

Abstract
Association of fibrosis with autoimmune pulmonary alveolar proteinosis (aPAP) is rare. However, prognoses of such cases are poor and the process of the formation of fibrosis is still unknown. In this study, we report a case of aPAP with progressive fibrosis occurring in a 46‐year‐old woman. She had undergone several repetitions of whole lung lavage (WLL) for 7 years and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) inhalation for 3 months; however, the progression of fibrosis was not hindered. Eventually, she was treated with bilateral lung transplantation. The computed tomography (CT) image suggested pulmonary fibrotic changes in her lung similar to usual interstitial pneumonia. However, the pathological analyses of explant lungs revealed that the fibrosis was not similar to ordinary interstitial pneumonias and suggested that the dysfunction of alveolar macrophage in removing the excess surfactant of alveolar spaces played an important role in the fibrogenesis in aPAP.
Keywords: Autoimmune pulmonary alveolar proteinosis, lung transplantation, pathology, pulmonary fibrosis
Introduction
The cause of autoimmune pulmonary alveolar proteinosis (aPAP) is the excessive production of a neutralizing autoantibody against granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) that impairs GM‐CSF‐dependent surfactant clearance mediated by alveolar macrophages (AMs) 1. In such situations, accumulation of intracellular debris in AMs and excess surfactants in alveolar spaces are observed. The prognoses of aPAP are generally good because whole lung lavage (WLL) and GM‐CSF inhalation therapy can achieve sufficient remission of the disease in the majority of patients 1. The frequency of fibrosis in courses of aPAP is rare (<2%); however, the prognoses of these cases are very poor. The mechanism of fibrogenesis in aPAP is still unknown and the treatment is not established 1. We report herein a case of aPAP associated with progressive fibrosis, the pathological features of explant lung, and our consideration about the process of fibrosis formation.
Case Report
A 46‐year‐old woman who had sinus sick syndrome visited another hospital in April 2006 for progressive cough and dyspnoea on effort. She received pacemaker implantation and was diagnosed simultaneously as having aPAP based on the crazy‐paving appearance on high‐resolution computed tomography (HRCT), "milky" appearance and the existence of foamy macrophage confirmed with Papanicolaou staining of broncho‐alveolar lavage fluid (BALF), and elevated serum levels of the anti‐GM‐CSF immunoglobulin G (IgG) antibody. Examination findings and laboratory data excluded other complications including haematological and connective tissue diseases that suggested secondary PAP (data not shown). She was treated with five repetitions of WLL between 2006 and 2013; however, aPAP gradually worsened. Furthermore, lung fibrosis appeared and progressed in the disease course (Fig. 1A). Then the patient visited our hospital on October 2013 and she received GM‐CSF inhalation for 3 months; however, the therapy was ineffective and hypoxia gradually progressed. Despite the considerable reduction of consolidation in lungs observed in chest CT, further WLL did not improve hypoxia. Although she received additional GM‐CSF inhalation therapy for 6 months, her fibrotic change progressed and bilateral pneumothorax occurred. Finally, she underwent bilateral lung transplantation on January 2015 (Fig. 1B). Pathological analyses of explant lungs were performed (Fig. 2). Macroscopic observation demonstrated the dilatation of bronchus and parenchymal fibrosis in all fields. In the whitish‐yellow coloured area, which is considered to be surfactant‐rich, the normal frameworks were relatively preserved (Fig. 2A). Histological observation revealed lymphocyte infiltration into the alveolar walls and eosinophilic surfactant material and macrophages in the alveolar lumina (Fig. 2B). A diversity of histological features of the explanted lung demonstrated normal architecture conserved in abundant surfactant materials (Fig. 2C) and fibrotic change in the areas with reduced surfactant materials (Fig. 2D). Membranous bronchioles with sub‐epithelial fibrosis adjacent clusters of cysts with collagen deposition and no epithelial lining were observed (Fig. 2E). These pathological findings were not similar to typical findings of idiopathic interstitial pneumonia and collagen vascular diseases. The cysts were not similar with those seen in usual interstitial pneumonia.
Figure 1.
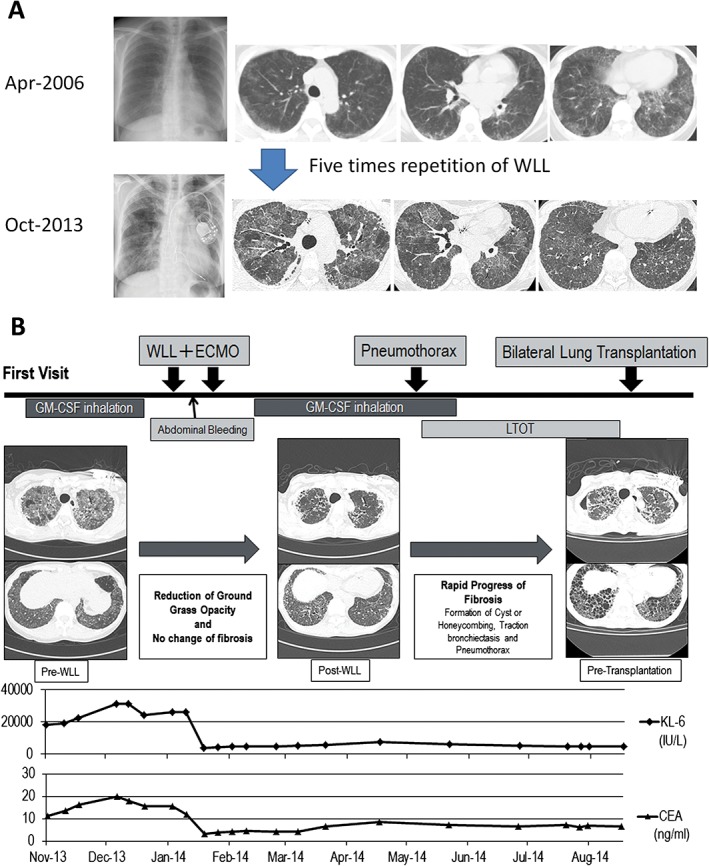
Clinical course of the patient. (A) Evolution of image findings from the first diagnosis until first visit at our hospital. (B) Clinical course of the patient from first granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) inhalation therapy until lung transplantation.
Figure 2.
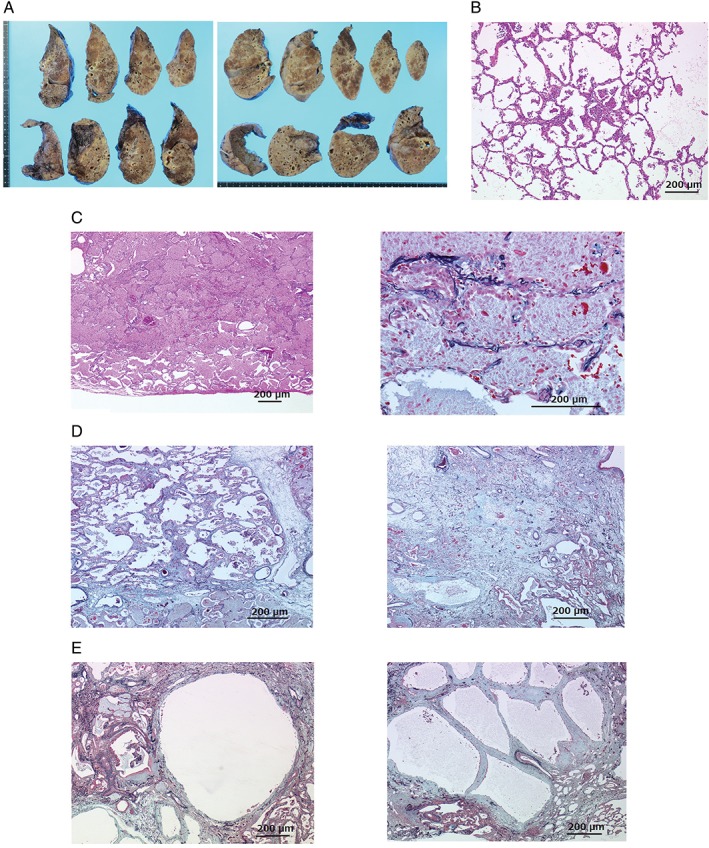
Pathological analyses of resected lungs of the patient. (A) Macroscopy of whole lungs demonstrated volume reduction and solid appearance in all lobes and cysts predominated in lower regions. (B) Initial pathological change within normal lung structure. (C) The normal lung structure was preserved in the areas with abundant eosinophilic granular surfactant proteins in alveolar spaces (right). The hyperplasia of the pneumocytes (type II alveolar epithelial cells) in these parts (left). (D) Fibrotic changes of intra‐alveolar, alveolar septum, and dilation of alveolar duct (right). Giant cells (macrophage originate) within alveolar fibrosis (left). (E) Clusters of cysts with collagen deposition and no epithelial lining were observed in the pulmonary lobule (right). A membranous bronchiole with sub‐epithelial fibrosis connected to the cystic lesion (left).
Discussion
Few cases report of aPAP associated with fibrosis; however, this report is the first one about whole lung pathological analysis. Surfactant homeostasis dysfunction is one of the causes of fibrosis initiation in some familial interstitial pneumonia 2. Our results suggest that the disturbance of surfactant homeostasis in alveolar spaces is associated with hyperplasia of type II pneumocyte cells induced by the lymphocyte alveolitis and that the fibrogenic processes are different from that of other interstitial pneumonias. Pathological findings that abundant surfactant material in the alveolar lumina maintained normal architecture, whereas interstitial fibrosis progressed in the nearby area of alveolar lumina with reduced surfactant material are significant. Surfactant materials may provide protection from injuries and fibrogenesis in some aPAP patients. Charbeneau reported that the deficiency of GM‐CSF resulted in the decrease of the anti‐fibrotic prostaglandin E2 (PGE2) derived from AMs which contributes to minimizing lung injury after insult 3. The repetitive procedure of WLL may induce mechanical stresses, representing a potent pro‐inflammatory stimulus. The repetitive procedure of WLL may remove anti‐fibrotic proteins and reduce protective effect from direct injury to alveolar cells. Kinehara reported that the accumulation of protein in alveolar spaces disappeared in the process of fibrogenesis of aPAP 4. The indication of WLL for patients with severe aPAP should be discussed in future cases. Macrophage transplantation should be considered as one of the treatment options in future cases 5.
Disclosure Statements
No conflict of interest declared.
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
Acknowledgements
We appreciate Kageaki Taima, Yoichiro Mitsuishi, Teruyuki Satou, Naoki Tode, Yasushi Hoshikawa, Hajime Kurosawa, and other members who joined the medical team for the treatment. This work was supported by a grant from the Ministry of Health Labour and Welfare, Japan (H26‐Itaku (Nan)‐Ippan‐077, 17ek0109079s0303) and by a grant from the Japan Society for the Promotion of Science (JSPS C‐15K0925).
Ono, M. , Saito, R. , Tominaga, J. , Okada, Y. , Ohkouchi, S. and Takemura, T. (2017) Pathological features of explant lungs with fibrosis in autoimmune pulmonary alveolar proteinosis. Respirology Case Reports, 5 (5), e00255. doi: 10.1002/rcr2.255.
Associate Editor: Trevor Williams
References
- 1. Inoue Y, Trapnell BC, Tazawa R, et al. 2008. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am. J. Respir. Crit. Care Med. 177:752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Whitsett JA, Wert SE, and Weaver TE. 2010. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 61:105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Charbeneau RP, Christensen PJ, Chrisman CJ, et al. 2003. Impaired synthesis of prostaglandin E2 by lung fibroblasts and alveolar epithelial cells from GM‐CSF−/− mice: implications for fibroproliferation. Am. J. Physiol. Lung Cell. Mol. Physiol. 284:L1103–L1111. [DOI] [PubMed] [Google Scholar]
- 4. Kinehara Y, Kida H, Inoue Y, et al. 2014. Development of microscopic polyangiitis‐related pulmonary fibrosis in a patient with autoimmune pulmonary alveolar proteinosis. BMC Pulm. Med. 14:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suzuki T, Arumugam P, Sakagami T, et al. 2014. Pulmonary macrophage transplantation therapy. Nature 514:450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]