

Abstract
Cyclic peptides are capable of binding and modulating challenging drug targets including protein-protein interactions. However, their lack of membrane permeability prevents their applications against intracellular targets. In this study, we show that it is possible to design a cell-permeable and biologically active cycloheptapeptide inhibitor against intracellular enzyme peptidyl-prolyl isomerase Pin1 by integrating cell-penetrating and target-binding sequences.
Graphical Abstract
Integration of Pin1-binding and cell-penetrating sequences results in a cell-permeable, biologically active cycloheptapeptide inhibitor against Pin1.
It is estimated that ~80% of therapeutically relevant drug targets are challenging for conventional drug modalities, namely small molecules and biologics.1 The most prominent examples of undruggable targets are proteins involved in intracellular protein-protein interactions (PPIs), which often possess large, flat binding sites. Small molecules generally do not bind to the flat binding sites with adequate affinity and/or specificity, while biologics (e.g., monoclonal antibodies) cannot cross the cell membrane to access these intracellular targets. Despite having well defined active-site pockets, some enzymes may also be challenging drug targets for small molecules. For example, the >100 human protein tyrosine phosphatases (PTPs) remain largely undruggable, because they all share a highly conserved active site structure and it has been difficult to develop inhibitors specific for a given PTP.2 In addition, the PTP active site is highly positively charged and requires negatively charged species for high-affinity binding, which are generally impermeable to the cell membrane. Another example is the peptidyl-prolyl cis-trans isomerase Pin1, which regulates the levels and functions of phosphoproteins by catalyzing phosphorylation-dependent cis/trans isomerization of peptidyl-prolyl bonds.3 Because of its critical roles in cell-cycle regulation and increased expression levels and activity in human cancers, Pin1 has been proposed as a potential target for the development of anticancer drugs.3 Thus far, however, the reported small-molecule Pin1 inhibitors either lack potency and specificity, or is impermeable to the cell membrane due to the presence of negatively charged phosphoamino acid mimetics.3,4
Cyclic peptides provide a potential solution for targeting these difficult proteins.5 Because of their larger sizes than conventional small molecules, cyclic peptides are capable of recognizing flat protein surfaces such as PPI interfaces with antibody-like affinity and specificity.6 By engaging in additional interactions with the less conserved surfaces outside the active site, cyclic peptides have the ability to differentiate structurally similar enzyme isoforms.7,8 A major limitation of cyclic peptides, however, is their general lack of membrane permeability. Researchers are currently exploring two different approaches to improving the membrane permeability of cyclic peptides. The first approach involves N-methylation of the peptide backbone and formation of intramolecular hydrogen bonds.9 An alternative and potentially more general approach is to conjugate the cyclic peptides to a cell-penetrating peptide (CPP).10 We have previously demonstrated that cyclic peptides of diverse sequences can be rendered cell-permeable by fusing them with a cyclic CPP such as cyclo(FΦRRRRQ) (cFΦR4, where Φ is L-2-naphthylalanine).8,11,12 Short peptidyl cargos have also been directly inserted into the cyclic CPP ring for cellular uptake (endocyclic delivery).13,14 The resulting mono- and bicyclic peptides were relatively large molecules, usually consisting of 10–20 amino acids. In this study, we intended to explore the possibility of designing cyclic peptides of smaller sizes as cell-permeable inhibitors against intracellular proteins. Smaller cyclic peptides have considerably greater proteolytic stability, due to greater conformational rigidity, and are less costly to produce than larger ones.
Through screening a combinatorial library, we previously discovered cyclo(D-Ala-Sar-D-pThr-Pip-Nal-Tyr-Gln) (where Sar is sarcosine, D-pThr is D-phosphothreonine, Pip is L-piperidine-2-carboxylic acid, and Nal is L-2-naphthylalanine) as a potent, selective Pin1 inhibitor (Fig. 1 and Table 1, peptide 1; IC50 = 31 nM).15 SAR studies revealed that only the D-pThr-Pip-Nal motif was required for Pin1 binding. Replacement of D-Ala, Sar, and Tyr with D-Arg and Arg residues resulted in a cycloheptapeptide that retained much of the Pin1 inhibitory activity (IC50 = 220 nM) but showed modest cellular entry into HeLa cells (Fig. 1 and Table 1, peptide 2).15 We selected peptide 2 as the starting point for optimization of its cellular uptake activities through medicinal chemistry efforts. We first replaced the Gln residue at the D-pThr+4 position (X2) with an arginine and the Arg residue at the D-pThr+3 position (X1) with phenylalanine, 4-fluorophenylalanine (Fpa), tryptophan, or Nal (Table 1, peptides 3–6). The resulting cyclic peptides resemble cFΦR4 in structure, each containing two hydrophobic aromatic residues followed by three arginine residues, and were expected to have improved cellular entry.13,16 Unfortunately, although the incorporation of a second aromatic hydrophobic residue at the D-pThr+3 position (X1) improved the cellular uptake of peptides 3–6 (data not shown), these modifications also reduced their Pin1-binding affinity by 4- to 16-fold, with IC50 values of 0.96 – 3.4 μM (Table 1). As a result, peptides 3–6 showed no significant cellular activity on HeLa or other cells.
Fig. 1.

Structures of previously reported cyclic peptidyl inhibitors against Pin1 (peptides 1 and 2).
Table 1.
Structures and Pin1 binding affinities of cyclic peptidesa
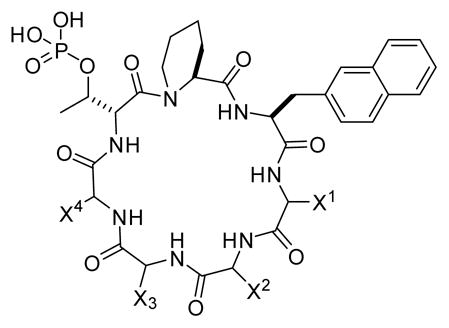
| |||||
|---|---|---|---|---|---|
|
| |||||
| Peptide | Sequence | IC50 (nM) | |||
|
| |||||
| X1 | X2 | X3 | X4 | ||
| 1 | Tyr | Gln | D-Ala | Sar | 31 ± 3b |
| 2 | Arg | Gln | D-Arg | D-Arg | 220 ± 40b |
| 3 | Phe | Arg | D-Arg | D-Arg | 1770 ± 280 |
| 4 | Fpa | Arg | D-Arg | D-Arg | 960 ± 210 |
| 5 | Trp | Arg | D-Arg | D-Arg | 1540 ± 270 |
| 6 | Nal | Arg | D-Arg | D-Arg | 3400 ± 810 |
| 7 | Nal | Arg | D-Arg | Arg | 2000 ± 590 |
| 8 | D-Nal | Arg | D-Arg | Arg | >10,000 |
| 9 | Fpa | Trp | D-Arg | Arg | 1810 ± 560 |
| 10 | Arg | D-Arg | Arg | Nal | 1550 ± 330 |
| 11 | Arg | Arg | D-Arg | D-Arg | 245 ± 86 |
| 12 | Arg | D-Arg | Arg | Arg | 5490 ± 160 |
| 13 | Arg | D-Arg | Arg | D-Arg | >10,000 |
| 14 | Arg | D-Arg | D-Arg | Arg | 680 ± 72 |
Data reported are mean ± SD of three independent experiments.
Data from ref. 15.
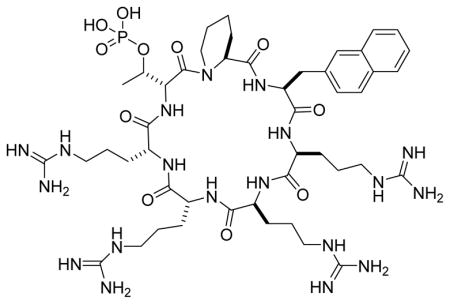
Our previous study showed that cyclic CPPs of different stereochemical configuration had very different cellular entry capabilities.16 We further modified the structure of peptide 6 by varying the stereochemical configuration of the D-pThr-1 (X4) and/or D-pThr+3 residues (X3), producing isomers 7 and 8 (Table 1). We also replaced the arginine at the X2 position of peptide 3 with a tryptophan (Table 1, peptide 9) or switched the side chains of X1 and X4 residues (peptide 10). However, peptides 7–10 failed to show significant improvement in Pin1 binding affinity or anti-proliferative activity against HeLa cells. Next, we reverted the Nal residue at the D-pThr+3 position of peptide 6 back to arginine to produce peptide 11 (Fig. 2A), because our previous library screening results indicated that among all of the residues tested at this position, arginine was the most preferred amino acid for Pin1 binding.15 Finally, we varied the stereochemistry of the four arginine residues of peptide 11 to generate stereoisomers 12–14 (Table 1). Interestingly, the four stereoisomers have Pin1 binding affinities that differ by >50-fold, with peptide 11 being most potent (IC50 = 245 nM) (Fig. 2B). We posit that the configuration of these arginine residues most likely perturbs the conformation of the D-pThr-Pip-Nal moiety and affects Pin1 binding indirectly, although we cannot rule out the possibility of their engaging in direct electrostatic interactions with the Pin1 protein surface.
Fig. 2.

Biochemical and cellular activities of peptide 11. (A) Structure of peptide 11. (B) Competition for binding to Pin1 (100 nM) between FITC-labeled peptide 1 (50 nM) and increasing concentrations of unlabelled peptide 11 (0–25 μM) as monitored by fluorescence anisotropy. (C) Effect of peptide 11 on the viability of HeLa (■), COS-7 (○), HEK293 (●), and HS68 cells (□) as determined by the MTT assay. (D) Anti-PML western blot analysis of cell lysates derived from HeLa cells after treatment with increasing concentrations of peptide 11 (or peptide 2). β-actin was used as loading control.
Peptide 11 was further evaluated for biological activity. Treatment of HeLa cells (which is a human cervical cancer cell line) for 72 h with peptide 11 resulted in dose-dependent reduction of cancer cell viability as monitored by the MTT assay, apparently due to inhibition of the proliferation of the cancer cells (Fig. 2C). Interestingly, peptide 11 did not reduce the viability of three non-cancerous cell lines tested, including COS-7 (monkey kidney fibroblast), HEK293 (human kidney epithelial), and HS68 (human skin fibroblast) cells. These results are consistent with the previous reports that the Pin1 activity is dispensable for normal cells (since Pin1−/− mice developed normally and had few defects at young ages17) but required for cancer cell proliferation.18 It has been hypothesized that cancer cells may have developed “addiction” to the high Pin1 activities. Liao et al. recently showed that a small-molecule Pin1 inhibitor, all-trans retinoic acid, selectively inhibited the growth of hepatocellular carcinoma cells in vitro and in a mouse xenograft model.18 To determine whether peptide 11 is capable of inhibiting the intracellular Pin1 activity, HeLa cells were treated with increasing concentrations of peptide 11 (0–10 μM) for 24 h and the intracellular level of promyelocytic leukaemia (PML), a well-established Pin1 substrate which undergoes Pin1-depedent proteasomal degradation,3,19 was monitored by western blot using a specific anti-PML antibody. Peptide 11 increased the PML protein levels in a dose-dependent manner, reaching a maximal PML level at 5–10 μM peptide 11 (Fig. 2D). As a comparison, treatment with peptide 2 did not significantly affect the PML level under the same conditions. These data suggest that peptide 11 entered the cytosol of HeLa cells and inhibited the intracellular Pin1 activity. Since peptides 2 and 11 have similar binding affinities to Pin1 (Table 1), the improved cellular activity of peptide 11 (relative to peptide 2) is likely the result of improved cellular uptake. To test this notion, we replaced the D-pThr residue of peptides 2 and 11 with a D-glutamic acid, added a dipeptide, Asp-Lys, to the D-Glu side chain, and labeled the lysine side chain with fluorescein isothiocyanate (FITC) to give peptides 15 and 16, respectively (see Fig. S1 in Supplementary Information for detailed structures). The Asp residue was intended to mimic D-pThr by providing a negative charge. Flow cytometry analysis of HeLa cells treated with the labeled peptides (5 μM) for 2 h at 37 °C showed that peptide 16 entered HeLa cells 2.5-fold more efficiently than peptide 15 (Fig. S2). Treatment of HeLa cells with peptide 16 (5 μM) for 2 h at 4 °C resulted in 3-fold lower uptake (or 67% reduction) relative to 37 °C, suggesting that cellular entry of these peptides was mediated by endocytosis followed by endosomal escape.16 Note that peptide 11 (and 16) differs from peptide 2 (and 15) by having an extra arginine (in place of Gln) at the X2 position (Table 1). This additional Arg residue increased the cellular uptake efficiency, apparently without adversely affecting the binding conformation of the D-pThr-Pip-Nal motif. The higher Pin1-binding affinity of peptide 11 relative to peptides 3–6 indicate that the arginine residue at the X1 position contributes significantly to Pin1 binding. This arginine, as well as the Nal residue immediately N-terminal to it, likely play the dual function of cellular entry and Pin1 binding.
Conclusions
In this work, we demonstrated that it is possible to design cell-permeable and biologically active cyclic peptides as small as a cycloheptapeptide by judicious integration of target-binding and cell-penetrating sequences. Although the mechanism by which these peptides enter cells remains to be defined, their highly hydrophilic nature renders them unlikely to cross the plasma membrane by passive diffusion; instead, their structural similarity to the reported cyclic CPPs and the reduced uptake at 4 °C suggest that they enter cells by endocytosis and subsequently exit from the endosome into the cytosol by a vesicle budding and collapsing mechanism.16 To the best of our knowledge, these are the smallest biologically active peptides that have been designed to enter cells by non-receptor-mediated endocytic mechanisms. This strategy should be applicable to developing small cyclic peptidyl ligands against other intracellular targets, providing an alternative to the more widely explored N-methylation/intramolecular hydrogen bond (passive diffusion) method.9
Supplementary Material
Acknowledgments
We thank Dr. Z. Qian for assistance with the cytometry analysis. This work was supported by the National Institutes of Health (GM110208). W.B. was financially supported by a predoctoral scholarship from the Egyptian government.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and supporting data and figures. See DOI: 10.1039/x0xx00000x
References
- 1.Verdine GL, Walensky LD. Clin Cancer Res. 2007;13:7264. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 2.Zhang ZY. Acc Chem Res. 2017;50:122. doi: 10.1021/acs.accounts.6b00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou XZ, Lu KP. Nat Rev Cancer. 2016;16:463. doi: 10.1038/nrc.2016.49. [DOI] [PubMed] [Google Scholar]
- 4.Moore JD, Potter A. Bioorg Med Chem Lett. 2013;23:4283. doi: 10.1016/j.bmcl.2013.05.088. [DOI] [PubMed] [Google Scholar]
- 5.For recent reviews, see: Cardote TAF, Ciulli A. ChemMedChem. 2016;11:787. doi: 10.1002/cmdc.201500450.Nevola L, Giralt E. Chem Commun. 2015;51:3302. doi: 10.1039/c4cc08565e.Dougherty PG, Qian Z, Pei D. Biochem J. 2017;474:1109. doi: 10.1042/BCJ20160619.
- 6.(a) Yamagishi Y, Shoji I, Miyagawa S, Kawakami T, Katoh T, Goto Y, et al. Chem Biol. 2011;18:1562. doi: 10.1016/j.chembiol.2011.09.013. [DOI] [PubMed] [Google Scholar]; (b) Shi ZD, Wei CQ, Lee K, Liu H, Zhang M, Araki T, Roberts LR, Worthy KM, Fisher RJ, Neel BG, Kelley JA, Yang D, Burke TR. J Med Chem. 2004;47:2166. doi: 10.1021/jm030510e. [DOI] [PubMed] [Google Scholar]; (c) Zhou H, Liu L, Huang J, Bernard D, Karatas H, Navarro A, Lei M, Wang S. J Med Chem. 2013;56:1113. doi: 10.1021/jm3015298. [DOI] [PubMed] [Google Scholar]; (d) Duncan, et al. J Med Chem. 2011;54:3854. doi: 10.1021/jm200156c. [DOI] [PubMed] [Google Scholar]
- 7.(a) Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Proc Natl Acad Sci U S A. 2001;98:87. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Angelini A, Cendron L, Chen S, Touati J, Winter G, Zanotti G, Heinis C. ACS Chem Biol. 2012;7:817. doi: 10.1021/cb200478t. [DOI] [PubMed] [Google Scholar]
- 8.Lian W, Jiang B, Qian Z, Pei D. J Am Chem Soc. 2014;136:9830. doi: 10.1021/ja503710n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Bockus AT, Schwochert JA, Pye CR, Townsend CE, Sok V, Bednarek MA, et al. J Med Chem. 2015;58:7409. doi: 10.1021/acs.jmedchem.5b00919. [DOI] [PubMed] [Google Scholar]; (b) Matsson P, Doak BC, Over B, Kihlberg J. Adv Drug Deliv Rev. 2016;101:42. doi: 10.1016/j.addr.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 10.(a) Zhang Y, Zhou S, Wavreille AS, DeWille J, Pei D. J Comb Chem. 2008;10:247. doi: 10.1021/cc700185g. [DOI] [PubMed] [Google Scholar]; (b) Desimmie BA, Humbert M, Lescrinier E, Hendrix J, Vets S, Gijsbers R, et al. Mol Ther. 2012;20:2064. doi: 10.1038/mt.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian Z, LaRochelle JR, Jiang B, Lian W, Hard RL, Selner N, Luechapanickhul R, Barrios AM, Pei D. Biochemistry. 2014;53:4034. doi: 10.1021/bi5004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trinh TB, Upadhyaya P, Qian Z, Pei D. ACS Comb Sci. 2016;18:75. doi: 10.1021/acscombsci.5b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qian Z, Liu T, Liu Y-Y, Briesewitz R, Barrios AM, Jhiang SM, Pei D. ACS Chem Biol. 2013;8:423. doi: 10.1021/cb3005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Upadhyaya P, Qian Z, Selner NG, Clippinger SR, Wu Z, Briesewitz R, Pei D. Angew Chem Int Ed. 2015;54:7602. doi: 10.1002/anie.201502763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu T, Liu Y, Kao H-Y, Pei D. J Med Chem. 2010;53:2494. doi: 10.1021/jm901778v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, Pei D. Biochemistry. 2016;55:2601. doi: 10.1021/acs.biochem.6b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimori F, Takahashi K, Uchida C, Uchida T. Biochem Biophys Res Commun. 1999;265:658. doi: 10.1006/bbrc.1999.1736. [DOI] [PubMed] [Google Scholar]
- 18.Liao X-H, Zhang AL, Zheng M, Li M-Q, Chen CP, Xu H, Chu Q-S, Yang D, Lu W, Tsai T-F, Liu H, Zhou XZ, Lu KP. Sci Reports. 2017;7:43639. doi: 10.1038/srep43639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reineke EL, Lam M, Liu O, Liu Y, Stanya KJ, Chang KS, Means AR, Kao HY. Mol Cell Biol. 2008;28:997. doi: 10.1128/MCB.01848-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.