

Abstract
CRISPR/Cas is a revolutionary gene editing technology with wide-ranging utility.[1] The safe, non-viral delivery of CRISPR/Cas components would greatly improve future therapeutic utility.[1e] We report the synthesis and development of zwitterionic amino lipids (ZALs) that are uniquely able to (co)deliver long RNAs including Cas9 mRNA and sgRNAs. ZAL nanoparticle (ZNP) delivery of low sgRNA doses (15 nM) reduces protein expression by >90% in cells. In contrast to transient therapies (e.g. RNAi), we show that ZNP delivery of sgRNA enables permanent DNA editing with an indefinitely sustained 95% decrease in protein expression. ZNP delivery of mRNA results in high protein expression at low doses in vitro (<600 pM) and in vivo (1 mg/kg). Intravenous co-delivery of Cas9 mRNA and sgLoxP induced expression of floxed tdTomato in the liver, kidneys, and lungs of engineered mice. ZNPs provide a chemical guide for rational design of long RNA carriers, and represent a promising step towards improving the safety and utility of gene editing.
Keywords: CRISPR/Cas, mRNA delivery, sgRNA delivery, Gene editing, Nanoparticles
Graphical abstract
We report the synthesis and development of zwitterionic amino lipids (ZALs) that are uniquely able to deliver long RNAs (Cas9 mRNA and targeted sgRNA) from a single ZAL nanoparticle (ZNP) to enable CRISPR/Cas gene editing.
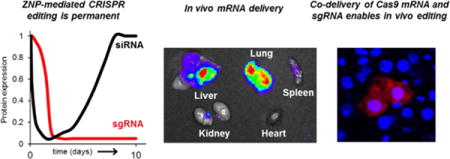
The CRISPR/Cas (clustered regularly interspaced short palindromic repeat / CRISPR-associated protein (Cas)) technology can edit the genome in a precise, sequence dependent manner, resulting in a permanent change.[1e] Because of the ability to target disease causing mutations, it holds incredible promise for one-time cures of genetic diseases. To date, successful editing has been mediated mainly by viral vectors, which require laborious customization for every target and present challenges for clinical translation due to immunogenicity, generation of antibodies that prevent repeat administration, and concerns about rare but dangerous integration events. There remains a clear need to accomplish CRISPR/Cas editing via synthetic nanoparticles (NPs) to expand the safe and effective applications of gene editing.
Herein we report the development of a new class of lipid-like materials termed zwitterionic amino lipids (ZALs) that are uniquely suitable for delivery of long nucleic acids, ~4,500 nucleotide (nt) Cas9 mRNA and ~100 nt sgRNAs, including co-delivery from the same NP. ZAL nanoparticles (ZNPs) are able to effectively induce permanent DNA editing in cells (Fig. 1). This approach simplifies CRISPR/Cas engineering because different sgRNAs can be easily designed, synthesized and packaged into these versatile synthetic carriers. To the best of our knowledge, this is the first successful non-viral system for in vitro and in vivo co-delivery of Cas9 mRNA and sgRNA.
Fig. 1. ZNPs enable permanent CRISPR/Cas-mediated DNA editing.
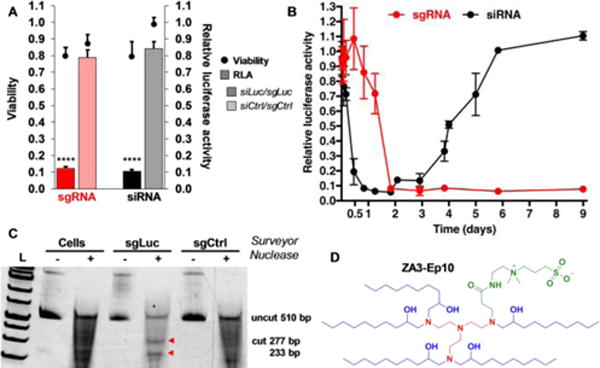
(A) Sequence specific silencing of luciferase by siRNA (9 nM) and editing by sgRNA (7 nM) in HeLa-Luc-Cas9 cells. N = 4 ± stdev, **** p < 0.0001 (B) Kinetically, silencing with siRNA is transient while sgRNA delivery results in permanent loss of luciferase signal after 2 days. (C) Sequence specific editing of luciferase was confirmed by the Surveyor assay. (D) The chemical structure of ZA3-Ep10.
CRISPR/Cas enables sequence-specific DNA editing by the RNA-guided CRISPR-associated protein 9 (Cas9) nuclease that forms double-strand breaks (DSBs) in genomic DNA.[1] Cas9 is guided by programmable RNA called single guide RNA (sgRNA).[1a] The Cas9/sgRNA complex recognizes the complementary genomic sequence with a 3′ protospacer adjacent motif (PAM) sequence. Following DNA cleavage, DSB repair pathways enable directed mutagenesis, or insertions / deletions (indels) that delete the targeted gene.[2] For therapeutic utility, transient Cas9 expression is preferred to limit off-target genomic alteration. Because both Cas9 protein and sgRNAs must be present in the same cells, co-delivery of Cas9 mRNA and sequence targeted sgRNA in one NP is an attractive method, particularly for in vivo use where tissue penetration and cellular uptake is more challenging. CRISPR/Cas editing using viruses,[2b, 2c, 3] membrane deformation,[4] ribonucleoprotein complex delivery,[5] and hydrodynamic injection[2d] are functional, but have limitations that could hinder in vivo therapeutic use in the clinic, including persistent expression of Cas9 and off target editing.
Although great advances have been made in the delivery of short RNAs (siRNA, miRNA) (~22 base pairs (bp) in length) by lipid nanoparticles (LNPs),[6] the ideal chemical and formulation composition is largely unknown for longer RNA cargo (mRNA, sgRNA). Highly effective LNPs are composed of a cationic lipid, zwitterionic phospholipid, cholesterol, and lipid poly(ethylene glycol) (PEG). Cationic lipids bind RNAs at low pH during mixing, and promote intracellular release as the pH decreases during endosomal maturation.[7] Computational modeling has shown that phospholipids function by solubilizing small RNAs inside of aqueous pockets within multi-component LNPs.[8] High cationic lipid density may thus minimize phospholipid stabilizing interactions with longer RNAs in LNPs. Cationic lipids also take up space within LNPs and could hinder inclusion of organized long RNAs at pH 7.4. Recent reports on mRNA delivery using alternative helper phospholipids (e.g. DOPE) further suggests that associated solubilizing forces may improve NP construction.[9] We therefore hypothesized combining the chemical and structural roles of zwitterionic lipids[10] and cationic lipids[9, 11] into a single lipid compound might improve delivery of longer RNAs by increasing molecular interactions within the LNP.
Zwitterionic amino lipids (ZALs) were rationally synthesized to contain a zwitterionic sulfobetaine head group, an amine rich linker region, and assorted hydrophobic tails (Fig. 2). A zwitterionic electrophilic precursor (SBAm) was prepared by the ring-opening reaction of 2-(dimethylamino)ethyl acrylamide with 1,3-propanesultone, which was easily isolated by in situ precipitation in acetone. Conjugate addition of different polyamines to SBAm afforded a series of zwitterionic amines that could be reacted with hydrophobic epoxides and acrylates to append 6 to 18 carbon alkyl tails and alcohol / ester groups to enhance ZAL-RNA interactions (Figs. S1–S4). To verify that ZNPs could generally bind and deliver RNA, the 72-member library was first screened for siRNA delivery to HeLa cells that stably expressed firefly luciferase (HeLa-Luc) (Fig. S5). This allowed structural identification of key amine cores, including ZA1, ZA3, and ZA6. Interestingly, epoxide-based ZALs (ZAx-Epn), were also more active than acrylate-based ZALs (ZAx-Acn) (Fig. S6). With lead compounds in hand, we focused on the delivery of sgRNAs and Cas9 mRNA. Both temporally staged and simultaneous co-delivery enabled fully exogenous gene editing.
Fig. 2.
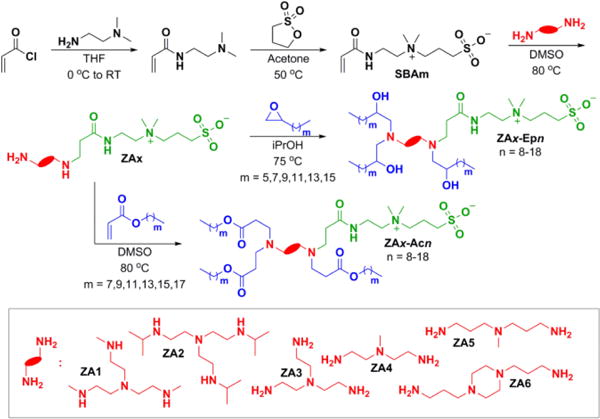
ZALs were designed to increase molecular interactions with longer RNAs by combining the chemical and structural roles of zwitterionic lipids and cationic lipids into a single lipid compound. High efficiency reactions provided access to a library of unique charge unbalanced lipids.
ZALs were evaluated for their ability to deliver CRISPR/Cas9 components using a stable cell line expressing both Cas9 and luciferase (HeLa-Luc-Cas9). A single HeLa-Luc-Cas9 cell clone was isolated following Cas9 lentiviral transduction of HeLa-Luc cells (Fig. S7). sgRNAs against luciferase were designed and generated according to previously reported methods targeting the first third of the gene (Table S1)[12] and evaluated by pDNA transfection (Fig. S8). The most active sgRNA against luciferase (sgLuc5, henceforth sgLuc) as well as control sgRNAs were synthesized by in vitro transcription. Next, lead ZNPs were loaded with sgLuc and evaluated for delivery to HeLa-Luc-Cas9 cells. Luciferase and viability[13] were measured after 48 hours (h) relative to untreated cells. As anticipated from the chemical design combining cationic and zwitterionic functionalities, ZNPs do not require inclusion of helper phospholipids (Fig. 3A).
Fig. 3. ZNPs enable delivery of long RNAs both in vitro and in vivo.
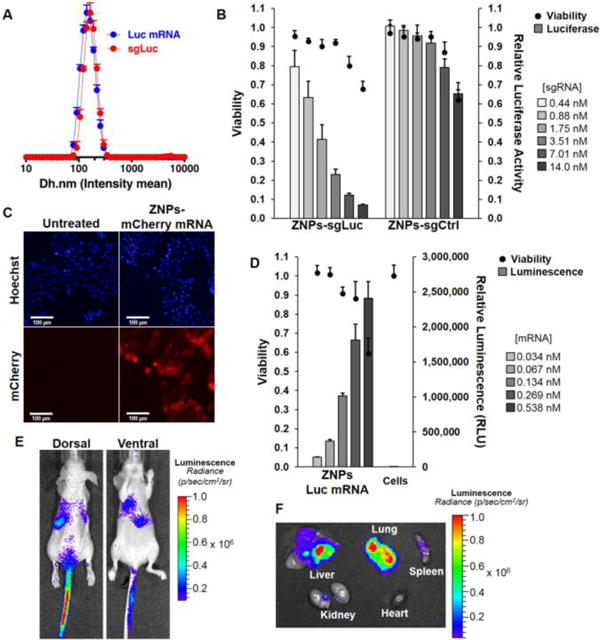
(A) ZA3-Ep10 ZNPs (ZAL:cholesterol:PEG-lipid = 100:77:1 (mol); ZAL:RNA = 7.5:1 (wt)) are uniform for both sgRNA and mRNA. (B) ZA3-Ep10 sgRNA ZNPs show dose-responsive Luc editing in HeLa-Luc-Cas9 cells. ZA3-Ep10 ZNPs can also deliver (C) mCherry mRNA (18h) and (D) luciferase mRNA (24h) to IGROV1 cells. (E) In vivo luciferase expression was achieved by systemic i.v. administration of ZA3-Ep10 Luc mRNA ZNPs (24h). Bioluminescence imaging both in vivo (E, athymic nude mice, 1 mg/kg) and ex vivo (F, C57BL/6 mice, 4 mg/kg) revealed expression of luciferase in liver, lung and spleen tissue.
Among the lead ZALs, ZA3-Ep10 was found to be the most efficacious for delivery of sgLuc (Fig. S9). Editing of luciferase DNA resulted in a dose-dependent decrease in luciferase expression (Fig. 3B). We verified CRISPR/Cas editing using the Surveyor nuclease assay,[14] which can detect indels (Fig. 1C). Given that sgRNAs require loading into Cas9 nucleases in cells and trafficking to the nucleus to perform sequence-guided editing, we wanted to understand the kinetics of this process, particularly in comparison to RNAi-mediated gene silencing. siLuc-mediated mRNA degradation is a fast process, where expression decreased by 40% within the first 4h. Luciferase was silenced by 92% by 20h and remained low for about 3 days. Thereafter, the protein expression steadily increased and reached baseline level 6 days after transfection (Fig. 1B and Fig. S10 (early time points)). In contrast, sgLuc-mediated DNA editing was kinetically slower, possibly due to the requirements to load into Cas9 and survey the DNA for PAMs. It took 20h for luciferase expression to decrease by 40%, ultimately going down by 95% after 2 days and remaining there indefinitely. The low luciferase expression (5%) persisted throughout the duration of the assay (9 days) due the permanent genomic change, even after multiple rounds of cellular division, suggesting that edited cells grew at the same rate of non-edited cells (Fig. 1B, Fig. S11).
Having demonstrated that ZA3-Ep10 ZNPs could effectively deliver sgRNAs (~100 nt), we next examined their ability to deliver even longer mRNA (1,000 to 4,500 nt). We delivered mRNA encoding mCherry mRNA (~1,000 nt) or luciferase mRNA (~2,000 nt) to IGROV1 human ovarian cancer cells. Bright mCherry expression was visible (Fig. 3C), and luciferase expression was observed to be dose-dependent (Fig. 3D). Notably, high expression required low mRNA doses (<600 pM). In contrast to sgRNA, which did not show a dependence on PEG lipid mole ratio in the formulation (Fig. S12, S13), delivery efficacy of mRNA decreased with higher PEG lipid ratios (Fig. S14), while there was only a modest change in ZNP size (Fig. S15). Optimization of PEGylation, particularly in view of in vitro to in vivo translation, is an ongoing challenge to be explored for each target disease, organ, and cell type.[15] Our report attempts to alleviate some of those concerns by examining different formulations in multiple cell types and mouse strains. Further supporting our design hypothesis, titration of a structurally analogous cationic lipid with increasing molar proportions of DOPE into the formulations showed an improvement in delivery of sgRNA and mRNA, while siRNA did not require additional zwitterionic content (Fig. S16). Moreover, efficacy of ZA3-Ep10 ZNPs was consistent across all RNA cargos, and outperformed the cationic analogue supplemented with phospholipid.
The optimal formulation was next evaluated in vivo through intravenous (i.v.) administration of ZA3-Ep10 mRNA ZNPs to multiple strains of mice. Bioluminescence imaging following Luc mRNA delivery in athymic nude mice (Fig. 3E, 1 mg/kg), C57BL/6 mice (Fig. 3F, 4 mg/kg), and NOD scid gamma (NSG) mice (Fig. S17, 1 mg/kg) resulted in expression of luciferase in liver, lung and spleen tissue 24h after injection which was quantified by ROI analysis (Fig. S18). Based on the high lung signal, we were motivated to explore co-delivery (one pot) CRISPR/Cas editing in lung cells.
Due the very long length of Cas9 mRNA (~4,500 nt), delivery using synthetic carriers is particularly challenging. Remarkably, we found the level of Cas9 mRNA in A549 lung cancer cells was very high after only 4h incubation with ZA3-Ep10 Cas9 mRNA ZNPs (Fig. 4A). Synthetically introduced mRNA decreased from >4 fold actin to 0.7 fold actin over the next 45h. Because translation of mRNA takes time, protein expression was low at 4h, increased considerably by 12h, and was the highest by 36h (Fig. 4A, B). It and was also dose dependent (Fig 4C). For ultimate in vivo utility, the use of synthetic NP carriers alleviates concerns of viral delivery. Moreover, delivery of Cas9 mRNA allows for transient expression of Cas9, minimizing persistence that can lead to off-target genomic alteration. This can reduce the significant therapeutic danger of incorporating an exogenous nuclease into the genome.
Fig. 4. ZNPs enable co-delivery of Cas9 mRNA and sgRNA for CRISPR/Cas editing.
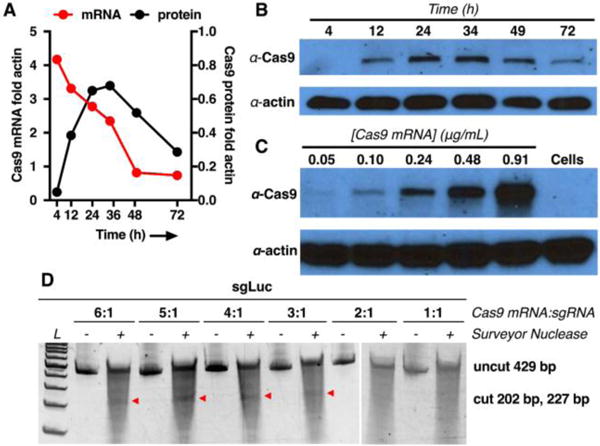
(A) The kinetics of mRNA and protein expression after ZNP delivery of Cas9 mRNA (0.48 ng/mL mRNA) to A549-Luc cells. Cas9 mRNA levels (A red curve) and protein expression (A black curve, B) were measured over time. (C) ZNPs enable dose responsive expression of Cas9, detectable as low as 0.05 μg/mL delivered mRNA. (D) Surveyor confirmed editing of the luciferase target at mRNA:sgRNA ratios of 3:1 or higher (wt). Co-delivery of Cas9 mRNA and sgCtrl showed no editing (Fig. S21).
As illustrated above, optimal delivery of mRNA and sgRNA is kinetically different. Indeed, we found that staged delivery in separate ZNPs was an effective treatment method. ZNP delivery of mRNA for 24h, to enable Cas9 protein expression, followed by sgRNA delivery in separate ZNPs enabled efficacious in vitro editing in both HeLa-Luc and A549-Luc cells (Figs. S20 and S21). However, when considering in vivo utility, Cas9 mRNA and sgRNA must be present in the same cell. We therefore reasoned that co-delivery of mRNA and sgRNA from a single NP would provide a greater editing efficiency since this method would guarantee delivery to the same individual cells. We explored a variety of conditions and found that effective editing of the target gene by ZNPs encapsulating both Cas9 mRNA and sgRNA required a ratio of mRNA:sgRNA greater than or equal to 3:1 (wt) as confirmed by the Surveyor assay (Fig. 4D), while control ZNPs did not show any editing (Fig. S21).
To examine co-delivery in vivo, we utilized genetically engineered mice containing a homozygous Rosa26 promoter Lox-Stop-Lox tdTomato (tdTO) cassette present in all cells.[3c] Co-delivery of Cas9-mRNA and sgRNA against LoxP[16] enabled deletion of the Stop cassette and induction of tdTO expression (Fig. 5A, Table S1). This is a challenging model for a synthetic carrier due to the need to make two cuts on the same allele for the tdTO to be expressed. ZNPs encapsulating Cas9 mRNA and sgLoxp at a 4:1 mRNA:sgRNA weight ratio were administered intravenously at a 5 mg/kg RNA dose (Fig. S22–S24). One week after administration, fluorescence signal from tdTO was detected in the liver and kidneys upon whole organ ex vivo imaging (Fig. 5B). Detailed examination of sectioned organs using confocal fluorescence microscopy showed tdTO-positive cells in liver, lung, and kidney tissues (Fig. 5C). Importantly tdTO positive cells were not detected when animals were treated with sgCtrl ZNPs (Fig. S25) and no significant change in body weight of treated animals was observed (Fig. S26). Primary hepatocytes were isolated from perfused livers and tdTO cells were counted by flow cytometry to quantify editing (Fig S27). To further confirm editing, tissues were harvested 2 months after ZNP sgLoxP treatment, which still exhibited strong fluorescent signal in the liver and kidneys (Fig. S28). This proof-of-principle data indicates that intravenous co-delivery of Cas9 mRNA and targeted sgRNA from a single ZNP can enable CRISPR/Cas editing in vivo.
Fig. 5. ZNPs enabled non-viral CRISPR/Cas editing in vivo.
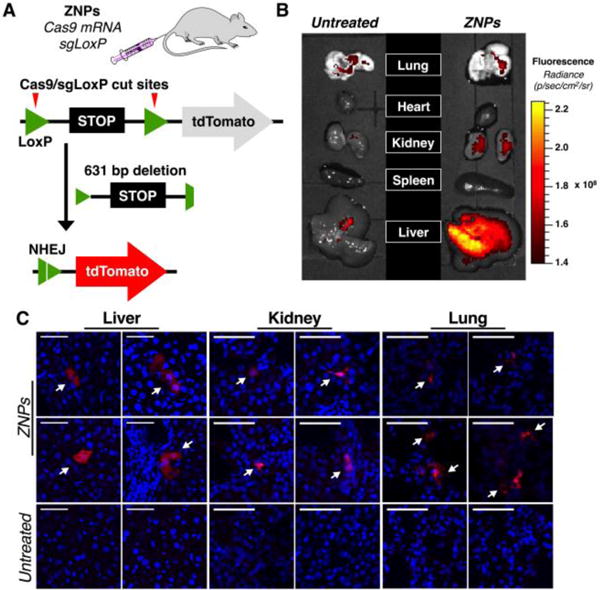
(A) Schematic representation shows that co-delivery of Cas9 mRNA and sgLoxP deletes the stop cassette and activates downstream tdTomato protein. (B) After administration of ZNPs encapsulating Cas9 mRNA:sgRNA (4:1, wt) at 5 mg/kg total RNA, tdTomato fluorescence was detected in the liver and kidney upon whole organ ex vivo imaging. (C) Confocal fluorescence microscopy of tissue sections showed tdTomato positive cells in liver, lung, and kidneys. Scale bars = 50 μm).
In summary, we report the development of the first non-viral delivery system for in vitro and in vivo co-delivery of Cas9 mRNA and targeted sgRNA. Given that multiple long RNAs can be packaged together, it is likely that ZNPs will be able to deliver DNA repair templates to mediate HDR gene correction. The use of scalable and translatable technologies, such as ZNPs, will provide powerful tools for in vivo gene editing to understand biology, create animal models, and treat diseases.
Supplementary Material
Acknowledgments
Support from Cancer Prevention and Research Institute of Texas (CPRIT) (R1212, DJS; RP140110, JBM, PK; R1209, HZ), Pollack Foundation (HZ), NIH (1R01CA190525, HZ), Burroughs Welcome (HZ), and Welch Foundation (I-1855, DJS) is acknowledged. We thank Prof. Neal Alto for helpful discussions.
Footnotes
Supporting information for this article can be found under: http://dx.doi.org/10.1002/anie.20X.
Contributor Information
Jason B. Miller, The University of Texas Southwestern Medical Center, Simmons Comprehensive Cancer Center, Department of Biochemistry, Dallas, Texas 75390 (USA).
Shuyuan Zhang, Children’s Research Institute, Departments of Pediatrics and Internal Medicine.
Petra Kos, The University of Texas Southwestern Medical Center, Simmons Comprehensive Cancer Center, Department of Biochemistry, Dallas, Texas 75390 (USA)
Hu Xiong, The University of Texas Southwestern Medical Center, Simmons Comprehensive Cancer Center, Department of Biochemistry, Dallas, Texas 75390 (USA)
Kejin Zhou, The University of Texas Southwestern Medical Center, Simmons Comprehensive Cancer Center, Department of Biochemistry, Dallas, Texas 75390 (USA)
Sofya S. Perelman, Department of Microbiology
Hao Zhu, Children’s Research Institute, Departments of Pediatrics and Internal Medicine.
Daniel J. Siegwart, The University of Texas Southwestern Medical Center, Simmons Comprehensive Cancer Center, Department of Biochemistry, Dallas, Texas 75390 (USA)
References
- 1.a) Jinek M, Chylinski K, Fonfara I, et al. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cong L, Ran FA, Cox D, et al. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mali P, Yang LH, Esvelt KM, et al. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sanchez-Rivera FJ, Jacks T. Nat Rev Cancer. 2015;15:387–395. doi: 10.1038/nrc3950. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sander JD, Joung JK. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Platt RJ, Chen SD, Zhou Y, et al. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R, et al. Nature. 2014;516:428–431. doi: 10.1038/nature13906. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xue W, Chen S, Yin H, et al. Nature. 2014;514:380–384. doi: 10.1038/nature13589. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yin H, Xue W, Chen S, et al. Nat Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chen SD, Sanjana NE, Zheng KJ, et al. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Long CZ, Amoasii L, Mireault AA, et al. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nelson CE, Hakim CH, Ousterout DG, et al. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tabebordbar M, Zhu KX, Cheng JKW, et al. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han X, Liu Z, Jo Mc, et al. Science Adv. 2015;1:e1500454. doi: 10.1126/sciadv.1500454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Zuris JA, Thompson DB, Shu Y, et al. Nat Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang M, Zuris JA, Meng FT, et al. Proc Natl Acad Sci USA. 2016;113:2868–2873. doi: 10.1073/pnas.1520244113. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sun WJ, Ji WY, Hall JM, et al. Angew Chem Int Ed. 2015;54:12029–12033. doi: 10.1002/anie.201506030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanasty R, Dorkin JR, Vegas A, et al. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 7.a) Wittrup A, Ai A, Liu X, et al. Nat Biotechnol. 2015;33:870–976. doi: 10.1038/nbt.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gilleron J, Querbes W, Zeigerer A, et al. Nat Biotechnol. 2013;31:638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 8.Leung AKK, Hafez IM, Baoukina S, et al. J Phys Chem C. 2012;116:22104–22104. [Google Scholar]
- 9.a) Fenton OS, Kauffman KJ, McClellan RL, et al. Adv Mater. 2016;28:2939–2943. doi: 10.1002/adma.201505822. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dong YZ, Dorkin JR, Wang WH, et al. Nano Lett. 2016;16:842–848. doi: 10.1021/acs.nanolett.5b02428. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jarzebinska A, Pasewald T, Lambrecht J, et al. Angew Chem Int Ed. 2016;55:9590–9594. doi: 10.1002/anie.201603648. [DOI] [PubMed] [Google Scholar]; d) Kauffman KJ, Dorkin JR, Yang JH, et al. Nano Lett. 2015;15:7300–7306. doi: 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]; e) Kaczmarek JC, Patel AK, Kauffman KJ, et al. Angew Chem Int Ed. 2016 [Google Scholar]
- 10.a) Venditto VJ, Dolor A, Kohli A, et al. Chem Commun. 2014;50:9109–9111. doi: 10.1039/c4cc02866j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kohli AG, Kierstead PH, Venditto VJ, et al. J Controlled Release. 2014;190:274–287. doi: 10.1016/j.jconrel.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Walsh CL, Nguyen J, Szoka FC. Chem Commun. 2012;48:5575–5577. doi: 10.1039/c2cc31710a. [DOI] [PubMed] [Google Scholar]
- 11.a) Akinc A, Zumbuehl A, Goldberg M, et al. Nat Biotechnol. 2008;26:561–569. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jayaraman M, Ansell SM, Mui BL, et al. Angew Chem Int Ed. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Love K, Mahon K, Levins C, et al. Proc Natl Acad Sci USA. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Semple SC, Akinc A, Chen JX, et al. Nat Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 12.Ran FA, Hsu PD, Wright J, et al. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Hao J, Kos P, Zhou K, et al. J Am Chem Soc. 2015;137:9206–9209. doi: 10.1021/jacs.5b03429. [DOI] [PubMed] [Google Scholar]; b) Zhou K, Nguyen LH, Miller JB, et al. Proc Natl Acad Sci USA. 2016;113:520–525. doi: 10.1073/pnas.1520756113. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yan Y, Liu L, Xiong H, et al. Proc Natl Acad Sci USA. 2016;113:E5702–E5710. doi: 10.1073/pnas.1606886113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guschin DY, Waite AJ, Katibah GE, et al. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 15.Whitehead KA, Matthews J, Chang PH, et al. ACS Nano. 2012;6:6922–6929. doi: 10.1021/nn301922x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Park AI, Mou H, et al. Genome Biol. 2015;16:111. doi: 10.1186/s13059-015-0680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.