate and expand available T cells from the patient or a donor (left) is giving way to the design and manufacture of engineered T cells with optimized properties (right). See text for abbreviations.
Phase 2: Engineered T cell therapies (autologous)
1. The need for genetic engineering tools
The implementation of T cell engineering begins with devising appropriate tools to genetically modify primary T cells. The first attempts to succeed made use of ecotropic γ-retroviral vectors to transduce mitogen-activated mouse splenocytes [23]. The same approach was subsequently adapted to human T lymphocytes [24–26]. Retroviral transduction was pivotal for launching mouse and human T cell engineering, which had been hitherto limited to transfection of surrogate leukemia cell lines or hybridomas, which do not recapitulate several critical features of normal T cell proliferation, function and survival. Receptors and signaling molecules could from thereon be studied in authentic T cells. These methods remain the foundation of many of today’s clinical trials based on T cell engineering [27–30]. There are by now an array of available T cell transduction methods, based on γ-retroviral, lentiviral, and nonviral DNA- or RNA-based vectors (reviewed in [31]). Gene editing techniques have recently been used to disrupt genes in primary T cells [32–34], further expanding the possibilities of T cell engineering. Gene editing may also be used to target CAR delivery, addressing limitations of randomly integrating vector systems [35].
2. Retargeting T cells: need for effective receptors for antigen
The genetic repurposing of a T cell requires the expression of a chosen receptor for antigen that serves as the targeting device and gatekeeper of T cell activation. This is achieved by expressing a T cell receptor (either αβ or γδ) [36] or an artificial receptor such as a CAR [37] The first CARs described by Eshhar and Brocker [38,39], initially called T-bodies, were TCR mimetics that aimed to mediate antigen recognition and T cell activation through a single chain rather than the 6 independent gene products that constitute the physiological TCR/CD3 complex (Figure 2). Upon introducing the CAR nomenclature, we referred to such molecules as first generation CARs [40]. Unlike the TCR, CARs bind cell surface antigens independently of HLA and are therefore not constrained by patient HLA haplotypes for their clinical utilization. After we and others revealed the shortcomings of ζ-chain-based TCR mimetics [41–43], we found that integrating costimulatory signals within the CAR itself enabled T cell expansion and preserved the function of human primary T cells upon repeated exposure to antigen [43,44]. These receptors, known as second-generation CARs [40], comprise an antigen binding domain and two signaling domains, one derived from a T cell–activating molecule, most commonly the ζ-chain of the CD3 complex and one derived from a costimulatory receptor, such as CD28 [44] or 4-1BB/CD137 [45], (Figure 2). CD28- and 4-1BB-based CARs are at present the best know CAR designs and have been extensively reviewed elsewhere [46,47]. The list of second generation CARs has vastly expanded over the past decade. Over 40 different specificities have been reported and various signaling domains have been evaluated include OX40, DAP10, DAP12, ICOS, NKG2D and more [48–50]. Further research is needed to better understand the respective properties of all these second generation CARs [46,47].
Figure 2.
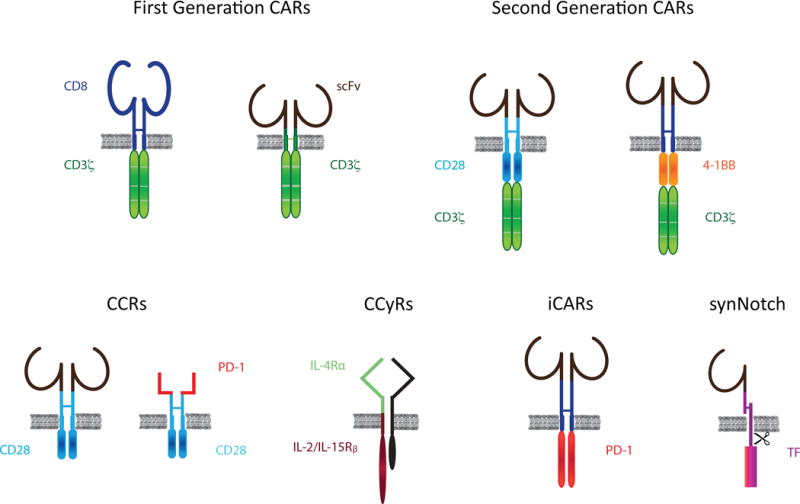
The expanding repertoire of synthetic immunoreceptors: CARs (first generation TCR mimetics, second generation providing integrated activating and costimulatory signals; CCRs, chimeric costimulatory receptor; CCyRs, chimeric cytokine receptors; iCARs, inhibitors of T cell activation; synNotch, synthetic Notch receptors. See text for definitions and references.
3. Third generation CARs and armored CARs
The CARs that have recently shown impressive clinical outcomes in patients with B cell malignancies are second generation CARs. The results obtained in patients with B cell malignancies are extensively reviewed elsewhere [43,51,52].
Third generation CARs are conceptually similar to second generation CARs, except for their use of multiple costimulatory components, for example 4-BB and CD28[53–55], or OX40 and CD28[56]. They have already been introduced in the clinic, but the first outcomes in clinical trials targeting Her2 or CD20 have not been very positive [57,58]. More investigation is needed to optimize and better define the therapeutic potential of this design. In a recent study evaluating novel CAR structures, third generation CARs did not perform as well as other designs [59].
The function of second generation CARs can however be enhanced through a variety of alternative strategies. Those based on the coexpression of an independent gene product co-transduced with the CAR may be referred to as armored CARs [60]. The purposes of this CAR + X approach are many (Figure 2). Some aim to further alter the function and/or persistence of the engineered T cell, while others act on the tumor cell or the tumor microenvironment; some approaches integrate the two goals. The molecules used in this setting include cytokines, such as interleukin-7, 12 and 15 [61–64], cytokine receptors [65,66], chimeric costimulatory receptors [67,68], costimulatory ligands [59,69] and other modulators of T cell activation [70,71]. A discussion of the rationale for each one of these designs is beyond the scope of this review, but this diversity reflects the enormous potential of combining immunological and synthetic biology principles for the further improvement of CAR T cells.
4. Control switches
CAR T cells are potent anti-tumor agents, as demonstrated in the context of various CD19+ malignancies, including NHL, CLL and ALL [72–74]. They can induce toxicities, such as B cell aplasia, severe cytokine release syndrome and neurotoxicity [72–74]. Several innovative strategies have been developed to counteract these occurrences, by either eliminating the engineered T cells or better constraining their function.
These strategies make use of remote or cell autonomous controls, utilizing small molecules, antibodies or synthetic receptors to regulate T cell activity. One approach is to activate a latent suicide switch, such as the inducible caspase-9 (iCasp9) enzyme, through the administration of a small molecule to induce T cell apoptosis [75]. Bifunctional small molecules that mediate the binding between antigen and CAR have also been developed to regulate target engagement [76]. A variation on this approach uses antibodies to mediate antigen recognition on target cells and binding of T cells expressing a synthetic Fc receptor [77].
Wu et al. [78] recently reported a design incorporating a remote control of CAR T cells, whereby a small molecule is used to dimerize antigen-binding and signaling domains. In contrast to the small molecule-controlled suicide switch, this ON-switch design represents a positive reversible regulation that does not eliminate T cells but rather restricts their activities (reviewed in [79]). Both the inducible caspase-9 approach and this one take advantage of well-established chemically induced dimerization (CID) modules developed in the 1990s, where two proteins bind only in the presence of a small molecule [80].
5. Combinatorial antigen targeting
Although the above safety switches allow remote temporal control of CAR T cell activity, they do not provide a means to improve spatial control of antigen engagement and tumor selectivity. To this end, combinatorial approaches integrating two autonomous antigen inputs to control CAR T cell functions have been developed to spatially discriminate between normal and tumor cells expressing a common target. Three strategies have emerged to address this challenge, all making use of different chimeric receptors: iCARs, CCRs and synNotch receptors (Figure 2).
iCARs are synthetic inhibitory receptors derived from the PD-1 or CTLA-4 receptors, intended to protect normal cells from targeted destruction based on the iCAR’s recognition of an antigen present in normal cells but not the tumor [81]. iCARs are neither CARs–as they do not trigger T cell activation–nor CCRs–as they do not enhance T cell function or persistence. Federov et al. showed that recombinant receptors mimicking two prototypic checkpoint blockade receptors, PD-1 and CTLA-4, could restrain T cell cytotoxicity and cytokine secretion in antigen-specific fashion[81]. The main interest in utilizing iCARs is to divert collateral damage to normal tissues without resorting to lymphotoxic means such as suicide genes and high dose corticosteroid therapy. This situation is in part analogous to T cell and natural killer cell immunoregulation, where inhibitory checkpoint blockade receptors or killer inhibitory receptors (KIRs), can reversibly block or limit immune responses.
Another approach utilizes complementary signals split between two receptors: a CAR for T cell activation and a chimeric co-stimulatory receptor (CCR) for costimulation [67]. Tumor selectivity can be enhanced by the use of two receptors with complementary, conditional signals. The two antigens need to be chosen in such a manner that that they are both expressed by the tumor cells but found alone on normal cells [82]. Acting in cell autonomous fashion, the required co-engagement of the CCR and the CAR upon recognition of two independent antigens reinforces tumor selectivity in vivo[82].
Wendell Lim and colleagues recently adapted the combinatorial principle in a temporal sequence, exploiting the unique mechanism and modular structure of the Notch-receptor (synthetic Notch receptor–synNotch) [83,84]. Notch engagement of its natural ligands leads to proteolytic release of an intracellular portion that possesses transcriptional regulatory function. Thus, a synNotch receptor specific for antigen A can secondarily induce expression of a CAR specific for antigen B [85]. In addition to spatial control of cellular behavior, the synNotch receptor systems has the potential to integrate multiple extracellular inputs in parallel, reviewed in [86] The ability to integrate combinatorial environmental cues may be useful to study specific cell-cell interaction in the tumor microenvironment.
6. Cell substrate optimization
A major question arising from the success and rise of CAR therapy is T cell subsets are optimal for best clinical results. Strikingly, the consistent clinical outcomes obtained in ALL patients have been obtained with bulk peripheral blood T cells comprising variable CD4+/CD8+ T cell ratios and variable proportions of naive and antigen-experienced T cells [72–74]. The quasi uniformity of those successful outcomes thus did not reveal whether an optimal CD4+/CD8+ T cell ratio or a superior T cell subset dominant contributes to the therapeutic outcome. However, we know from murine studies that, although CD8+ or CD4+ CAR T cells alone can exert significant therapeutic effects [87,88], a mixture of both subsets displays superior efficacy [88,89]. Much remains to be learned about the potential defined T cell subsets, in particular the central memory (TCM) and the stem cell–like memory (TSCM) subsets [90–94]. It is likely that the selection of optimal T cell subsets for CAR therapy will impact the efficacy, consistency, and safety of CAR therapy [95,96].
Phase 3: Engineered T cell therapies (heterologous)
Current CAR therapies make use of autologous T lymphocytes, obliging to manufacture T cells in personalized fashion. In principle, the use of allogeneic cell sources could reduce such manufacturing needs. However, a reliable path to generating safe and effective T cells in a non-autologous setting still remains to be established (reviewed in [97]). The use of heterologous cells poses two major additional challenges: donor T cells may attack the recipient, causing GVHD (a well know obstacle in the transplantation field) and the recipient may reject the incoming T cells, limiting their persistence and therapeutic efficacy (another well known obstacle in the transplantation field). Nonetheless, some first clinical experiences are emerging, shedding some light on the nature and magnitude of the obstacles to overcome. These few studies were conducted in the context of substantially immunocompromised recipients.
To avoid allorecognition of the recipient and GVHD, one may either utilize VSTs with reduced alloreactive potential or delete the endogenous TCR. The clinical experience with these approaches is limited. CAR-expressing VSTs have not shown much anti-tumor activity in a trial utilizing EBV-specific T cells expressing a CD19 CAR [98,99]. A case report of the compassionate use of a CD19 CAR T cell edited at the TRCα locus has been difficult to interpret, but was useful in showing that a very small fraction of T cells escaping TCR deletion greatly expanded and induced GVHD [100]. Interestingly, however, CD19 CAR DLI in selected patients with relapsed lymphoma after an allogeneic transplant without GVHD, has shown encouraging tumor responses without GVHD [101], consistent with prior studies in murine models [102].
Yet another alternative approach is to generate T cells with optimal features from precursors that are amenable to substantive genetic engineering and thorough safety testing, such as pluripotent stem cells. The first successes in the in vitro generation of human T cells from embryonic stem cells and induced pluripotent stem cells have been recently reported [103–105]. Themeli et al demonstrated that CAR T cells derived from human iPS cells could induce significant tumor regressions in a xenogeneic lymphoma model [105], providing the first evidence in support of the feasibility of generating “synthetic T cells” for therapeutic purposes (reviewed in [97]).
Thus, while the challenges to developing off-the-shelf strategies remain substantial, it is intriguing to think about the possible advent of “Phase 3” in the historical progression of T cell-based immunotherapies.
Conclusions and outlook
This article summarizes what T cell therapies have achieved to date and lays out directions for their further evolution. The crux of the exposé is that immunological principles will find a new embodiment and purpose thanks to advances that are extraneous to the field of immunology: genetic engineering, synthetic biology and cell manufacturing sciences.
Immunologists have defined the principles governing immune responses, from antigen processing and presentation to antigen recognition, from T cell ontogeny and T cell priming to memory formation, and from T cell subset definition to regulatory network integration. On the operational front, immunotherapists have tested the limits of this knowledge in the context of human pathology. To me, the acid test of fundamental knowledge is the ability to alleviate patient suffering and enable curative medical interventions.
In this regards, natural adoptive cell therapies – Phase 1 – have provided numerous critical insights into the potential and the limitations of the natural immune system to defeat cancer: adoptively transferred allogeneic T cells can direct profound anti-tumor responses (e.g., the graft-versus-leukemia effect) and protect against viral infections (e.g., against herpes viruses), provided that enough T cells of the right specificity and fitness are administered to the patient. Unselected allogeneic T can be therapeutic but also mediate severe, potentially lethal, toxicities (e.g., GVHD induced by T cell replete marrow grafts or DLI). In a few settings, autologous T cells occasionally mediate tumor regressions upon adoptive transfer (e.g., using TILs against melanoma), provided that they persist and are not inactivated by the tumor microenvironment, which often requires aggressive conditioning.
Genetic engineering technologies enable to redesign T cells, and extend their capabilities and performance. Combined with the use of synthetic biology tools such as chimeric antigen receptors, chimeric costimulatory receptors, chimeric cytokine receptors, iCARs, synNotch and safety switches, T cells may eventually overcome the resistance that tumors successfully oppose to the action of natural T cells. The optimal T cell substrate for such manipulations will likely be defined soon and reinforce these approaches. This is the current state of the art – “Phase 2” – which has only just begun.
In the future, the combined use of ever-improving genetic tools, synthetic biology tools, stem cell biology and manufacturing sciences, may enable the use of T cells prepared from alternative, ie non-autologous, sources. This next phase would represent another groundbreaking leap for cell therapy. The biological challenges to reach this stage, however, should not be underestimated. For the present, the main priorities are to demonstrate the efficacy of CAR therapy beyond the CD19 paradigm, especially against solid tumors, and further advance T cell manufacturing, which will hopefully allay the logistical and economic questions some have about the potential to implement cell therapies on a very large scale.
Highlights.
The CD19 paradigm has established CARs as a novel immunotherapy in oncology immunotherapy
The synthetic tool box is not limited to CARs and includes CCRs, iCARs and synNotch
The use of alternative T cell sources poses substantial challenges but shows promise
Genetic engineering and synthetic biology are transforming adoptive T cell therapy
Second generation CARs have ushered in a new era of T cell-based cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature citations
- 1.Mitchison NA. Studies on the immunological response to foreign tumor transplants in the mouse. I. The role of lymph node cells in conferring immunity by adoptive transfer. J Exp Med. 1955;102:157–177. doi: 10.1084/jem.102.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winn HJ. Nat Cancer Inst. 1959:113–137. [Google Scholar]
- 3.Klein G, Sjogren HO, Klein E, Hellstrom KE. Demonstration of resistance against methylcholanthrene-induced sarcomas in the primary autochthonous host. Cancer Res. 1960;20:1561–1572. [PubMed] [Google Scholar]
- 4.Rosenberg SA, Terry WD. Passive immunotherapy of cancer in animals and man. Adv Cancer Res. 1977;25:323–388. doi: 10.1016/s0065-230x(08)60637-5. [DOI] [PubMed] [Google Scholar]
- 5.Miller JF, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell. 2015;27:439–449. doi: 10.1016/j.ccell.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Butturini A, Bortin MM, Gale RP. Graft-versus-leukemia following bone marrow transplantation. Bone Marrow Transplant. 1987;2:233–242. [PubMed] [Google Scholar]
- 7.Champlin R. Immunobiology of bone marrow transplantation as treatment for hematologic malignancies. Transplant Proc. 1991;23:2123–2127. [PubMed] [Google Scholar]
- 8.Korngold R, Sprent J. Graft-versus-host disease in experimental allogeneic bone marrow transplantation. Proc Soc Exp Biol Med. 1991;197:12–18. doi: 10.3181/00379727-197-43217a. [DOI] [PubMed] [Google Scholar]
- 9.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–1561. doi: 10.1016/S0140-6736(09)60237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenberg PD. Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells. Adv Immunol. 1991;49:281–355. doi: 10.1016/s0065-2776(08)60778-6. [DOI] [PubMed] [Google Scholar]
- 11.Old LJ. Tumor immunology: the first century. Curr Opin Immunol. 1992;4:603–607. doi: 10.1016/0952-7915(92)90034-c. [DOI] [PubMed] [Google Scholar]
- 12.Melief CJ. Tumor eradication by adoptive transfer of cytotoxic T lymphocytes. Adv Cancer Res. 1992;58:143–175. doi: 10.1016/s0065-230x(08)60294-8. [DOI] [PubMed] [Google Scholar]
- 13.Sadelain M. From adoptive immunity to CAR therapy: an evolutionary perspective. Encyl Immunol. 2016 in press. [Google Scholar]
- 14.Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med. 1982;155:1823–1841. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grimm EA, Robb RJ, Roth JA, Neckers LM, Lachman LB, Wilson DJ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. III. Evidence that IL-2 is sufficient for direct activation of peripheral blood lymphocytes into lymphokine-activated killer cells. J Exp Med. 1983;158:1356–1361. doi: 10.1084/jem.158.4.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hromas R, Cornetta K, Srour E, Blanke C, Broun ER. Donor leukocyte infusion as therapy of life-threatening adenoviral infections after T-cell-depleted bone marrow transplantation. Blood. 1994;84:1689–1690. [PubMed] [Google Scholar]
- 17.Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, Ljungman P, Ferrant A, Verdonck L, Niederwieser D, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995;86:2041–2050. [PubMed] [Google Scholar]
- 18.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 19.Wu R, Forget MA, Chacon J, Bernatchez C, Haymaker C, Chen JQ, Hwu P, Radvanyi LG. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J. 2012;18:160–175. doi: 10.1097/PPO.0b013e31824d4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heslop HE, Brenner MK, Rooney CM. Donor T cells to treat EBV-associated lymphoma. N Engl J Med. 1994;331:679–680. doi: 10.1056/NEJM199409083311017. [DOI] [PubMed] [Google Scholar]
- 21.Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, Castro-Malaspina H, Childs BH, Gillio AP, Small TN, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994;330:1185–1191. doi: 10.1056/NEJM199404283301703. [DOI] [PubMed] [Google Scholar]
- 22.Melenhorst JJ, Castillo P, Hanley PJ, Keller MD, Krance RA, Margolin J, Leen AM, Heslop HE, Barrett AJ, Rooney CM, et al. Graft versus leukemia response without graft-versus-host disease elicited by adoptively transferred multivirus-specific T-cells. Mol Ther. 2015;23:179–183. doi: 10.1038/mt.2014.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadelain M, Mulligan RC. Efficient-retroviral-mediated gene transfer into murine primary lymphocytes [abstract no. 34] In: International Congress of Immunology, Magyar Immunológiai Társaság, International Union of Immunological Societies (IUIS), editor. Abstracts : 8th International Congress of Immunology, Budapest, Hungary, August 23–28, 1992. Budapest; New York: Springer-Verlag; 1992. [Google Scholar]
- 24.Mavilio F, Ferrari G, Rossini S, Nobili N, Bonini C, Casorati G, Traversari C, Bordignon C. Peripheral blood lymphocytes as target cells of retroviral vector-mediated gene transfer. Blood. 1994;83:1988–1997. [PubMed] [Google Scholar]
- 25.Bunnell BA, Muul LM, Donahue RE, Blaese RM, Morgan RA. High-efficiency retroviral-mediated gene transfer into human and nonhuman primate peripheral blood lymphocytes. Proc Natl Acad Sci U S A. 1995;92:7739–7743. doi: 10.1073/pnas.92.17.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallardo HF, Tan C, Ory D, Sadelain M. Recombinant retroviruses pseudotyped with the vesicular stomatitis virus G glycoprotein mediate both stable gene transfer and pseudotransduction in human peripheral blood lymphocytes. Blood. 1997;90:952–957. [PubMed] [Google Scholar]
- 27.Riviere I, Brose K, Mulligan RC. Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc Natl Acad Sci U S A. 1995;92:6733–6737. doi: 10.1073/pnas.92.15.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, Yeh R, Capacio V, Olszewska M, Hosey J, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. Journal of Immunotherapy. 2009;32:169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. Journal of Clinical Investigation. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Riviere I. Clinical Manufacturing of CAR T Cells: Foundation of a Promising Therapy. Molecular Therapy Oncolytics. 2016 doi: 10.1038/mto.2016.15. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, Bas C, Lemaire L, Galetto R, et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 33.Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna JA, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menger L, Gouble A, Marzolini MA, Pachnio A, Bergerhoff K, Henry JY, Smith J, Pule M, Moss P, Riddell SR, et al. TALEN-mediated genetic inactivation of the glucocorticoid receptor in cytomegalovirus-specific T cells. Blood. 2015;126:2781–2789. doi: 10.1182/blood-2015-08-664755. [DOI] [PubMed] [Google Scholar]
- 35.Eyquem J, Mansilla-Soto J, Odak A, Sadelain M. One-step generation of universal CAR T cells. American Society of Gene & Cell Therapy 19th Annual Meeting; Washington, D.C., USA. May 4–7, 2016. [Google Scholar]
- 36.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: Engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–437. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 37.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 38.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brocker T, Peter A, Traunecker A, Karjalainen K. New simplified molecular design for functional T cell receptor. European Journal of Immunology. 1993;23:1435–1439. doi: 10.1002/eji.1830230705. [DOI] [PubMed] [Google Scholar]
- 40.Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Current Opinion in Immunology. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong MC, Latouche JB, Krause A, Heston WDW, Bander NH, Sadelain M. Cancer Patient T Cells Genetically Targeted to Prostate-Specific Membrane Antigen Specifically Lyse Prostate Cancer Cells and Release Cytokines in Response to Prostate-Specific Membrane Antigen. Neoplasia. 1999;1:123–127. doi: 10.1038/sj.neo.7900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brocker T. Chimeric Fv-ζ or Fv-ε receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood. 2000;96:1999–2001. [PubMed] [Google Scholar]
- 43.Sadelain M. CAR therapy: the CD19 paradigm. J Clin Invest. 2015;125:3392–3400. doi: 10.1172/JCI80010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nature Biotechnology. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 45.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 46.Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Current Opinion in Immunology. 2015;33:9–15. doi: 10.1016/j.coi.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Der Stegen SJC, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nature Reviews Drug Discovery. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. doi: 10.1038/nbt.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016;6:133–146. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kochenderfer JN. Genetic engineering of T cells in leukemia and lymphoma. Clin Adv Hematol Oncol. 2014;12:190–192. [PubMed] [Google Scholar]
- 52.Ramos CA, Savoldo B, Dotti G. CD19-CAR trials. Cancer J. 2014;20:112–118. doi: 10.1097/PPO.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, Till B, Raubitschek A, Forman SJ, Qian X, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–725. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 54.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 57.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pegram HJ, Park JH, Brentjens RJ. CD28z CARs and armored CARs. Cancer J. 2014;20:127–133. doi: 10.1097/PPO.0000000000000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115:3508–3519. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, Rosenberg SA. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18:1672–1683. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, Spicer JF, Davies DM, Maher J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285:25538–25544. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, Dotti G. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res. 2014;20:131–139. doi: 10.1158/1078-0432.CCR-13-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krause A, Guo HF, Latouche JB, Tan C, Cheung NKV, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. Journal of Experimental Medicine. 1998;188:619–626. doi: 10.1084/jem.188.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol. 2012;51:263–272. doi: 10.1016/j.molimm.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 69.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, Sadelain M. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 70.Riese MJ, Wang LC, Moon EK, Joshi RP, Ranganathan A, June CH, Koretzky GA, Albelda SM. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013;73:3566–3577. doi: 10.1158/0008-5472.CAN-12-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, O’Brien S, Lo A, Liu X, Zhao Y, Albelda SM. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin Cancer Res. 2016;22:436–447. doi: 10.1158/1078-0432.CCR-15-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RPT, Carpenter RO, Maryalice SS, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. Journal of Clinical Oncology. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303r–a139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sadelain M, Brentjens R, Riviere I, Park J. CD19 CAR Therapy for Acute Lymphoblastic Leukemia. Am Soc Clin Oncol Educ Book. 2015;35:e360–363. doi: 10.14694/EdBook_AM.2015.35.e360. [DOI] [PubMed] [Google Scholar]
- 75.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015;137:2832–2835. doi: 10.1021/jacs.5b00106. [DOI] [PubMed] [Google Scholar]
- 77.Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, Campana D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014;74:93–103. doi: 10.1158/0008-5472.CAN-13-1365. [DOI] [PubMed] [Google Scholar]
- 78.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350:aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun J, Sadelain M. The quest for spatio-temporal control of CAR T cells. Cell Res. 2015;25:1281–1282. doi: 10.1038/cr.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 81.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra–172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gordon WR, Zimmerman B, He L, Miles LJ, Huang J, Tiyanont K, McArthur DG, Aster JC, Perrimon N, Loparo JJ, et al. Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch. Dev Cell. 2015;33:729–736. doi: 10.1016/j.devcel.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, Lim WA. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell. 2016;164:780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Themeli M, Sadelain M. Combinatorial Antigen Targeting: Ideal T-Cell Sensing and Anti-Tumor Response. Trends Mol Med. 2016 doi: 10.1016/j.molmed.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, King PD, Larson S, Weiss M, Rivière I, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nature Medicine. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 88.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261r–a151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moeller M, Kershaw MH, Cameron R, Westwood JA, Trapani JA, Smyth MJ, Darcy PK. Sustained antigen-specific antitumor recall response mediated by gene-modified CD4+ T helper-1 and CD8+ T cells. Cancer Res. 2007;67:11428–11437. doi: 10.1158/0008-5472.CAN-07-1141. [DOI] [PubMed] [Google Scholar]
- 90.Oliveira G, Ruggiero E, Stanghellini MT, Cieri N, D’Agostino M, Fronza R, Lulay C, Dionisio F, Mastaglio S, Greco R, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med. 2015;7:317r–a198. doi: 10.1126/scitranslmed.aac8265. [DOI] [PubMed] [Google Scholar]
- 91.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. 2012;35:651–660. doi: 10.1097/CJI.0b013e31827806e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, Drexler I, Hofer T, Riddell SR, Busch DH. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity. 2014;41:116–126. doi: 10.1016/j.immuni.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 94.Busch DH, Frassle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. 2016 doi: 10.1016/j.smim.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, Salter A, Hudecek M, Maloney DG, Turtle CJ. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014;20:141–144. doi: 10.1097/PPO.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR. Chimeric antigen receptor-modified T cells derived from defined CD8(+) and CD4(+) subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Themeli M, Riviere I, Sadelain M. New Cell Sources for T Cell Engineering and Adoptive Immunotherapy. Cell Stem Cell. 2015;16:357–366. doi: 10.1016/j.stem.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–2973. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nature Medicine. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qasim W, Persis J, Samarasinghe S, Ghorashian S, Zhan H, Stafford S, Butler K, Ahsan G, Gilmour K, Pule M, et al. First Clinical Application of Talen Engineered Universal CAR19 T Cells in B-ALL [Abstract/Program: Oral and Poster Abstracts]. Presented at the 2015 American Society of Hematology Conference; Orlando, Florida USA. December 5, 2015; Available from: https://ash.confex.com/ash/2015/webprogram/Paper81653.html. 2015. [Google Scholar]
- 101.Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, Gea-Banacloche JC, Pavletic SZ, Hickstein DD, Lu TL, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol. 2016 doi: 10.1200/JCO.2015.64.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arnab G, Davila ML, Young LF, Kloss C, Gunset G, Smith OM, West ML, Singer NV, Holland AM, Jeng RR, et al. CD19-Targeted Donor T Cells Exert Potent Graft Versus Lymphoma Activity and Attenuated Gvhd [Abstract/Program: Oral and Poster Abstracts]. Presented at the 54th American Society of Hematology Annual Meeting and Exposition; Atlanta, Georgia USA. December 10, 2012; Available from: https://ash.confex.com/ash/2012/webprogram/Paper51817.html 2012. [Google Scholar]
- 103.Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, Nakayama-Hosoya K, Iriguchi S, Uemura Y, Shimizu T, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 104.Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S, Koseki H, Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013;12:31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 105•.Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, Sadelain M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31:928–933. doi: 10.1038/nbt.2678. CAR T cells derived from human iPS cells induce tumor regression after adoptive transfer to tumor-bearing mice. [DOI] [PMC free article] [PubMed] [Google Scholar]