

Abstract
Both high sugar and fat diets can induce prosteatotic genes, leading to obesity and obesity-associated diseases, including hepatic steatosis. Unsaturated fat/fatty acid (USFA) reduces high sugar-induced hepatic steatosis by inhibiting the induced prosteatotic genes. In contrast, it is still unclear how USFA ameliorates saturated fat/fatty acid (SFA)-induced hepatic steatosis. As sugar and fat have different transport and metabolic pathways, we hypothesized that USFA suppressed SFA-induced hepatic steatosis via a different set of prosteatotic genes. To test this, we implemented high SFA vs USFA diets and a control diet in C57BL/6 mice for 16 weeks. Severe hepatic steatosis was induced in mice fed the SFA diet. Among a nearly complete set of prosteatotic genes, only the stearoyl-coenzyme a desaturase 1 (Scd1), cluster of differentiation 36 (Cd36), and peroxisome proliferator-activated receptor γ (Pparγ) genes that were differentially expressed in the liver could contribute to SFA-induced steatosis or the alleviative effect of USFA. That is, the SFA diet induced the expression of Cd36 and Pparγ but not Scd1, and the USFA diet suppressed Scd1 expression and the induction of Cd36 and Pparγ. These findings were mainly recapitulated in cultured hepatocytes. The essential roles of SCD1 and CD36 were confirmed by the observation that the suppression of SCD1 and CD36 with small interfering RNA or drug treatment ameliorated SFA-induced lipid accumulation in hepatocytes. We thus concluded that SCD1, CD36, and PPARγ were essential to the suppression of SFA-induced hepatic steatosis by main dietary USFA, which may provide different therapeutic targets for reducing high-fat vs sugar-induced hepatic steatosis.
Keywords: Saturated fatty acid, Unsaturated fatty acid, Obese mice, Hepatic steatosis, Hepatic cells
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) begins with hepatic steatosis and may progress to nonalcoholic steatohepatitis, an inflammatory state leading to cirrhosis and liver failure. Nonalcoholic fatty liver disease constitutes a silent epidemic and has increased dramatically in recent years due to the rising incidence of obesity and metabolic syndrome [1]. A majority of patients with NAFLD meet the National Cholesterol Education Program criteria of the United States for metabolic syndrome, and, therefore, NAFLD is often considered to be the hepatic manifestation of metabolic syndrome [2,3]. Based on these findings, that hepatic steatosis is a definitive step in the progression of NAFLD and is strongly associated with metabolic syndrome and other obesity-related disorders; alleviating hepatic steatosis is a promising approach to prevent the progression of NAFLD and treat obesity-related metabolic diseases.
Although physical activity and weight loss are effective measures for reducing liver fat [4], emerging data indicate that compared with saturated fat/fatty acids (SFA), dietary unsaturated fat high in n-6 polyunsaturated fatty acid (PUFA) can reduce liver fat and improve the metabolic status without weight loss [5]. This benefit together with the effect of PUFA on the reduction of coronary artery disease events [6,7] and diabetes [8] makes dietary PUFA a potential therapy for NAFLD and other obesity-associated metabolic diseases. Similar to the beneficial effect of PUFA, diets rich in monounsaturated fatty acid also reduce the risk of coronary artery disease [9-11] and prevent SFA-induced insulin resistance by increasing mitochondrial β oxidation [12], channeling SFA into triacylglycerols (TAGs) [13], preventing ceramide synthesis [14], and reducing endoplasmic reticulum stress [15].
It is well documented that both high sugar and fat diets can lead to obesity and obesity-associated diseases, including hepatic steatosis. Some progress has been made in the identification of mechanisms underlying the alleviative effect of PUFA in the context of high sugar-induced hepatic steatosis. For example, PUFAs with chains of 20 carbon or longer reduce high sugar-induced hepatic lipogenesis by suppressing the induction of prosteatotic genes (e.g., fatty acid synthase [FASN], stearoyl-coenzyme a desaturase 1 [SCD1], and sterol regulatory element-binding protein [SREBP1]) [16-18]. In contrast, it remains unclear whether high-fat–induced hepatic steatosis is inhibited by unsaturated fat/fatty acid (USFA) (especially the main dietary monounsaturated fatty acid, oleate [OLE], and the main dietary PUFA, linoleate [LO]) via the same set of prosteatotic genes.
To promote the understanding of USFA inhibition in the context of SFA-induced hepatic steatosis, we recently developed a milk fat–based diet (MD) that was lower in USFA and higher in SFA, in contrast to the classic lard-based (LD) high-fat diet (HFD) [15]. Using these diets, we revealed a novel mechanism by which USFA ameliorated SFA-induced insulin resistance [15]. However, it is unclear whether the MD induces more severe hepatic steatosis in mice than the LD. A previous study demonstrated that, in humans, consumption of dietary SFA could result in hepatic steatosis, which was reduced by supplemented LO [5]. We thus speculated that our animal model with SFA and USFA diets could recapitulate these observations in humans and that the use of these models might shed light on the mechanisms by which SFA vs USFA differentially regulate human hepatic steatosis. Indeed, severe steatosis was present in the livers of MD but not LD-fed mice. From this observation, we hypothesized that the differential regulation of hepatic steatosis by SFA vs USFA was mediated by a set of differentially expressed prosteatotic genes. The objectives of this study were thus to identify the genes using our unique animal model as well as to demonstrate whether the genes were essential to the USFA suppression of SFA-induced hepatic steatosis in an in vitro model. The experimental data indicated that the expression of Scd1, cluster of differentiation 36 (Cd36), and peroxisome proliferator-activated receptor γ (Pparγ) in the liver was differentially regulated by the MD vs the LD compared with the control diet (CD). The difference between the MD and LD treatments could be due to the different dietary/plasma fatty acid profiles. The in vitro studies confirmed that main dietary SFA (palmitate [PAL]) vs USFA (LO/OLE) differentially regulated the expression of Scd1, Cd36, and Pparγ and that these genes played an essential role in fatty acid-regulated lipid accumulation. This study thus sheds light on how main dietary USFA inhibits SFA-induced haptic steatosis.
2. Methods and materials
2.1. Mice
Six-week-old C57BL/6 male mice purchased from Jackson Laboratory (Bar Harbor, ME, USA) were housed in light- (12 h:12 h light-dark cycle) and temperature-controlled quarters (21°C) and provided with water and normal chow (no. 5001 PMI Nutrition, Brentwood, MO, USA) ad libitum. After 1 week of acclimation, they were randomly divided into 3 groups and were administered test diets (Harlan Laboratories, Inc, Indianapolis, IN, USA), including the LD (TD.06414, ~60% kJ from lard), the MD (TD.09766, ~60% kJ from anhydrous butter oil), and an isoenergetic low-fat diet (TD.08810, 10% kJ from fat). The diet compositions are shown in Table 1, and the composition of mineral (AIN-93G-MX) and vitamin mixtures (AIN-93-VX) was described previously [19]. Using our preliminary data, the number of mice per group was justified by statistical power analysis, that is, 6 mice for each HFD group and 3 mice for the CD group. During treatment, the mice gradually gained weight, and they appeared to be healthy. The mice were euthanized by isoflurane (Hospira, Inc, Lake Forest, IL, USA) anesthesia and underwent cervical dislocation after 16 weeks of feeding. The livers were harvested and flash-frozen in liquid nitrogen for further analyses. This study was approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina and the VA Medical Center in accordance with the Guide for the care and use of laboratory animals (National Institutes of Health Publication No. 86-23, revised 1996).
Table 1.
Ingredient composition of the diets fed to mice
| Diet a
|
|||
|---|---|---|---|
| CD | LD
|
MD | |
| g/kg Diet | |||
| Ingredient | |||
| Casein | 210 | 265 | 265 |
| l-cystine | 3 | 4 | 4 |
| Maltodextrin | 100 | 160 | 160 |
| Sucrose | 39.14 | 90 | 90 |
| Anhydrous milk fat | 20 | 0 | 310 |
| Lard | 20 | 310 | 0 |
| Soybean oil | 20 | 30 | 30 |
| Hi-Maize 220 (resistant starch) | 500 | 0 | 0 |
| Cellulose | 35 | 65.5 | 65.5 |
| Mineral mix, AIN-93G-MX | 35 | 48 | 48 |
| Calcium phosphate, dibasic | 0 | 3.4 | 3.4 |
| Vitamin mix, AIN-93-VX | 15 | 21 | 21 |
| Choline bitartrate | 2.75 | 3 | 3 |
| Tertiary butylhydroquinone, antioxidant | 0.01 | 0 | 0 |
| Red food color | 0 | 0 | 0.1 |
| Blue food color | 0 | 0.1 | 0 |
| Yellow food color | 0.1 | 0 | 0 |
The diets used in this study include LD HFD, MD HFD, and an isoenergetic low-fat CD purchased from Harlan Laboratories, Inc. Their catalog numbers are D.06414 (LD), TD.09766 (MD), and TD.08810 (CD), respectively.
2.2. Cell culture
HepG2 cells from the American Tissue Culture Collection (Manassas, VA, USA) were cultured according to the protocols provided. Briefly, cells were grown (2 × 105 per well in 12-well dishes) at 37°C in Dulbecco minimum essential medium (catalog [cat] no. 30-2002; American Tissue Culture Collection) containing 10% fetal bovine serum (cat no. 10082; Invitrogen, Grand Island, NY, USA) for 1 to 2 days before treatment. Free fatty acids (FFA) were purchased from Sigma, and stock solutions were made with ethanol/water. The working solution was made by mixing the stock solution with 2% fatty acid-free bovine serum albumin (BSA) in Dulbecco minimum essential medium. The control was a 2% BSA solution supplemented with vehicle. For the FFA treatment, HepG2 cells were treated with 2% BSA-conjugated 0.625 mmol/L OLE, LO, and PAL and were supplemented with or without 0.25 mmol/L OLE or LO for 14 hours. For treatment with SCD1 inhibitor (no. 1716-1; BioVision, Milpitas, CA, USA), the cells were first pretreated with 200 nmol/L SCD1 inhibitor or vehicle (dimethyl ulphoxide) for 1 hour, followed by 12 hours of treatment with PAL.
2.3. Oil red O staining
The oil red O (ORO) protocol has been described previously [20]. In brief, frozen tissue sections placed on clean glass slides were fixed in 10% formalin for 1 hour, followed by 3 rinses in distilled water. The slides were immersed in propylene glycol for 5 minutes twice before staining with ORO for 30 minutes at room temperature. The slides were then immersed in 85% propylene glycol for 3 minutes, followed by 3 rinses in distilled water. For the ORO staining of cultured cells, the procedure is as follows: wash the cultured cells in a dish with phosphate buffered saline, fix the cells in 10% formalin, incubate for 10 minutes at room temperature, discard the old formalin, add fresh 10% formalin, incubate for 1 hour, remove formalin and rinse with distilled water 3 times, air dry for at least 10 minutes, place in absolute propylene glycol and incubate for 5 minutes, stain in prewarmed ORO for 10 minutes in an oven at 60°C, differentiate in 85% propylene glycol solution for 5 minutes, and rinse in twice with distilled water. Images were acquired under a microscope at ×200 magnification. After image acquisition, the stained cells were air dried, followed by dissolving the ORO in 100% isopropanol. Quantification of the ORO staining was expressed as absorbance readings at 550 nm.
2.4. Protein assay
The protein content in a sample was determined using the Bio-Rad RC DC protein assay kit (BioRad, cat no. 500-0119) according to the manufacturer’s instructions. Briefly, 2 μL of protein sample in buffer solution was mixed with 100 μL of reagent I, followed by spinning the mixture in a microfuge at room temperature for 5 minutes. After discarding the supernatant, the precipitate was suspended in 30 μL of mixed reagent solution (made by mixing 1 mL of reagent A with 20 μL of reagent S) and incubated for 5 minutes. Subsequently, 200 μL of reagent B was added and mixed. After 15 minutes of incubation at room temperature, the optical density was acquired at 650 nm in a spectrophotometer.
2.5. Triacylglycerol analysis
Frozen liver samples were pulverized and homogenized in a solution of 1-butanol, Triton X-100, and methanol (4:1:1) using a probe sonicator (4 times for 10 seconds each); incubated for 1 hour at room temperature; and centrifuged for 5 minutes at 4500g. The protein concentration of the supernatant was determined by Micro BCA protein assay reagents (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer’s instructions. The TAG content was determined using a Serum Triglyceride Determination Kit (Sigma-Aldrich, St Louis, MO, USA). The values were normalized to total protein.
2.6. RNA interference
All small interfering RNAs [siRNAs], including the CD36 (cat no. SI03065195), Scd1 (cat no. SI04184488) and negative control (cat no. 1027416), were from Qiagen (Valencia, CA, USA). Their target sequences are CD36, 5’-CAGCAACATTCAAGTTAAGCA and SCD1, 5’-AGGAGATAGGAAGCCAGACTA. HepG2 cells were transfected with these siRNAs using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. In brief, a 20 pmol/μL stock solution of siRNA was used. At the time of transfection, 4 μL of the siRNA stock solution was mixed with 100 μL serum-free Opti-MEM I medium (31985; Gibco), after which this mixture was combined with another mixture (containing 4 μL of Lipofectamine 2000 and 100 μL of Opti-MEM I medium). After 20 minutes of incubation, 208 μL of the mixture was added to the treated cells in 1 mL of regular cell culture media (containing 10% fetal bovine serum). The treatment with different fatty acids was conducted after 48 to 72 hours of transfection, followed by the subsequent evaluation of the knockdown effect of the siRNAs. Each RNA interference assay was performed in triplicate or more.
2.7. RNA analysis
Total RNA isolation, complementary DNA synthesis, and real-time polymerase chain reaction were described previously [21]. The following gene-specific primers were from SABioscience (Frederick, MA, USA) (Table 2) or were custom designed: human PPARγ, 5’-GCG ATT CCT TCA CTG ATA C and 5’-CTT CCA TTA CCG AGA GAT CC and human actin, 5’-ATT GGC AAT GAG CGG TTC C and 5’-GGT AGT TTC GTG GAT GCC ACA. Expression was normalized to actin messenger RNA and calculated relative to the baseline control using the comparative ∆∆Ct method.
Table 2.
Catalog numbers of commercial primers from SABiosciences.com
| Primer name | Catalog no. |
|---|---|
| mouse Scd1 | PPM05664E-200 |
| mouse cyp3a11 | PPM03917E-200 |
| mouse Cyp7a1 | PPM03986B-200 |
| mouse Fabp1 | PPM26259A-200 |
| mouse Mttp | PPM24881A-200 |
| mouse Acadl | PPM04368A-200 |
| mouse Acox1 | PPM04407A-200 |
| mouse Cs | PPM29621A-200 |
| mouse Pparg | PPM05108B-200 |
| mouse Fasn | PPM03816E-200 |
| mouse Cpt1 | PPM25930B-200 |
| mouse Acc1 | PPM05109E-200 |
| mouse Fatp2 | PPM37730E-200 |
| human CD36 | PPH01356A-200 |
| human SCD1 | PPH00015A-200 |
| human CYP7A1 | PPH01231A-200 |
| human PPARG | PPH02291F-200 |
Abbreviations: ACC1, acetyl-CoA carboxylase alpha; Acox1, acyl CoA oxidase 1; ACADL, acyl-CoA dehydrogenase, long chain; CPT1, carnitine palmitoyltransferase 1; CS, citrate synthase; FABP1, fatty acid binding protein 1; Fatp2, fatty acid transport protein 2.
2.8. Immunoblot analysis
Protein lysates from cultured cells or mouse tissues were analyzed by standard Western blot protocols [14] using the following primary antibodies: SCD1 (cat no.2438; Cell Signaling, Danvers, MA, USA), CD36 (cat no. PA1-46480; Thermo Fisher Scientific, Rockford, IL, USA), PPARγ (cat no. PA1-27585; Thermo Fisher Scientific), glyceraldehyde-3-phosphate dehydrogenase (cat no.2118; Cell Signaling), and actin (cat no. sc-1616; Santa Cruz Biotechnology, Inc, Santa Cruz, CA). Quantification of the proteins was performed using ImageJ software.
2.9. Statistical analyses
All values are expressed as the means ± SEM. One-way analysis of variance with treatment as a fixed effect was performed to calculate the F value using SPSS software. If this value was larger than the F value at α = .05, we considered there to be a significant difference among the different treatments. Subsequently, the statistical significance for 2-treatment comparisons was determined by Tukey-Kramer post hoc tests. A value of P < .05 was chosen a priori to be indicative of statistical significance.
3. Results
3.1. Severe hepatic steatosis was induced by SFA rather than USFA diets in mice
A recent study indicates that, in humans, an SFA diet leads to hepatic steatosis and that supplement with n-6 PUFA (ie, LO) suppresses it [5]. To determine whether a mouse model of diet-induced obesity could recapitulate this finding, we fed 3 groups of 6-week-old C57bl/6J male mice for 16 weeks with an MD SFA-rich diet (60% kJ from fat), a classic LD USFA-rich diet (60% kJ from fat), and an isoenergetic CD (10% kJ from fat) (the dietary components are presented in Table 1). During treatment, average body weights for the CD, LD, and MD groups gradually increased from 21.4 ± 0.36 to 31.4 ± 1.3 g, 21.8 ± 0.35 to 36.7 ± 1.3 g, and 22.2 ± 0.47 to 43.0 ± 1.9 g, respectively. The mice appeared to be healthy. There was no significant difference in feed consumption among groups. Fatty acid profile analysis found that the percentages of dietary fatty acids were in parallel with the profile of plasma fatty acids (Tables 3 and 4) [15]. Compared with the LD, the MD induced severe hepatic steatosis in mice, which was indicated by a heavier liver weight (Fig. 1A) and higher content of liver triacylglyceride (Fig. 1B and C). This suggests that MD-induced hepatic steatosis may better mimic human liver disease than LD-induced hepatic steatosis.
Table 3.
Fatty acyls in the diets fed to mice
| Diet a
|
|||
|---|---|---|---|
| CD | LD | MD | |
| Fatty acyl | % | ||
| 14:0 | 4.3 ± 0.01 | 1.3 ± 0.00 | 10.0 ± 0.02 |
| 16:0 | 21.4 ± 0.01 | 22.3 ± 0.04 | 27.9 ± 0.06 |
| 18:0 | 9.9 ± 0.01 | 12.7 ± 0.05 | 10.6 ± 0.02 |
| 18:1 | 29.0 ± 0.13 | 38.0 ± 0.05 | 24.3 ± 0.14 |
| 18:2n-6 | 23.3 ± 0.08 | 19.3 ± 0.03 | 7.6 ± 0.02 |
Denotes different diets including CD, LD, and MD. n = 3. Data are presented as means ± SEM.
Table 4.
Fatty acid composition of plasma of mice after 16 weeks of dietary treatment
| Diet a
|
|||
|---|---|---|---|
| CD | LD | MD | |
| Fatty acyl | % | ||
| 14:0 | 1.8 ± 0.10 | 2.8 ± 0.61 | 7.8 ± 0.17 |
| 16:0 | 9.3 ± 0.57 | 13.1 ± 1.7 | 16.0 ± 0.35 |
| 18:0 | 4.0 ± 0.35 | 5.6 ± 1.2 | 6.9 ± 0.08 |
| 18:1 | 11.5 ± 1.3 | 19.7 ± 1.3 | 14.7 ± 0.99 |
| 18:2n-6 | 8.6 ± 1.8 | 22.1 ± 0.86 | 10.7 ± 0.97 |
Denotes the mice were treated with different diets including CD, LD, and MD. n = 3 to 6. Data are presented as means ± SEM.
Fig. 1.
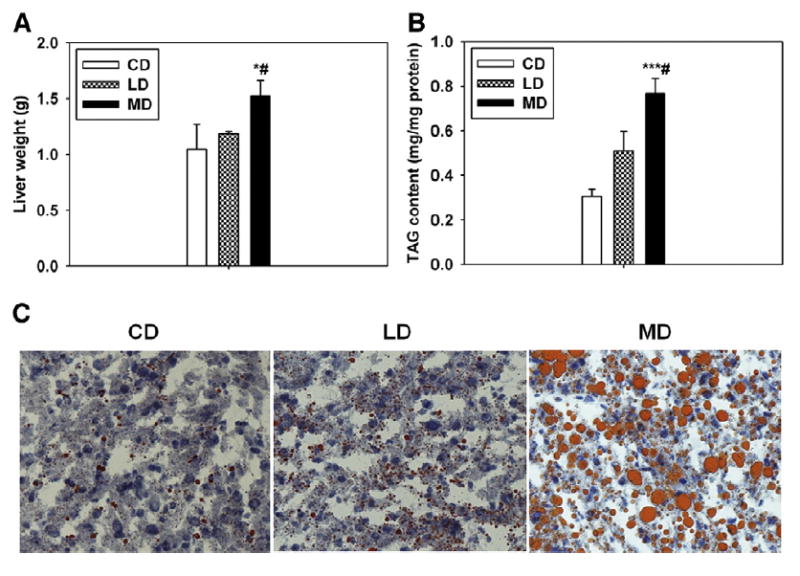
Dietary fat composition determines the severity of hepatic steatosis in mice. Liver weights (A) and TAG content in the livers of mice fed with different diets at 16 weeks of feeding (B). n = 3 to 6. C, Representative micrographs of the ORO staining. n = 3. *P < .05 and ***P < .001 vs CD, respectively; #P < .05 vs LD. The results are presented as the means ± SEM.
3.2. Prosteatotic genes were differentially regulated in the livers of SFA vs USFA-fed mice
Liver samples harvested from the mice fed with different diets were used to determine the expression of genes known to play roles in hepatic steatosis. The results indicated that the suppression of Scd1 was only observed in the LD- but not the CD- or MD-fed mice (Fig. 2A-C), potentially explaining the limited steatosis in LD-fed mice. Moreover, the MD-fed mice had elevated levels of Pparγ and Cd36, a major fatty acid transporter [22-25], which occurred to a much lesser degree in the LD-fed mice (Fig. 2A-C). Among other genes, Fasn was down-regulated by the MD but not the LD, whereas acyl CoA oxidase 1 and fatty acid transport protein 2 were up-regulated by both the MD and LD in contrast to the CD, and there were no changes in the expression of the rest of the genes (Fig. 3A-C).
Fig. 2.
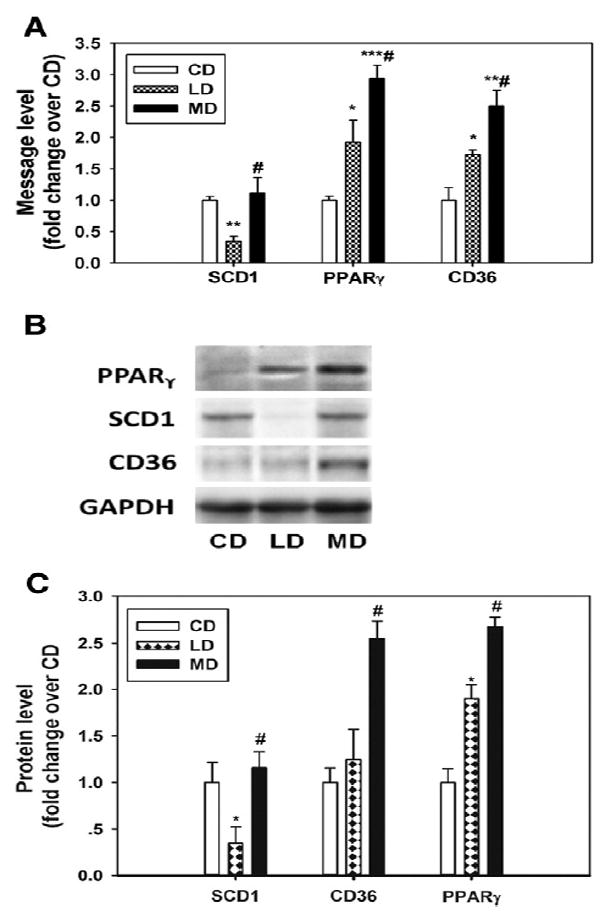
Dietary fat composition determines the severity of steatosis through the regulation of prosteatotic genes, including Scd1, Pparγ, and Cd36, in the liver. The message levels (A) and representative images and quantification of the immunoblots for Scd1, Pparγ, and Cd36 in the livers of mice at 16 weeks of feeding (B and C). n = 3 to 6; *P < .05, **P < .01, ***P < .001 vs CD, respectively; #P < .05 vs LD. The results are presented as the means ± SEM.
Fig. 3.
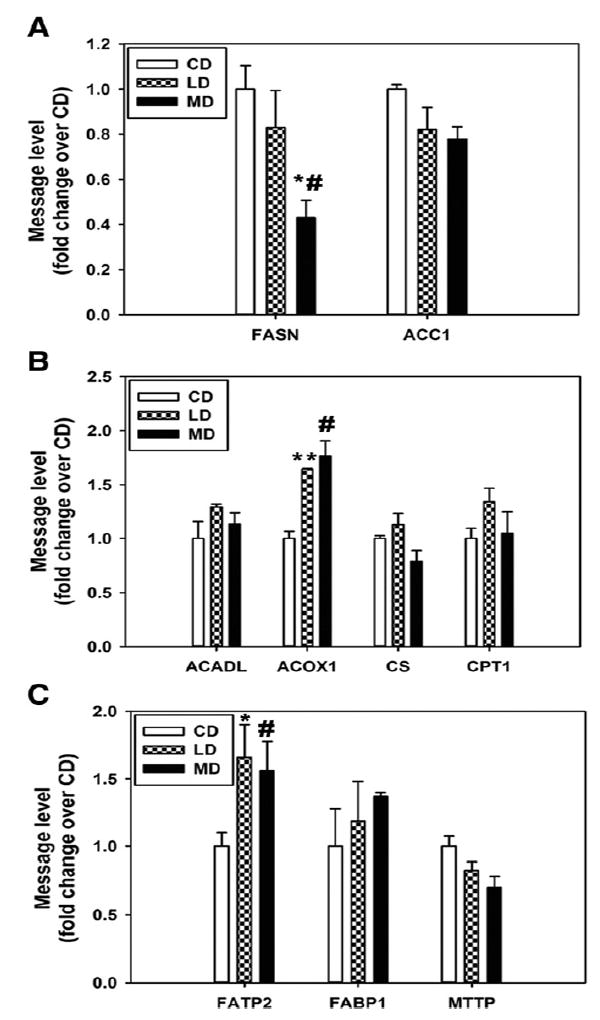
The message levels of some prosteatotic genes are not regulated in a manner favoring the milk fat diet induction of hepatic steatosis in mice. These genes include those involved in de novo lipogenesis (A), fatty acid catabolism (B), and fatty acid uptake and release (C). RNA samples for quantitative polymerase chain reaction analysis were isolated from the livers of mice at 16 weeks of feeding. n = 3 to 6.; *P < .05 and **P < .001 vs CD, respectively; #P < .05 vs LD. The results are presented as the means ± SEM.
3.3. Fatty acids differentially regulated SCD1, PPARγ, and CD36 in HepG2 cells
To test whether plasma FFA mediated the differential regulation of prosteatotic genes by the HFDs, HepG2 cells were treated with PAL with or without main dietary USFA (ie, LO or OLE) cotreatment. The results indicated that the expression patterns of SCD1, PPARγ, and CD36 recapitulated the in vivo data (Fig. 4A-C), that is, SFA induced the expression of PPARγ and CD36 but not SCD1, and USFA inhibited SCD1 expression and the induction of PPARγ and CD36.
Fig. 4.
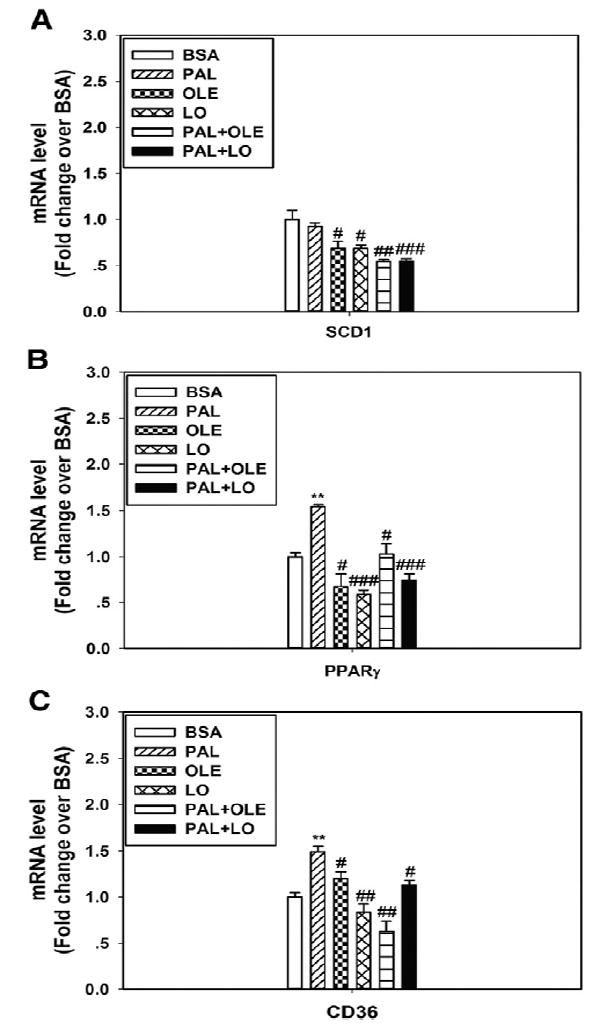
Fatty acids differentially regulate prosteatotic gene expression in HepG2 cells. Message levels of SCD1 (A), PPARγ (B), and CD36 (C) were differentially regulated by fatty acids including PAL, OLE, and LO. n = 3. **P < .01 vs BSA; #P < .05, ##P < .01, ###P < .001 vs PAL, respectively. The results are presented as the means ± SEM.
3.4. Inhibition of SCD1 and CD36 reduced lipid accumulation in cultured hepatocytes
To test whether the down-regulation of SCD1 could potentially explain the limited steatosis in the LD liver, HepG2 cells were treated with PAL in the presence of the SCD1 inhibitor, which almost completely prevented steatosis (Fig. 5A and B), supporting the key role for SCD1 in SFA-induced hepatic steatosis. This was further supported by transfection with SCD1 siRNA, which reduced the message to nearly 50% relative to the control siRNA (Fig. S1A) and completely blocked SFA-induced steatosis (Fig. 5C and D). Together, these data support the requirement for SCD1 in steatosis and, moreover, reveal the underlying mechanism by which LD-fed Scd1 deletion mice displayed little difference to LD-fed wild-type mice, that is, that the LD suppressed Scd1 expression.
Fig. 5.
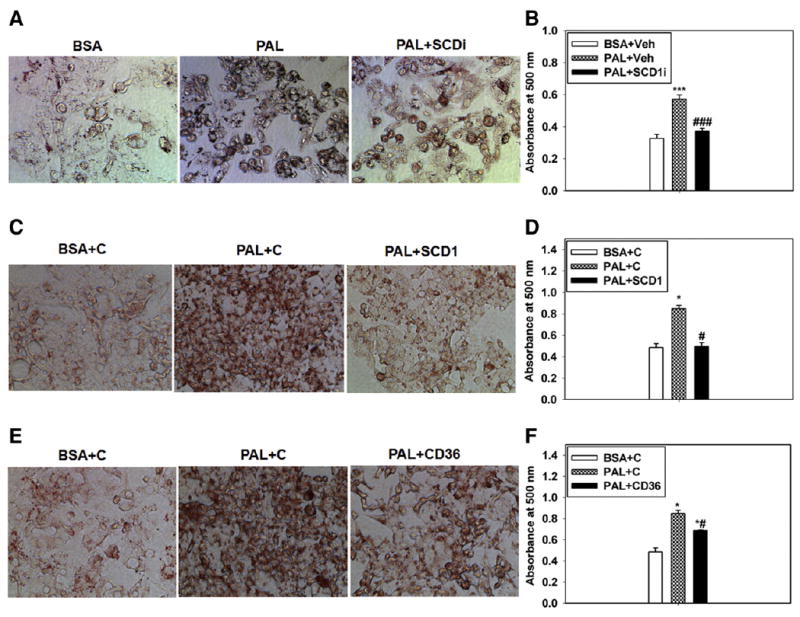
Inhibition of SCD1/CD36 prevents PAL-induced lipid accumulation in HepG2 cells. Representative images of the ORO staining and the quantification of lipid accumulation by absorbance readings (which is normalized to total cell protein per well of the culture dish) in cells treated with BSA or PAL plus the vehicle or SCD1 inhibitor (A and B), BSA or PAL plus the negative control siRNA or SCD1 siRNA (C and D), and BSA or PAL plus the negative control siRNA or CD36 siRNA (E and F). Veh, dimethyl sulphoxide; SCD1i, SCD1 inhibitor; C, negative control siRNA. n = 3. *P < .05 and ***P < .001 vs BSA, respectively. #P < .05, ###P < .001 vs PAL, respectively. The results are presented as the means ± SEM.
To test whether CD36 played a role in SFA-induced steatosis, HepG2 cells were transfected with siRNA directed toward CD36, which reduced the message by approximately 70% (Fig. S1B). This prevented PAL-induced steatosis to a similar degree (Fig. 5E and F). Together with the observed down-regulation of Scd1 by USFA, we concluded that the different fatty acid profiles of the 2 diets profoundly impacted steatosis at least partially due to their differential effects on the messages for genes with key roles in steatosis.
3.5. Potential mechanism by which dietary fatty acids differentially regulate the expression of PPARγ and CD36
To understand how these genes were differentially regulated by dietary SFA vs USFA, we examined the liver X receptor (LXR) and pregnane X receptor (PXR) pathways, as previous studies indicated that PPARγ and CD36 are target genes of PXR [26] and that LXR may regulate PXR via its unique target gene, Cyp7a1 [27,28]. Our data indicated that Cyp3a11, a PXR-specific target gene [26,29,30], was differentially regulated by the HFDs (Fig. 6A). Moreover, Cyp7a1 was also differentially regulated by the HFDs in mice (Fig. 6B) and by FFAs in cultured HepG2 cells (Fig. 6C). These findings suggested that the LXR/PXR axis probably mediated the differential regulation of PPARγ and CD36 by the HFDs.
Fig. 6.
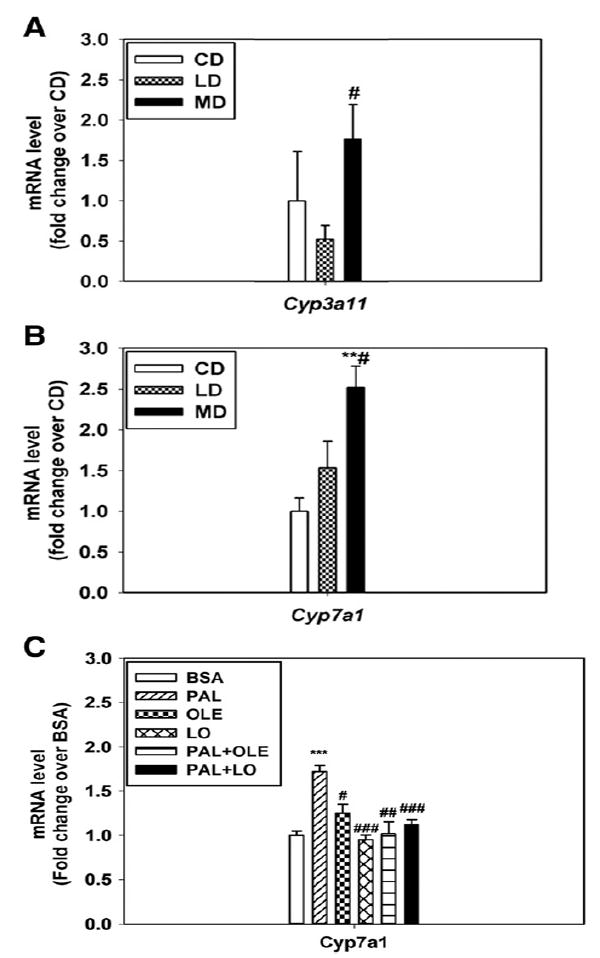
Differential expression of CYP3a11 and CYP7a1 in the livers of mice or HepG2 cells treated with SFA vs UFA. Message levels of CYP3a11 (A) and CYP7a1 (B) were differentially expressed in the livers of mice fed with LD vs MD; n = 3 to 6; **P < .01 vs CD, #P < .05 vs LD. C, Message levels of CYP7a1 were differentially regulated in cultured HepG2 cells treated with 0.625 mmol/L PAL with or without OLE/LO. Cells treated with BSA were used as the control. n = 5. ***P < .001 vs BSA; ##P < .01, ###P < .001 vs PAL, respectively. The results are presented as the means ± SEM.
4. Discussion
It is well known that USFA can reduce the incidence of coronary artery disease and improve the metabolic status of patients with diabetes [6-8]. These benefits, together with the recent finding that main dietary n-6 PUFA (LO) ameliorates hepatic steatosis [5], suggest that dietary USFA could be used as a potential treatment for NAFLD and other related metabolic diseases. In this study, we confirmed that dietary USFA (including LO) could diminish SFA-induced hepatic steatosis. To uncover the molecular mechanism underlying this phenomenon, we studied an almost whole set of genes that is known to play roles in hepatic steatosis, including de novo lipogenesis, fatty acid catabolism, and fatty acid uptake, in addition to PPARγ, a key regulator of steatosis [31-34]. The data indicated that main dietary SFA (PAL) induced the expression of Cd36 and Pparγ and that main dietary USFA (LO/OLE) suppressed Scd1 expression and the SFA induction of Cd36 and Pparγ. In other words, Scd1, Pparγ, and Cd36, rather than other prosteatotic genes, were regulated in the manner favoring the MD induction of hepatic steatosis, suggesting that only Scd1, Pparγ, and Cd36 contributed to the differential effect of SFA vs USFA on hepatic steatosis. From these findings, we propose that SFA-induced hepatic steatosis in mice is better suited for the study of human NAFLD and that the caveat of an LD-based mouse model may prevent the discovery of the mechanism underlying NAFLD.
In this study, the essential roles of SCD1 and CD36 in SFA-induced lipid accumulation/hepatic steatosis were demonstrated through molecular biology and pharmaceutical approaches. Based on these findings, together with the plasma FFA profiles reflecting dietary fatty acyl profiles [15], we concluded that dietary USFA alleviated hepatic steatosis/lipid accumulation, at least partially, via SCD1, CD36, and PPARγ and that plasma FFA probably mediated the differential regulation of these genes and hepatic steatosis through the dietary fatty acid composition. This study gives insight into the molecular mechanism by which the SFA diet induces severe hepatic steatosis, whereas the USFA diet suppresses this induction.
Mammalian triglycerides largely contain unsaturated fatty acyls at the Sn2 position, and stearoyl-CoA desaturase, which introduces a double bond into the saturated fatty acids, can function as a rate-limiting enzyme to regulate the biosynthesis of TAGs [35-37]. Cluster of differentiation 36 has emerged as a key fatty acid transporter in diverse cell types, including macrophages, cardiomyocytes, and hepatocytes. Peroxisome proliferator-activated receptor γ is a key regulator involved in adipocyte differentiation and other obesity-associated diseases. The roles of these genes in lipid accumulation/hepatic steatosis were evaluated previously with the deletion or transgenic overexpression of these genes [31,34,37-40]; however, their roles in the context of SFA-induced lipid accumulation were uncertain. Despite no induction of SCD1 by SFA, this study showed that the lipid accumulation induced by SFA was also inhibited by SCD1 inhibitor or siRNAs targeting Scd1/Cd36, indicating that SCD1 and CD36 are required for SFA-induced steatosis. Intriguingly, the knockdown efficiency for SCD1 (the SCD1 message was reduced to 50%) was lower than that for CD36 (the CD36 message was reduced to 70%). However, the strong effect on the phenotype (or lipid accumulation) was observed in the cells treated with SCD1 siRNA rather than CD36 siRNA. This phenomenon is probably due to a lack of induction of SCD1 by the PAL in the cells, whereas CD36 was strongly induced by PAL. The 50% reduction of SCD1 that was not induced by PAL may significantly impair the synthesis of TAG, leading to the strong suppression of lipid accumulation in the treated cells. In addition, it should be emphasized here that, in spite of the suppression of messenger RNA, the amount of lingering protein may not be reflective of the transcript abundance. The requirement for SCD1 in steatosis may explain the previously perplexing result that LD-fed Scd1 deletion mice displayed little difference to LD-fed wild-type mice [37], that is, USFA in the LD suppressed hepatic Scd1 expression and thus steatosis in wild-type mice. With regard to the mechanism by which SCD1 expression was inhibited by USFA, we speculated that SREBP1 mediated the suppression of SCD1 by USFA, as many studies have shown that SCD1 is a target gene of SREBP1 in obesity and that its expression is inhibited by PUFA [17,41-43].
It should be mentioned that FASN and SCD1, the key players in de novo lipogenesis, were unregulated or down-regulated by the MD, which is different from the observation that these genes are up-regulated in the livers of mice fed high sugar diets [18], suggesting that the mechanism underlying the induction of hepatic steatosis by SFA is at least partially different from that of the high sugar diet. In contrast, inhibition of SCD1 by PUFA was seen in the livers of mice treated with either obesogenic diet, which indicates that SCD1 is critical for the PUFA suppression of hepatic steatosis [17,44-47].
With regard to the mechanism by which these genes were differentially regulated by dietary SFA vs USFA, this study provided some useful information. Cyp7a1 is an LXR-specific target gene [27,28], and fatty acids and their derivatives can regulate LXR activity and the expression of its target genes [48]. This study showed that Cyp7a1, the rate-limiting enzyme in the synthesis of bile acid [49,50], was differentially regulated by the HFDs in mice and by FFAs in cultured HepG2 cells. This finding suggested that the LXR pathway was activated in our model system. Moreover, bile acids are endogenous ligands of PXR [51], and we found that Cyp3a11, a PXR-specific target gene [26,29,30], was differentially regulated by the HFDs. This finding suggested that the PXR pathway was also activated in the model system. As previous studies have indicated that PPARγ and CD36 are target genes of the PXR [26], we thus concluded that the LXR/PXR axis probably mediated the differential regulation of PPARγ and CD36 by the HFDs, which is consistent with the roles of LXR and PXR in hepatic steatosis reviewed by Lee et al [52].
In this study, we noticed that the weight gains of the mice fed different diets varied at the end of the feeding experiment (20.9 g for the MD group, 14.9 g for the LD group, and 10.0 g for the CD group) and that the ratio of the liver to body weight for the MD, LD, and CD groups was 4.2%, 2.8%, and 3.3%, respectively. This suggests that although body weight gain was greater in the MD group than in the CD and LD groups, the liver gained weight more rapidly than the body. As the TAG content of the livers of MD-treated mice was significantly higher than those of the CD and LD-treated mice, we concluded that the liver weight gain in the MD group was primarily due to increased liver fat, which is consistent with the notion that the MD promotes fatty liver compared with the LD.
In the present study, we used an in vitro model to extrapolate in vivo findings. Our cell studies only investigated the direct effect of SFA vs USFA on hepatic steatosis and the related gene expression. Because of the complexities of the diet formulations, we could not rule out the effects of other dietary components on these phenotypes. More work is needed to completely understand the effects induced by the MD vs the LD.
In summary, this study reveals the potential mechanisms by which SFA promotes hepatic steatosis, whereas the USFA diet alleviates SFA-induced hepatic steatosis via the differential regulation of key prosteatotic genes including SCD1, CD36, and PPARγ. These genes may serve as therapeutic targets in the context of SFA-induced hepatic steatosis, as they do not completely overlap with the set of prosteatotic genes (eg, FASN) induced by high sugar diets. The strategy to treat HFD-induced hepatic steatosis should be distinguished from that to treat high sugar diet-induced hepatic steatosis. Based on our findings, we accept the hypothesis that the differential regulation of hepatic steatosis by SFA vs USFA was mediated by a set of differentially expressed prosteatotic genes. In addition, this study presents a potential caveat of the classic LD HFD because USFA suppresses and/or fails to up-regulate prosteatotic genes that mediate hepatic steatotis in vivo, and the discovery of mechanisms underlying NAFLD may be precluded by generating obesity in mice fed with LD HFD. Thus, this study presents a novel diet-induced obesity mouse model with increased suitability for the study of human NAFLD and other obesity-related diseases.
Supplementary Material
Acknowledgments
This work was financially supported by the Natural Sciences Foundation of China (31472086) and the Priority Academic Program Development of Jiangsu Higher Education Institutions through awards to TG as well as by a VA Merit Award and 1R01HL117233 awarded to LAC.
Abbreviations
- BSA
bovine serum albumin
- CD
control diet
- CD36
cluster of differentiation 36
- cat
catalog
- FASN
fatty acid synthase
- FFA
free fatty acid
- HFD
high-fat diet
- LD
lard-based high-fat diet
- LXR
liver X receptor
- MD
milk fat–based high-fat diet
- NAFLD
nonalcoholic fatty liver disease
- ORO
oil red O
- PPAR
peroxisome proliferator-activated receptor
- PUFA
polyunsaturated fatty acid
- PXR
pregnane X receptor
- SCD1
stearoyl-coenzyme a desaturase 1
- SFA
saturated fat/fatty acid
- siRNA
small interfering RNA
- SREBP
sterol regulatory element-binding protein
- TAG
triacylglycerol
- USFA
unsaturated fat/fatty acid
Footnotes
Conflict of interest: none declared.
TG and LAC designed the research and wrote the article; TG, LX, and SBR conducted the research and analyzed the data. DK contributed to writing and editing of the article.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nutres.2015.06.012.
References
- 1.Schattenberg JM, Schuppan D. Nonalcoholic steatohepatitis: the therapeutic challenge of a global epidemic. Curr Opin Lipidol. 2011;22:479–88. doi: 10.1097/MOL.0b013e32834c7cfc. [DOI] [PubMed] [Google Scholar]
- 2.Neuschwander-Tetri BA, Clark JM, Bass NM, Van Natta ML, Unalp-Arida A, Tonascia J, et al. Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology. 2010;52:913–24. doi: 10.1002/hep.23784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–85. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazo M, Solga SF, Horska A, Bonekamp S, Diehl AM, Brancati FL, et al. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes Care. 2010;33:2156–63. doi: 10.2337/dc10-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjermo H, Iggman D, Kullberg J, Dahlman I, Johansson L, Persson L, et al. Effects of n-6 PUFAs compared with SFAs on liver fat, lipoproteins, and inflammation in abdominal obesity: a randomized controlled trial. Am J Clin Nutr. 2012;95:1003–12. doi: 10.3945/ajcn.111.030114. [DOI] [PubMed] [Google Scholar]
- 6.Mozaffarian D, Micha R, Wallace S. Effects on coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7:e1000252. doi: 10.1371/journal.pmed.1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Astrup A, Dyerberg J, Elwood P, Hermansen K, Hu FB, Jakobsen MU, et al. The role of reducing intakes of saturated fat in the prevention of cardiovascular disease: where does the evidence stand in 2010? Am J Clin Nutr. 2011;93:684–8. doi: 10.3945/ajcn.110.004622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riserus U, Willett WC, Hu FB. Dietary fats and prevention of type 2 diabetes. Prog Lipid Res. 2009;48:44–51. doi: 10.1016/j.plipres.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ros E. Dietary cis-monounsaturated fatty acids and metabolic control in type 2 diabetes. Am J Clin Nutr. 2003;78:617S–25S. doi: 10.1093/ajcn/78.3.617S. [DOI] [PubMed] [Google Scholar]
- 10.Gillingham LG, Harris-Janz S, Jones PJ. Dietary monounsaturated fatty acids are protective against metabolic syndrome and cardiovascular disease risk factors. Lipids. 2011;46:209–28. doi: 10.1007/s11745-010-3524-y. [DOI] [PubMed] [Google Scholar]
- 11.Rasmussen BM, Vessby B, Uusitupa M, Berglund L, Pedersen E, Riccardi G, et al. Effects of dietary saturated, monounsaturated, and n-3 fatty acids on blood pressure in healthy subjects. Am J Clin Nutr. 2006;83:221–6. doi: 10.1093/ajcn/83.2.221. [DOI] [PubMed] [Google Scholar]
- 12.Henique C, Mansouri A, Fumey G, Lenoir V, Girard J, Bouillaud F, et al. Increased mitochondrial fatty acid oxidation is sufficient to protect skeletal muscle cells from palmitate-induced apoptosis. J Biol Chem. 2010;285:36818–27. doi: 10.1074/jbc.M110.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coll T, Eyre E, Rodriguez-Calvo R, Palomer X, Sanchez RM, Merlos M, et al. Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J Biol Chem. 2008;283:11107–16. doi: 10.1074/jbc.M708700200. [DOI] [PubMed] [Google Scholar]
- 14.Hu W, Ross J, Geng T, Brice SE, Cowart LA. Differential regulation of dihydroceramide desaturase by palmitate versus monounsaturated fatty acids: implications for insulin resistance. J Biol Chem. 2011;286:16596–605. doi: 10.1074/jbc.M110.186916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geng T, Hu W, Broadwater MH, Snider JM, Bielawski J, Russo SB, et al. Fatty acids differentially regulate insulin resistance through endoplasm reticulum stress-mediated induction of tribbles homologue 3: a potential link between dietary fat composition and the pathophysiological outcomes of obesity. Diabetologia. 2013;56:2078–87. doi: 10.1007/s00125-013-2973-2. [DOI] [PubMed] [Google Scholar]
- 16.Xu J, Nakamura MT, Cho HP, Clarke SD. Sterol regulatory element binding protein-1 expression is suppressed by dietary polyunsaturated fatty acids. A mechanism for the coordinate suppression of lipogenic genes by polyunsaturated fats. J Biol Chem. 1999;274:23577–83. doi: 10.1074/jbc.274.33.23577. [DOI] [PubMed] [Google Scholar]
- 17.Sekiya M, Yahagi N, Matsuzaka T, Najima Y, Nakakuki M, Nagai R, et al. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology. 2003;38:1529–39. doi: 10.1016/j.hep.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 18.Miyazaki M, Dobrzyn A, Man WC, Chu K, Sampath H, Kim HJ, et al. Stearoyl-CoA desaturase 1 gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J Biol Chem. 2004;279:25164–71. doi: 10.1074/jbc.M402781200. [DOI] [PubMed] [Google Scholar]
- 19.Reeves PG, Nielsen FH, Fahey GC., Jr AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–51. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 20.Fiorini RN, Kirtz J, Periyasamy B, Evans Z, Haines JK, Cheng G, et al. Development of an unbiased method for the estimation of liver steatosis. Clin Transplant. 2004;18:700–6. doi: 10.1111/j.1399-0012.2004.00282.x. [DOI] [PubMed] [Google Scholar]
- 21.Hu W, Bielawski J, Samad F, Merrill AH, Jr, Cowart LA. Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via upregulation of sphingosine kinase message and activity. J Lipid Res. 2009;50:1852–62. doi: 10.1194/jlr.M800635-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. 2000;275:14501–8. doi: 10.1074/jbc.275.19.14501. [DOI] [PubMed] [Google Scholar]
- 23.Luiken JJ, Glatz JF, Bonen A. Fatty acid transport proteins facilitate fatty acid uptake in skeletal muscle. Can J Appl Physiol. 2000;25:333–52. [PubMed] [Google Scholar]
- 24.Luiken JJ, Schaap FG, van Nieuwenhoven FA, van der Vusse GJ, Bonen A, Glatz JF. Cellular fatty acid transport in heart and skeletal muscle as facilitated by proteins. Lipids. 1999;34:S169–75. doi: 10.1007/BF02562278. [Suppl.] [DOI] [PubMed] [Google Scholar]
- 25.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–91. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, et al. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem. 2006;281:15013–20. doi: 10.1074/jbc.M511116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiang JY, Kimmel R, Stroup D. Regulation of cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription by the liver orphan receptor (LXRalpha) Gene. 2001;262:257–65. doi: 10.1016/s0378-1119(00)00518-7. [DOI] [PubMed] [Google Scholar]
- 28.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 29.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–74. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A. 2001;98:3375–80. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, Gonzalez-Periz A, et al. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–50. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- 32.Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–76. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 33.Zhang YL, Hernandez-Ono A, Siri P, Weisberg S, Conlon D, Graham MJ, et al. Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J Biol Chem. 2006;281:37603–15. doi: 10.1074/jbc.M604709200. [DOI] [PubMed] [Google Scholar]
- 34.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem. 2003;278:498–505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 35.Cohen P, Ntambi JM, Friedman JM. Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:271–80. doi: 10.2174/1568008033340117. [DOI] [PubMed] [Google Scholar]
- 36.Ntambi JM. The regulation of stearoyl-CoA desaturase (SCD) Prog Lipid Res. 1995;34:139–50. doi: 10.1016/0163-7827(94)00010-j. [DOI] [PubMed] [Google Scholar]
- 37.Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X, et al. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 2007;6:484–96. doi: 10.1016/j.cmet.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 38.Coburn CT, Knapp FF, Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem. 2000;275:32523–9. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- 39.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, et al. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–62. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 40.Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, et al. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737–47. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waters KM, Ntambi JM. Polyunsaturated fatty acids inhibit hepatic stearoyl-CoA desaturase-1 gene in diabetic mice. Lipids. 1996;31:S33–6. doi: 10.1007/BF02637047. [Suppl.] [DOI] [PubMed] [Google Scholar]
- 43.Ntambi JM, Sessler AM, Takova T. A model cell line to study regulation of stearoyl-CoA desaturase gene 1 expression by insulin and polyunsaturated fatty acids. Biochem Biophys Res Commun. 1996;220:990–5. doi: 10.1006/bbrc.1996.0520. [DOI] [PubMed] [Google Scholar]
- 44.Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, et al. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A. 2001;98:6027–32. doi: 10.1073/pnas.111138698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kajikawa S, Harada T, Kawashima A, Imada K, Mizuguchi K. Highly purified eicosapentaenoic acid prevents the progression of hepatic steatosis by repressing monounsaturated fatty acid synthesis in high-fat/high-sucrose diet-fed mice. Prostaglandins Leukot Essent Fat Acids. 2009;80:229–38. doi: 10.1016/j.plefa.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 46.Moon YS, Latasa MJ, Griffin MJ, Sul HS. Suppression of fatty acid synthase promoter by polyunsaturated fatty acids. J Lipid Res. 2002;43:691–8. [PubMed] [Google Scholar]
- 47.Paulauskis JD, Sul HS. Hormonal regulation of mouse fatty acid synthase gene transcription in liver. J Biol Chem. 1989;264:574–7. [PubMed] [Google Scholar]
- 48.Johnson TE, Ledwith BJ. Peroxisome proliferators and fatty acids negatively regulate liver X receptor-mediated activity and sterol biosynthesis. J Steroid Biochem Mol Biol. 2001;77:59–71. doi: 10.1016/s0960-0760(01)00027-9. [DOI] [PubMed] [Google Scholar]
- 49.De Fabiani E, Mitro N, Anzulovich AC, Pinelli A, Galli G, Crestani M. The negative effects of bile acids and tumor necrosis factor-alpha on the transcription of cholesterol 7alpha-hydroxylase gene (CYP7A1) converge to hepatic nuclear factor-4: a novel mechanism of feedback regulation of bile acid synthesis mediated by nuclear receptors. J Biol Chem. 2001;276:30708–16. doi: 10.1074/jbc.M103270200. [DOI] [PubMed] [Google Scholar]
- 50.Chiang JY. Regulation of bile acid synthesis. Front Biosci. 1998;3:d176–93. doi: 10.2741/a273. [DOI] [PubMed] [Google Scholar]
- 51.Kliewer SA, Willson TM. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J Lipid Res. 2002;43:359–64. [PubMed] [Google Scholar]
- 52.Lee JH, Zhou J, Xie W. PXR and LXR in hepatic steatosis: a new dog and an old dog with new tricks. Mol Pharm. 2008;5:60–6. doi: 10.1021/mp700121u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.