

Abstract
Esophageal cancer is the sixth most common cause of cancer-related death worldwide. Current chemotherapy regimens include a combination of 5-fluorouracil (5-FU) and cisplatin, but more efficient therapy strategies are needed to increase 5-year survival. Alterations in the signaling pathway of the tumor suppressor gene Rb-1, which encodes a phosphoprotein (pRB) that negatively regulates the G1/S transition of the cell cycle, are present in 70% of all tumors, but its role in esophageal cancer is still unclear. Most of these are alterations leading to up-regulation of the activity of cyclin-dependent kinases (CDKs) to phosphorylate pRB, which suggests that keeping the wild type pRB phosphorylated might be advantageous. Besides proliferation, pRB also regulates apoptosis induced by tumor necrosis factor-alpha (TNF-α) and DNA-damage. We investigated the status of phosphorylation of pRB along esophageal tumorigenesis stages, as well as whether hyperphosphorylation of pRB could suppress apoptosis induced by cisplatin, 5-FU, or TNF-α in esophageal cancer cells. pRB phosphorylation increased progressively from normal esophageal tissue to metaplasia and adenocarcinoma, suggesting that pRB phosphorylation increases along esophageal tumor stages. When RB-1 was knocked down or CDK inhibitors reduced the levels of phosphorylated pRB, opposite apoptotic effects were observed, depending on the combination of drugs tested: whereas TNF-α- and cisplatin-induced apoptosis increased, 5-FU-induced apoptosis decreased. Taken together, these data suggest that pRB plays a role in esophageal adenocarcinoma and that, depending on the type of anti-cancer treatment, combining CDK inhibitors and chemotherapy has the potential to increase the sensitivity of esophageal cancer cells to cell death.
Introduction
Esophageal cancer is the eighth most common cancer worldwide, as well as the sixth most common cause of cancer-related death [1] due to its late diagnosis. The two main histological types of esophageal cancer are adenocarcinoma and squamous cell carcinoma [2]. Adenocarcinoma tumorigenesis is associated with an inflammatory process related to gastroesophagic reflux disease [3], [4], involving a rise in cytokine tumor necrosis factor-alpha (TNF-α) as the disease progresses from metaplasia (Barrett's esophagus) to adenocarcinoma [5], [6]. Squamous cell carcinoma tumorigenesis is less well defined, but is characterized by an increase in the proliferation of esophageal epithelial cells, followed by a dysplastic alteration, with onset usually related to the consumption of alcohol and smoking [7], [8]. Treatment options for esophageal cancer include surgical resection, chemotherapy, radiotherapy, or a combination of modalities [8]. The current chemotherapy protocol consists of a combination of cisplatin (CIS) and 5-fluorouracil (5-FU), sometimes together with other agents such as irinotecan and oxaliplatin [9], [10], [11]. Unfortunately, apart from therapeutic efforts, esophageal cancer still has an associated average five-year survival of only 18%, highlighting the need for a better understanding of tumor biology and its response to therapy [12].
The retinoblastoma protein (pRB, encoded by the Rb-1 gene, is a tumor suppressor that negatively regulates cell cycle progression, as well as coordinating other physiological processes, such as apoptosis [13], [14], [15], [16], [17], [18]. pRB hyperphosphorylation by cyclin/CDK complexes (mainly cyclin D and CDK 4/6) is a key event for the G1 to S phase transition [19], [20]. Previous studies from our research group and others [21], [22], [23], [24] have demonstrated that pRb has an anti-apoptotic effect in specific conditions: expression of a non-cleavable form of pRb prevented cell death induced by TNF-R1 activation in cultured fibroblasts and by lipopolysaccharide (LPS) and TNF-α exposure in gastrointestinal mucosa of mice [21], [22]. Seventy percent of all tumor types have changes in the pRB pathway, with the nature of the change varying across types [14], [25], [26]. Mutations in Rb-1 occur in childhood retina tumor, in 90% of small cell lung cancers (SCLC), and in 70% of bladder cancers. Conversely, pRB inactivation by hyperphosphorylation, caused by alterations in upstream pathway components (such as the cyclin/CDK complexes), mostly happens in glioblastoma; colon cancer; non-small cell lung cancer (NSCLC); and pancreatic, liver, and breast cancers [25], [27], making CDK inhibition a good target to block proliferation — indeed, several CDK inhibitors are currently in clinical trials [28], [29], [30]. However, the literature on the status of the pRb pathway in esophageal tumors is scarce. A study by Davelaar and colleagues in 2015 showed that adenocarcinoma cell lines and tissue samples have increased pRB phosphorylation at its serine 795 residue compared to normal esophageal tissue samples [31]. This finding suggests that, in esophageal adenocarcinoma, as in colon cancers and glioblastomas, the pRB pathway may be altered by pRB hyperphosphorylation, rather than by the loss of Rb-1. Indeed, cyclin D1 overexpression is a predictor of poor prognosis for esophageal carcinoma patients [32], [33]. Additionally, Sarbia and colleagues showed that loss of pRb expression is not a common event in esophageal adenocarcinoma. Rather, they observed an increase in pRB expression in adenocarcinoma samples compared to samples of esophageal pre-cancer (Barrett's esophagus) [31], [34].
In this work, we investigated whether phosphorylated pRB regulates apoptosis sensitivity in esophageal cancer cells. We pharmacologically manipulated the levels of phosphorylated pRB using CDK inhibitors or reduced pRB levels by using siRNA and assessed the effect of these manipulations on the survival of esophageal cancer cell lines exposed to chemotherapeutic agents 5-FU or cisplatin, or to TNF-α, a cytokine frequently present in the tumor microenvironment. In addition, we evaluated whether pRB expression increases in esophageal metaplasia and adenocarcinoma specimens compared to normal esophageal tissue. Our results suggest that combining CDK inhibitors and currently used chemotherapeutic agents can block proliferation and increase cancer cell death and is a promising strategy to treat esophagus cancer. Importantly, this cell death sensitization is drug combination-specific.
Material and Methods
Human Biopsies
Human biopsies taken from patients treated at Clementino Fraga Filho University Hospital (HUCFF), Rio de Janeiro, Brazil, were used to detect phospho-pRB, in accordance with the Helsinki Declaration of 1975. The analyses were performed in samples from control patients (n = 8), patients with Barrett's esophagus (n = 10), esophageal squamous cells carcinoma (n = 7), and adenocarcinoma (n = 5). The paraffin-embedded tissues were cross-sectioned (5 μm), and the slides were deparaffinized and hydrated. Antigen retrieval was performed by boiling the slides in 10 mM citrate buffer at pH 6.0 for 10 min.
Immunofluorescence Analysis
For the detection of phosphorylated pRB, slides were incubated with anti-phosphorylated RB at Ser807/811 (1:100; Cell Signaling Technology, Danvers, MA, USA). The secondary antibody used was donkey anti-rabbit conjugated to Alexa Fluor® 594 dye (1:500; Invitrogen, Carlsbad, CA, USA). Nuclei were stained with DAPI (49–6-diamidino-2-phenylindole). Slides were mounted using Biomeda Gel Mount aqueous mounting medium. For each sample, the percentage of phospho-pRB-positive cells was calculated by counting at least 500 cells in five representative fields under a Nikon TE300 fluorescence microscope (Nikon, Tokyo, Japan) at 400× magnification. The immunofluorescence assay was performed three times for each sample. For normal esophagus samples, analysis was only performed if the sample included the basal zone with basophilic proliferative epithelial cells.
Cell Culture
We used three commercially available human esophageal cancer cell lines: OE19, OE21, and TE-13 (Sigma-Aldrich, St. Louis, MO, USA). OE19 is derived from a stage III adenocarcinoma of gastric cardia/esophageal gastric junction. OE21 was established from a stage II squamous carcinoma of mid esophagus and TE-13 is also a squamous cell carcinoma cell line. Cells were maintained in RPMI medium (GIBCO; Sigma-Aldrich) supplemented with L-glutamine, 10% fetal bovine serum (FBS), 100 μ/ml penicillin, and 50 mg/ml streptomycin. Cells were kept under 5% CO2 at 37 °C. The cell lines were genetically tested by STR-PCR (the investigated loci were D2S1338, D19S433, CSF1PO, TPOX, TH01, vWA, d16S539, d7s820, d13s317, D5S818, FGA, D3S1358, D18S51, D8S1179, D21S11, and amelogenia) to confirm origin and rule out cross-contamination.
For immunohistochemical analysis and cell death assays, cells were grown in 24-well plates (initial density: 5 × 104 cells/well) for 48 h. For TNF-α treatments, cells were treated with human recombinant TNF-α (10 ng/ml; PeproTech, Rocky Hill, NJ, USA) and the protein synthesis inhibitor cycloheximide (CHX 2.5 μg/ml, Sigma-Aldrich) in the cell culture medium for 8 h. To inhibit CDKs, cells were treated with roscovitine, a CDK 1, 2, 5, and 7 inhibitor [35] (Ros, 20 μM; Sigma-Aldrich) for 12 h or pretreated with Ros for 4 h followed by additional treatment with TNF/CHX for 8 h. For flavopiridol (Flavo, 300 nM; Sigma-Aldrich), a potent CDK 1, 2, 4, and 7 inhibitor [35], the incubation period was 8 h for the experiments involving TNF. The concentration of CDK inhibitors was determined by their effectiveness to reduce pRB phosphorylation in esophageal cell lines (supplementary information, Figure S2A).
Figure S2.

Representative Western blot images of three independent experiments showing the levels of phosphorylated pRB (ppRB SER807/811) in OE-19 and OE-21 cells after treatment with roscovitine (Ros, 20 μM) or flavopiridol (Flavo, 300 nM) compared to the control (CTL) (A), or 24 h after pRB silencing with siRNA (siRB) compared to the transfection with non-targeting RNA (siNeg) (B). α-Tubulin (α-Tub) was used as loading control.
To evaluate chemotherapy resistance, cells were treated with 5-FU (5-fluorouracil, 60 μM) or cisplatin (40 μM) and Ros (20 μM) or flavopiridol (300 nM) for 24 h. No working concentrations of the drugs used in this study induced more than 60% of viability loss in all cell lines tested with MTT assays (Figure S1). The levels of phosphorylated pRB in the esophageal cell lines tested were reduced by 60% after 24 h of Ros treatment and by 90% after flavopiridol treatment (Figure S2A).
Figure S1.

Survival curves (percent cell survival for each drug concentration tested) of OE-21, TE-13, and OE-19 cells exposed to a range of TNF-α (A), 5-FU (B), or cisplatin (CIS) (C) concentrations for 24 h. Cell survival was evaluated by MTT assay. Data represent the mean and standard error of three independent experiments done in triplicates.
Cell Viability
Cell viability of OE-19, OE-21, and TE-13 cell lines was evaluated by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay based on the colorimetric measurement of formazan dye formed from MTT by mitochondrial dehydrogenases. Exponentially growing cells were plated in 96-well plates at a seeding density of 2 × 104 cells/well. After 24 h, cells were treated with increasing concentrations of TNF, 5-FU, or cisplatin for 22 h. MTT (Sigma-Aldrich) was added at a final concentration of 0.5 mg/mL and the plates were incubated at 37 °C for another 2 h. At the end of the incubation period, the medium was removed, and cells were lysed with DMSO. The absorbance of the solubilized product was measured at 570 nm.
RB-1 Knockdown
To silence RB-1 in the cell lines, cells were plated at 1.5 × 104 cells/well in 96-well plates with RPMI medium supplemented with 10% FBS. Next, cells were transfected using RNAiFect (Ambion, Austin, TX, USA) and 30 nM siRNA-RB (sequence GUUGAUAAUGCUAUGUCAA, synthesized by Ambion) according to a previous paper [13]. BLOCK-iT™ Fluorescent Oligo (Thermo Fisher Scientific, Waltham, MA, USA) was used as RNAi Transfection Control. 24 h post-transfection, pRB levels were reduced by 80% to 90% in the esophageal cell lines used, and phosphorylated pRB levels were also reduced (Figure S2B).
Caspase 3/7 Activity Assay
Analysis of cell death by apoptosis was performed using CellEvent® Caspase-3/7 Green Detection Reagent kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. Nuclei were stained with 1 μM Hoechst 33,342 (Sigma-Aldrich) in PBS for 20 min. Cells were plated (1.5 × 104 per well) in 96-well μClear® black Cellstar® plates (Greiner Bio-One, Kremsmünster, Austria). After treatment incubation, cells were imaged (200× magnification, at least 15 fields per sample in triplicates) on an Operetta® High-Content Imaging System (PerkinElmer, Waltham, MA, USA). Nuclei were classified as viable or pyknotic based on nuclei morphology and staining intensity. The percentage of caspase-positive cells in each condition was calculated using Harmony® High-Content Imaging and Analysis Software (PerkinElmer).
Western Blotting
For Western blotting, cells were washed with cold PBS and protein was isolated by extraction in RIPA buffer (50 mM Tris–HCl, pH 7.4; 1% NP-40; 0.25% sodium deoxycholate; 15 mM NaCl; 1 mM EDTA; 1 mM PMSF; 5 μg/ml aprotinin; 5 μg/ml pepstatin; leupeptin 5 μg/ml; 1 mM NaF; and 1 mM Na3VO4). The extracted protein (containing 15–40 μg of protein per sample) was diluted in sample buffer (10% SDS; 10 mM β-mercaptoethanol; 20% glycerol; 0.2 M Tris–HCl, pH 6.8; and 0.05% bromophenol blue) and electrophoresed in polyacrylamide gel (SDS-PAGE) according to our previous paper [13]. The antibodies used were polyclonal anti-RB 851 (1:5000, kindly given by Dr. Jean Y. J. Wang), phospho-RB at Ser807/811 (1:1000, Cell Signaling Technology), and anti-α-tubulin (1:10,000, Sigma-Aldrich). Protein bands were visualized using the West Pico and Femto chemiluminescence system (Thermo Fisher Scientific), and band densities (related to α-tubulin density in each sample) were quantified using Image J software (National Institutes of Health, Bethesda, MD, USA).
Statistical Analysis
The data were analyzed using one-way analysis of variance (ANOVA) and Tukey's post-test and analysis was performed on GraphPad Prism5 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Phosphorylated pRB Levels Correlate with Barrett's Esophagus and Adenocarcinoma
To investigate the importance of the pRB pathway in esophageal cancer, we first evaluated the phosphorylation status of pRB in esophageal cancer samples. Overall, 30 human esophageal samples — eight control samples (no esophageal lesion), 10 Barrett's esophagus samples, five adenocarcinoma samples, and seven esophageal squamous cell carcinoma samples — were analyzed by immunofluorescence, using an antibody that recognizes pRB phosphorylated in the serine 807/811 residues (phospho-pRB). Control samples had 16 ± 1% of phospho-pRB-positive cells, whereas samples from patients with Barrett's esophagus had 53 ± 3%, and samples from esophageal adenocarcinoma patients showed 76 ± 3% of phospho-pRB-positive cells (Figure 1). On the other hand, phospho-pRB staining of the seven samples of squamous cell carcinoma yielded conflicting results, with three negative samples, three positive samples (>80% of positive cells), and one sample with zones with nearly 100% of labeling and others without any labeling.
Figure 1.
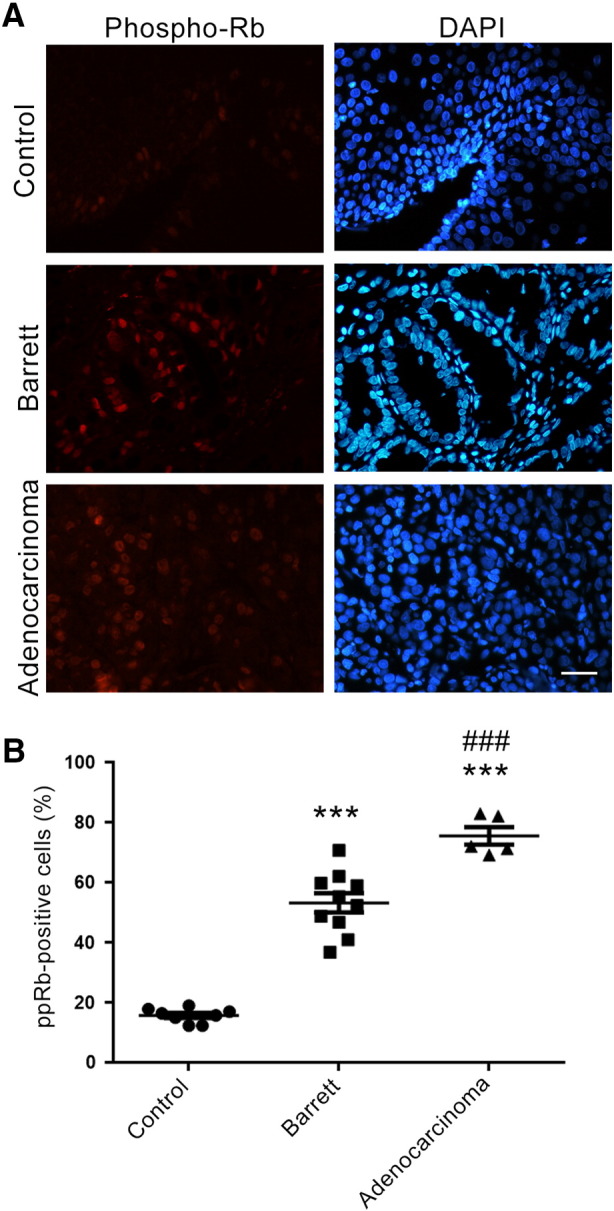
Phospho-pRB immunofluorescence staining in esophageal biopsies.
A. Representative images of pRB phosphorylated at serine 807/811 residues (Phospho-Rb, red) in esophageal biopsy samples: control (healthy esophagus, no lesions), Barret (Barrett's esophagus), and esophageal adenocarcinoma. Nuclei were stained with DAPI (blue). B. Histogram showing the percentage of phospho-pRB-positive cells in control (circles), Barrett's esophagus (squares), and adenocarcinomas (triangles) samples. The histogram shows the average for each group and each point represents an individual case within the groups. ***P < .001 compared to the control group. ###P < .001 compared to the Barrett's esophagus group (ANOVA and Tukey's post-test). Control: n = 8; Barrett's esophagus: n = 10; and adenocarcinoma: n = 5.
Reduction of Phosphorylated pRB Increases Esophageal Cancer Cell Death Induced by TNF-α
We then tested whether pRB hyperphosphorylation can contribute to esophageal cancer resistance to cell death induction by TNF-α. To do that, we compared apoptosis induced by this treatment in cancer cell lines (measured by in vitro caspase 3/7 activity assay and quantification of the number of pyknotic nuclei), in the presence or absence of the pharmacological CDK inhibitors roscovitine (Ros) or flavopiridol (Flavo), which inhibit pRB phosphorylation (Figure S2A), or after silencing pRB through RNA interference (RNAi, Figure S2B). In OE-19 adenocarcinoma cells, siRNA-mediated silencing of pRB increased the proportion of TNF/CHX-induced caspase-positive cells threefold (Figure 2A). TNF/CHX treatments started 24 h post-transfection with siRNA, when pRB levels in the esophageal cell lines had decreased by 80–90% (Figure S2B).
Figure 2.

Effect of pRB manipulation on TNF-α- and TNF-α + cycloheximide-induced apoptosis.
A. Representative images of three independent caspase 3/7 activity experiments with OE-19 cells for each experimental group, acquired on an Operetta® multi-analysis platform (PerkinElmer). Green staining (Caspase) represents active caspase, blue staining (Hoechst) represents nuclei stained with Hoechst. The third row (Merge) shows the superimposition of the Caspase and Hoechst staining. B, C, and D. Histograms showing the percentage of active caspase-positive cells after 8-h treatment with TNF-α (10 mg/mL) combined with cycloheximide (CHX) with or without roscovitine (ROS, 20 μM), flavopiridol (Flavo, 300 nM), or pRB silencing (RNA RB) in OE-19 (B), OE-21 (C), and TE-13 (D) cells. CTL-control untreated, RNA NEG – transfected with negative control siRNA.
Data represent the mean and standard error of three independent experiments. * Indicates comparison to the TNF/CHX group; # indicates comparison to TNF/CHX + RNA-Neg, * indicates comparison to TNF/CHX. *P < .05, **P < .01, ***P < .001, and ##P < .01 by ANOVA and Tukey's post- test.
Pharmacological inhibition of pRB phosphorylation by treatment with Ros or Flavo increased TNF/CHX-induced cell death (percentage of caspase-positive esophageal cancer cells) in the OE19 adenoma cell line three- and eightfold, respectively (Figure 2A, B). In the TE-13 squamous cell carcinoma cell line, pRB inhibition, either through silencing, Ros, or Flavo treatment, also increased TNF/CHX-induced cell death (P < .001 for pRB silencing or Ros pretreatment and P < .001 for Flavo pretreatment; Figure 2D). However, only Flavo increased the percentage of TNF/CHX-induced caspase-positive cells in the OE-21 squamous cell carcinoma cells (Figure 2C).
Because protein synthesis inhibitors are too toxic for use in cancer therapy, we also tested whether CDK inhibitors or pRB silencing could enhance TNF-α-induced cell death without cycloheximide. CDK inhibitors did increase the TNF-α-induced activated caspase 3/7 levels, but the effectiveness of each inhibitor depended on the cell line. In OE-19 Flavo (but not Ros) increased, by twofold, the percentage of caspase-positive cells after TNF-α treatment, which were limited to up to 10%, whereas in OE-21 cells, the percentage of caspase-positive cells after TNF-α increased only after Ros pretreatment (P < .05) (twofold increase). However, inactivation of pRB using siRNA-mediated silencing failed to change the levels of activated caspase 3/7 induced by the TNF-α treatment alone in OE-19 and OE-21. In TE-13 cells, Flavo, Ros, or pRB knockdown induced a small but significant increase in the percentage of caspase-positive cells after TNF-α treatment. The highest increase to 15% of caspase-positive cells (a two-fold increase of the TNF-α -induced cell death) was obtained with Flavo + TNF-α.
Inhibition of pRB Phosphorylation Decreases Esophageal Cancer Cell Death Induced by 5-fluorouracil
Inhibiting pRB decreased cell death induced by 5-fluorouracil (5-FU, a first-line chemotherapy agent for esophageal cancer), on esophageal cancer cell lines (Figure 3). In OE-19 cells, the effect observed was a fourfold decrease in 5-FU-induced cell death, upon pharmacological inhibition of pRB phosphorylation by Ros (P < .001) and after pRB silencing with siRNA (P < .001), whereas Flavo treatment had no effect on 5-FU-induced cell death (Figure 3A). In OE-21 cells (Figure 3B), the percentage of 5-FU-induced caspase-positive cells dropped from 19% to 10% after Ros pretreatment (P < .001), to 8% after pRB silencing (P < .001), whereas Flavo treatment had no effect on 5-FU-induced cell death. In TE-13 cells, 5-FU-induced cell death declined from 22% to 13% after pRB silencing (P < .001), and to 14% after Ros pretreatment (P < .001), with Flavo having no significant effect.
Figure 3.
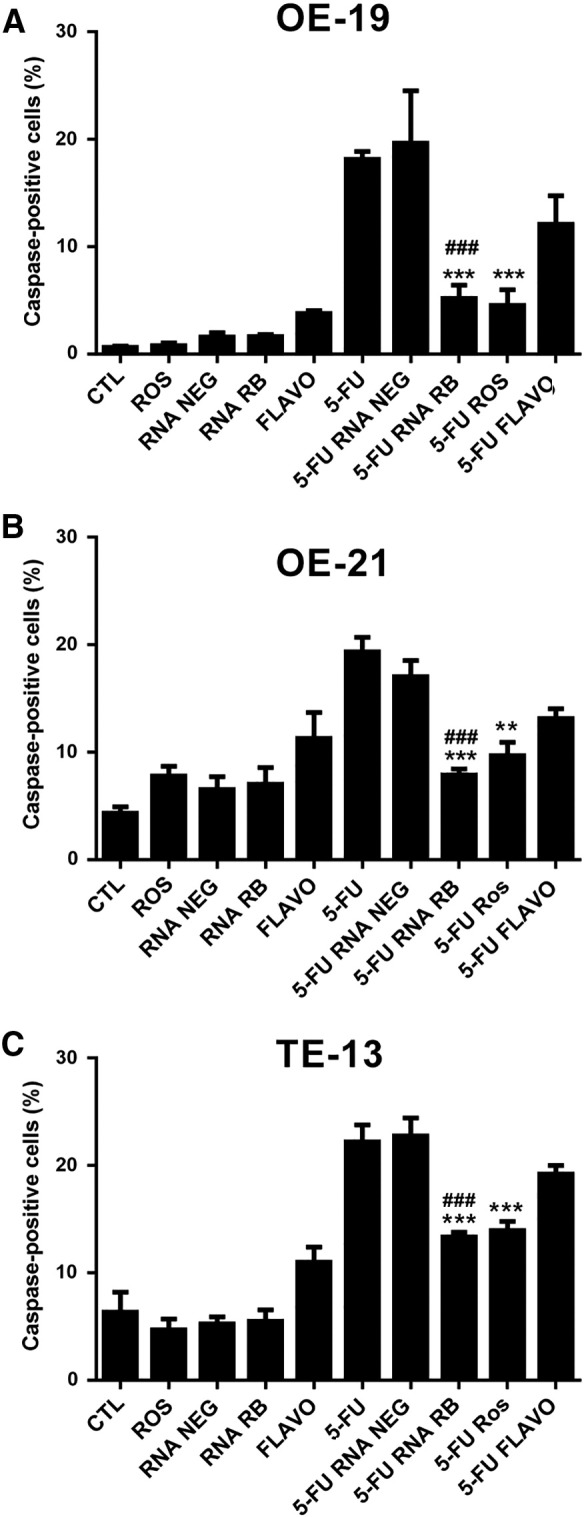
Effect of pRB manipulation on 5-fluorouracil -induced apoptosis.
A–C. Histograms showing the percentage of active caspase-positive cells for OE-19, OE21, and TE-13 cells, respectively, after 24-h treatment with 5-FU (60 μM) with or without roscovitine (ROS, 20 μM), flavopiridol (Flavo, 300 nM), or pRB silencing (RNA RB). CTL-control untreated, RNA NEG – transfected with negative control siRNA. Data represent the mean and standard error of three independent experiments. * Indicates comparison to the 5-FU group; # indicates comparison to the 5-FU + RNA-Neg group. *P < .05, ***P < .001, ##P < .01 and ###P < .001 by ANOVA and Tukey's post-test.
Inhibition of pRB Phosphorylation Increases Esophageal Cancer Cell Death Induced by Cisplatin
pRB inhibition had the opposite effect on the sensitivity of esophageal cancer cells to cisplatin (CIS), compared to 5-FU (Figure 4). In OE-19 cells, pretreatment with Flavo increased fourfold the percentage of caspase-positive cells after cisplatin treatment, although neither Ros nor pRB silencing had any effect (Figure 4A). In OE-21 and TE-13 cells, all forms of pRB inhibition (pRB silencing, Ros or Flavo treatment) resulted in increased CIS-induced caspase activity (Figure 4B and C).
Figure 4.
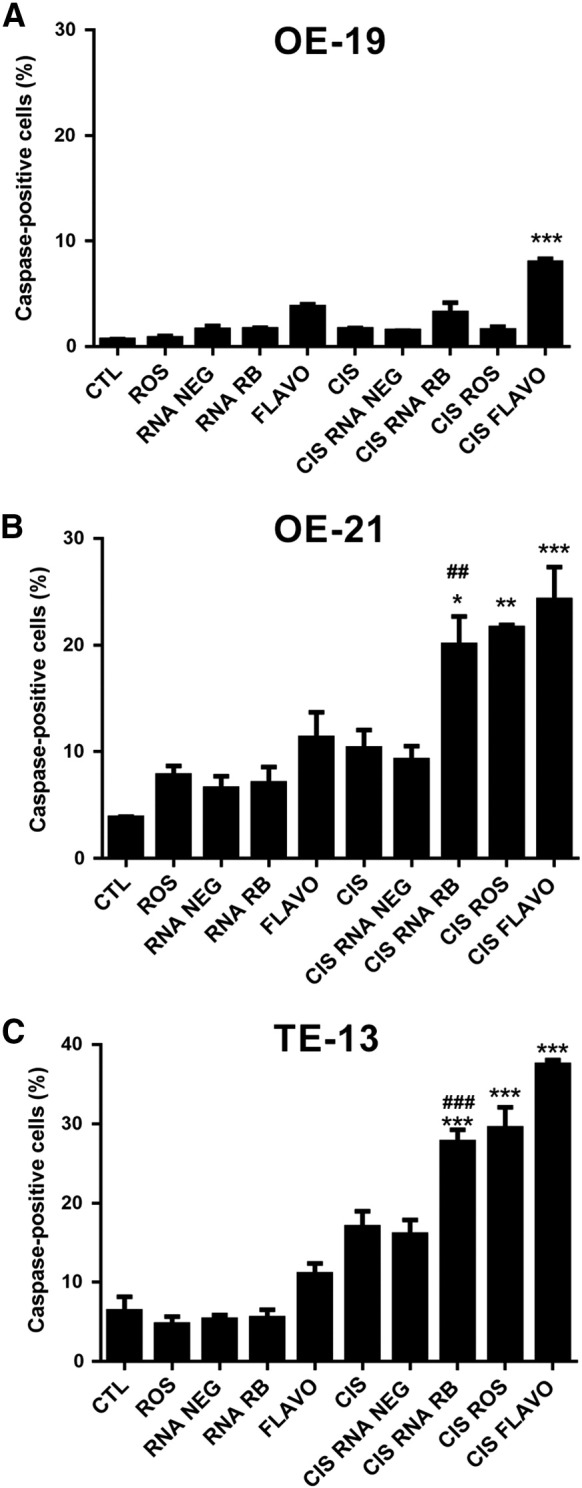
Effect of pRB manipulation on cisplatin-induced apoptosis.
A–C. Histograms showing the percentage of active caspase-positive cells for OE-19, OE21, and TE-13 cells, respectively, after 24-h treatment with cisplatin (CIS, 40 μM) with or without roscovitine (ROS, 20 μM), flavopiridol (Flavo, 300 nM), or pRB silencing (RNA RB). CTL-control untreated, RNA NEG – transfected with negative control siRNA. Data represent the mean and standard error of three independent experiments. * Indicates comparison to the cisplatin (CIS) group; # indicates comparison to the CIS + RNA-Neg group. *P < .05, **P < .01, ***P < .001, ##P < .01, ###P < .01, and by ANOVA and Tukey's post-test.
Table 1 summarizes the effect of different combinations of pRB inactivation methods and chemotherapeutic agents on human esophageal cancer cell death. In cells with low phospho-pRB levels (induced experimentally, either by Ros or Flavo treatment or by pRB silencing), 5-FU induced less cell death, favoring tumor survival. Conversely, treatment of pRB-deficient cells with cisplatin increased cell death, especially in esophageal squamous cell carcinoma cell lines OE-21 and TE-13.
Table 1.
Effect of Combination Drug Therapy on Esophageal Cancer Cell Lines Using Chemotherapeutics and Phospho-pRB Inhibition
| Condition | OE-19 | OE-21 | TE-13 |
|---|---|---|---|
| TNF10 vs. TNF10 + siRNA RB | 0 | 0 | + |
| TNF10 vs. TNF10 + Ros | 0 | + | +++ |
| TNF10 vs. TNF10 + Flavo | +++ | 0 | +++ |
| TNF10 + CHX vs. TNF10 + CHX + siRNA RB | + | 0 | +++ |
| TNF10 + CHX vs. TNF10 + CHX + Ros | + | 0 | +++ |
| TNF10 + CHX vs. TNF10 + CHX + Flavo | +++ | ++ | +++ |
| 5-FU vs. 5-FU + siRNA RB | − | − | − |
| 5-FU vs. 5-FU + Ros | − | − | − |
| 5-FU vs. 5-FU + Flavo | 0 | 0 | 0 |
| CIS vs. CIS + siRNA RB | 0 | + | +++ |
| CIS vs. CIS + Ros | 0 | ++ | +++ |
| CIS vs. CIS + Flavo | +++ | +++ | +++ |
(−) Indicates a reduction in the apoptosis rate: −P < .05, −−P < .01, and −−−P < .001.
(+) Indicates an increase in the apoptosis rate: +P < .05, ++P < .01, and +++P < .001.
(0) Indicates no statistical significance.
One-way ANOVA and Tukey's post-test.
Discussion
In this work, we sought to establish whether inhibition of the pRB pathway, necessary for TNF-α- and DNA-damage-induced cell death [21], [22], [36], could be a target to improve esophageal cancer response to therapy, a question of particular relevance as CDK inhibitors, which prevent pRB phosphorylation, have been used extensively in clinical trials [28], [37], [38], [39].
Firstly, we showed that pRB phosphorylation progressively increases along the stages of esophageal adenocarcinoma tumorigenesis, reaching the highest levels in esophageal adenocarcinomas. This finding contributes to the scarce literature on the status of the pRB pathway in esophageal cancers, indicating that pRB hyperphosphorylation is important in esophageal adenocarcinoma tumor progression [31], [34]. In contrast, our analysis of the pRB phosphorylation status on squamous cell carcinoma yielded no consistent results among the different samples. This fact may be due to an actual biological characteristic of this type of esophageal tumor: esophageal carcinoma tumorigenesis is not as established as adenocarcinomagenesis, and the molecular pathways involved are still unknown. However, the number of available samples was limited, and it remains possible that a statistically significant occurrence of pRB hyperphosphorylation in squamous carcinoma samples could be observed in a larger sample.
We also showed that, in general, down-regulation of pRB phosphorylation by CDK inhibitors, or silencing of pRB with siRNA, sensitizes both adenocarcinoma and squamous cell carcinoma esophageal cell lines to the chemotherapeutic agent cisplatin and to the cytokine TNF-α. However, inhibition of pRB phosphorylation increased the resistance of esophageal cancer cell lines to 5-FU, an anticancer drug used worldwide, regardless of whether the cell death was detected by caspase 3/7 activity (Figure 2, Figure 3, Figure 4) or pyknotic nuclei (data not shown). These results indicate, specifically, that 5-FU treatment may have limited cytotoxic effects in esophageal cancer cells with low pRB levels and, more broadly, that pRB phosphorylation status is critical for chemotherapy-induced cell death in esophageal squamous cell carcinoma and adenocarcinoma, and might be of value for guiding therapeutic interventions.
At least part of the anti-apoptotic role of pRB is attributed to its participation in the crosstalk between apoptosis and autophagy, a molecular recycling process believed to be involved in resistance to chemotherapeutic agents [13], [16], [18]. However, the autophagy induced by 5-FU can function either as a survival or as a death mechanism, apparently depending on the combination of genetic alterations in each cell [40], [41], [42]. We and others have previously shown that pRB regulates the completion of the autophagic process induced by anticancer drugs [13], [18], [21], [43].
Thus, it may be the case that, in the esophageal cell lines analyzed in our study, autophagy works as a death mechanism induced by 5-FU, which would explain the increased cell resistance observed when pRB function was modulated [13]. Additionally, because pRB can have anti- and pro-apoptotic roles depending on the stimulus [13], [18], [21], [22], [44], [45], the inhibition of pRB phosphorylation by CDK inhibitors in our study had different effects on the esophageal cell lines tested for different drug treatments. A recent review by Indovina and co-workers [46] suggested that the role of pRB on chemotherapy treatment outcomes could vary depending on whether the chemotherapeutic agent is cytotoxic or cytostatic. Because pRB functions as a major cell cycle blocker, it is believed to enhance the effects of cytostatic agents. Here the contrasting results obtained after cytotoxic cisplatin or 5-FU treatment showed additional complexity in predicting the outcome of loss of hyperphosphorylated pRB. Accordingly, the phosphorylation resistant form of RB1 causes contrasting effects in response to different apoptotic stimuli [47]. Thus, the use of CDK inhibitors in an attempt to sensitize cancer cells to chemotherapy (by modulating the pRB pathway) need to be evaluated on a case-by-case basis, depending on the chemotherapeutic agent used [46].
TNF-α, applied directly to the tumor, has been used to treat cancer patients. In one study, the injection of human recombinant TNF-α in the tumor cavity after surgical resection of glioblastoma showed no toxicity or major side effects and improved survival time in some patients [48]. In a multicentric phase I clinical trial with esophageal adenocarcinoma patients, intratumoral injections of TNFerade biologic, an adenovirus containing the human TNF-α gene, in combination with radiotherapy and standard chemotherapy (cisplatin and 5-FU), was associated with a median 34-month increase in survival, compared to patients who received radiotherapy and chemotherapy alone [49]. In our study, the combination of pharmacological CDK inhibitors with TNF-α increased the levels of apoptotic cells. These results suggest that pRB phosphorylation protects some esophageal cancer cells against TNF-α-induced cell death and, more importantly, that this combination may have the potential to increase the survival of esophageal cancer patients. It is interesting to point that this effect of the Flavopiridol was particularly significant regardless whether TNF-α treatment was combined with the protein synthesis inhibitor cycloheximide, which blocks the activation of the NF-κB survival pathway.
The two CDK inhibitors used in this study, roscovitine (seliciclib) and flavopiridol (alvocidib), have been tested in several phase I and II clinical trials, both as monotherapy and as combination therapy, for several human cancers [50]. In our hands, flavopiridol and roscovitine had some effects that were somewhat dissimilar to pRB-silencing, inducing apoptosis regardless of pRB inhibition, which indicates that these CDK inhibitors may have additional targets. A recent review on the selectivity of protein kinase inhibitors [35] showed that roscovitine inhibits not only cell cycle-related CDKs but also other kinases such as ERK2 and pyridoxal kinase, an enzyme that participates in the metabolism of vitamin B6, whereas flavopiridol also inhibits GSK-3α, GSK-3β, and RNA synthesis [28], [35], [37], [39]. In addition, flavopiridol can inhibit transcription and sensitize cells [38]. Accordingly, flavopiridol enhanced radiation-induced apoptosis and inhibition of transcription activity in human esophageal adenocarcinoma cells, suggesting that CDK inhibitors could potentially be combined with radiation therapy as radiosensitizers [51]. Recently, more specific CDK-4 and -6 inhibitors such as Palbociclib and Abemaciclib have been tested [30] showing promise as either mono or combined therapy [29], [52], [53]. It would be interesting to test whether interference with CDK 4 and 6 activity alone would sensitize esophageal cells or increase their resistance to 5-FU.
Here, we showed that combining current chemotherapy regimens used to treat esophageal cancer via pRB inhibition with either pharmacological or genetic tools shows promise for improving treatment of this lethal disease. Importantly, the effects observed were due not only to blockade of cell proliferation, but also increased cell death. However, drug combinations must be carefully investigated because of their possible opposite effects. Our findings indicate that modulation of the pRB pathway and/or the use of CDK inhibitors should be considered for optimal and, possibly, tailored treatment of esophageal cancer.
Acknowledgements
We thank the National Council for Scientific and Technological Development (CNPq), Brazilian Cancer Foundation (Oncobiology Program of the UFRJ) and the Rio de Janeiro Research Foundation (FAPERJ) for financial support. We thank Dr. Jean Wang (University of California San Diego) for donating the RB-851 antibody.
The following are the supplementary data related to this article.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.tranon.2017.06.008.
Contributor Information
Rossana C. Soletti, Email: rossanasoletti@gmail.com.
Deborah Biasoli, Email: deborahbiasoli@gmail.com.
Nathassya A.L.V. Rodrigues, Email: nathassya.accioly@gmail.com.
João M.A. Delou, Email: jmdelou@gmail.com.
Renata Maciel, Email: rmmdsantos@gmail.com.
Vera L.A. Chagas, Email: veraachagas@gmail.com.
Rodrigo A.P. Martins, Email: rodrigomartins@ufrj.br.
Stevens K. Rehen, Email: srehen@lance-ufrj.org.
Helena L. Borges, Email: hborges@icb.ufrj.br.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Daly JM, Fry WA, Little AG, Winchester DP, McKee RF, Stewart AK, Fremgen AM. Esophageal cancer: results of an American College of Surgeons Patient Care Evaluation Study. J Am Coll Surg. 2000;190:562–572. doi: 10.1016/s1072-7515(00)00238-6. [discussion 572-563] [DOI] [PubMed] [Google Scholar]
- 3.Chang JT, Katzka DA. Gastroesophageal reflux disease, Barrett esophagus, and esophageal adenocarcinoma. Arch Intern Med. 2004;164:1482–1488. doi: 10.1001/archinte.164.14.1482. [DOI] [PubMed] [Google Scholar]
- 4.Dulai GS, Guha S, Kahn KL, Gornbein J, Weinstein WM. Preoperative prevalence of Barrett's esophagus in esophageal adenocarcinoma: a systematic review. Gastroenterology. 2002;122:26–33. doi: 10.1053/gast.2002.30297. [DOI] [PubMed] [Google Scholar]
- 5.Tselepis C, Perry I, Dawson C, Hardy R, Darnton SJ, McConkey C, Stuart RC, Wright N, Harrison R, Jankowski JA. Tumour necrosis factor-alpha in Barrett's oesophagus: a potential novel mechanism of action. Oncogene. 2002;21:6071–6081. doi: 10.1038/sj.onc.1205731. [DOI] [PubMed] [Google Scholar]
- 6.Solaymani-Dodaran M, Logan RF, West J, Card T, Coupland C. Risk of oesophageal cancer in Barrett's oesophagus and gastro-oesophageal reflux. Gut. 2004;53:1070–1074. doi: 10.1136/gut.2003.028076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 8.Napier KJ, Scheerer M, Misra S. Esophageal cancer: a Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol. 2014;6:112–120. doi: 10.4251/wjgo.v6.i5.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ilson DH, Saltz L, Enzinger P, Huang Y, Kornblith A, Gollub M, O'Reilly E, Schwartz G, DeGroff J, Gonzalez G. Phase II trial of weekly irinotecan plus cisplatin in advanced esophageal cancer. J Clin Oncol. 1999;17:3270–3275. doi: 10.1200/JCO.1999.17.10.3270. [DOI] [PubMed] [Google Scholar]
- 10.Mohammad NH, ter Veer E, Ngai L, Mali R, van Oijen MG, van Laarhoven HW. Optimal first-line chemotherapeutic treatment in patients with locally advanced or metastatic esophagogastric carcinoma: triplet versus doublet chemotherapy: a systematic literature review and meta-analysis. Cancer Metastasis Rev. 2015;34:429–441. doi: 10.1007/s10555-015-9576-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakajima M, Kato H. Treatment options for esophageal squamous cell carcinoma. Expert Opin Pharmacother. 2013;14:1345–1354. doi: 10.1517/14656566.2013.801454. [DOI] [PubMed] [Google Scholar]
- 12.Society AC . 2016. Cancer facts and figures 2016. [Editor (ed)^(eds): City] [Google Scholar]
- 13.Biasoli D, Kahn SA, Cornelio TA, Furtado M, Campanati L, Chneiweiss H, Moura-Neto V, Borges HL. Retinoblastoma protein regulates the crosstalk between autophagy and apoptosis, and favors glioblastoma resistance to etoposide. Cell Death Dis. 2013;4:e767. doi: 10.1038/cddis.2013.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–138. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 16.Delou JM, Biasoli D, Borges HL. The complex link between apoptosis and autophagy: a promising new role for RB. An Acad Bras Ciênc. 2016;88:2257–2275. doi: 10.1590/0001-3765201620160127. [DOI] [PubMed] [Google Scholar]
- 17.Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Sun K, Wang H, Dai Y. Knockdown of retinoblastoma protein may sensitize glioma cells to cisplatin through inhibition of autophagy. Neurosci Lett. 2016;620:137–142. doi: 10.1016/j.neulet.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 20.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 21.Chau BN, Borges HL, Chen TT, Masselli A, Hunton IC, Wang JY. Signal-dependent protection from apoptosis in mice expressing caspase-resistant Rb. Nat Cell Biol. 2002;4:757–765. doi: 10.1038/ncb853. [DOI] [PubMed] [Google Scholar]
- 22.Han J, Soletti RC, Sadarangani A, Sridevi P, Ramirez ME, Eckmann L, Borges HL, Wang JY. Nuclear expression of beta-catenin promotes RB stability and resistance to TNF-induced apoptosis in colon cancer cells. Mol Cancer Res. 2013;11:207–218. doi: 10.1158/1541-7786.MCR-12-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boutillier AL, Trinh E, Loeffler JP. Caspase-dependent cleavage of the retinoblastoma protein is an early step in neuronal apoptosis. Oncogene. 2000;19:2171–2178. doi: 10.1038/sj.onc.1203532. [DOI] [PubMed] [Google Scholar]
- 24.Collard TJ, Urban BC, Patsos HA, Hague A, Townsend PA, Paraskeva C, Williams AC. The retinoblastoma protein (Rb) as an anti-apoptotic factor: expression of Rb is required for the anti-apoptotic function of BAG-1 protein in colorectal tumour cells. Cell Death Dis. 2012;3:e408. doi: 10.1038/cddis.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viatour P, Sage J. Newly identified aspects of tumor suppression by RB. Dis Model Mech. 2011;4:581–585. doi: 10.1242/dmm.008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 28.Cicenas J, Kalyan K, Sorokinas A, Jatulyte A, Valiunas D, Kaupinis A, Valius M. Highlights of the latest advances in research on CDK inhibitors. Cancers (Basel) 2014;6:2224–2242. doi: 10.3390/cancers6042224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 30.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, Torres R, Ajamie RT, Wishart GN, Flack RS. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig New Drugs. 2014;32:825–837. doi: 10.1007/s10637-014-0120-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davelaar AL, Straub D, Parikh KB, Lau L, Fockens P, Krishnadath KK. Increased phosphorylation on residue S795 of the retinoblastoma protein in esophageal adenocarcinoma. Int J Oncol. 2015;47:583–591. doi: 10.3892/ijo.2015.3040. [DOI] [PubMed] [Google Scholar]
- 32.Jiang W, Zhang YJ, Kahn SM, Hollstein MC, Santella RM, Lu SH, Harris CC, Montesano R, Weinstein IB. Altered expression of the cyclin D1 and retinoblastoma genes in human esophageal cancer. Proc Natl Acad Sci U S A. 1993;90:9026–9030. doi: 10.1073/pnas.90.19.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang MT, Chen G, An SJ, Chen ZH, Huang ZM, Xiao P, Ben XS, Xie Z, Chen SL, Luo DL. Prognostic significance of cyclinD1 amplification and the co-alteration of cyclinD1/pRb/ppRb in patients with esophageal squamous cell carcinoma. Dis Esophagus. 2012;25:664–670. doi: 10.1111/j.1442-2050.2011.01291.x. [DOI] [PubMed] [Google Scholar]
- 34.Sarbia M, Tekin U, Zeriouh M, Donner A, Gabbert HE. Expression of the RB protein, allelic imbalance of the RB gene and amplification of the CDK4 gene in metaplasias, dysplasias and carcinomas in Barrett's oesophagus. Anticancer Res. 2001;21:387–392. [PubMed] [Google Scholar]
- 35.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang X, Masselli A, Frisch SM, Hunton IC, Jiang Y, Wang JY. Blockade of tumor necrosis factor-induced Bid cleavage by caspase-resistant Rb. J Biol Chem. 2007;282:29401–29413. doi: 10.1074/jbc.M702261200. [DOI] [PubMed] [Google Scholar]
- 37.Bible KC, Peethambaram PP, Oberg AL, Maples W, Groteluschen DL, Boente M, Burton JK, Gomez Dahl LC, Tibodeau JD, Isham CR. A phase 2 trial of flavopiridol (Alvocidib) and cisplatin in platin-resistant ovarian and primary peritoneal carcinoma: MC0261. Gynecol Oncol. 2012;127:55–62. doi: 10.1016/j.ygyno.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blagosklonny MV. Flavopiridol, an inhibitor of transcription: implications, problems and solutions. Cell Cycle. 2004;3:1537–1542. doi: 10.4161/cc.3.12.1278. [DOI] [PubMed] [Google Scholar]
- 39.Cicenas J, Kalyan K, Sorokinas A, Stankunas E, Levy J, Meskinyte I, Stankevicius V, Kaupinis A, Valius M. Roscovitine in cancer and other diseases. Ann Transl Med. 2015;3:135–147. doi: 10.3978/j.issn.2305-5839.2015.03.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Hou N, Faried A, Tsutsumi S, Kuwano H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur J Cancer. 2010;46:1900–1909. doi: 10.1016/j.ejca.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 41.O'Donovan TR, O'Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7:509–524. doi: 10.4161/auto.7.6.15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong HY, Guo XL, Bu XX, Zhang SS, Ma NN, Song JR, Hu F, Tao SF, Sun K, Li R. Autophagic cell death induced by 5-FU in Bax or PUMA deficient human colon cancer cell. Cancer Lett. 2010;288:68–74. doi: 10.1016/j.canlet.2009.06.039. [DOI] [PubMed] [Google Scholar]
- 43.Jiang H, Martin V, Gomez-Manzano C, Johnson DG, Alonso M, White E, Xu J, McDonnell TJ, Shinojima N, Fueyo J. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010;70:7882–7893. doi: 10.1158/0008-5472.CAN-10-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, Walensky LD, Lees JA. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. 2013;27:1003–1015. doi: 10.1101/gad.211326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, Haigis K, Gulino A, Lees JA. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009;15:184–194. doi: 10.1016/j.ccr.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Indovina P, Pentimalli F, Casini N, Vocca I, Giordano A. RB1 dual role in proliferation and apoptosis: cell fate control and implications for cancer therapy. Oncotarget. 2015;6:17873–17890. doi: 10.18632/oncotarget.4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masselli A, Wang JY. Phosphorylation site mutated RB exerts contrasting effects on apoptotic response to different stimuli. Oncogene. 2006;25:1290–1298. doi: 10.1038/sj.onc.1209161. [DOI] [PubMed] [Google Scholar]
- 48.Oshiro S, Tsugu H, Komatsu F, Ohnishi H, Ueno Y, Sakamoto S, Fukushima T, Soma G. Evaluation of intratumoral administration of tumor necrosis factor-alpha in patients with malignant glioma. Anticancer Res. 2006;26:4027–4032. [PubMed] [Google Scholar]
- 49.Chang KJ, Reid T, Senzer N, Swisher S, Pinto H, Hanna N, Chak A, Soetikno R. Phase I evaluation of TNFerade biologic plus chemoradiotherapy before esophagectomy for locally advanced resectable esophageal cancer. Gastrointest Endosc. 2012;75:1139–1146.e1132. doi: 10.1016/j.gie.2012.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krystof V, Uldrijan S. Cyclin-dependent kinase inhibitors as anticancer drugs. Curr Drug Targets. 2010;11:291–302. doi: 10.2174/138945010790711950. [DOI] [PubMed] [Google Scholar]
- 51.Raju U, Ariga H, Koto M, Lu X, Pickett J, Valdecanas D, Mason KA, Milas L. Improvement of esophageal adenocarcinoma cell and xenograft responses to radiation by targeting cyclin-dependent kinases. Radiother Oncol. 2006;80:185–191. doi: 10.1016/j.radonc.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 52.Amita Patnaik LSR, Tolaney Sara M, Tolcher Anthony W, Goldman Jonathan Wade, Gandhi Leena, Papadopoulos Kyriakos P, Beeram Muralidhar, Rasco Drew Warren, Myrand Scott P, Kulanthaivel Palaniappan. 2014. LY2835219, a novel cell cycle inhibitor selective for CDK4/6, in combination with fulvestrant for patients with hormone receptor positive (HR+) metastatic breast cancer. [Editor (ed)^(eds): City] [Google Scholar]
- 53.Tamura K, Mukai H, Naito Y, Yonemori K, Kodaira M, Tanabe Y, Yamamoto N, Osera S, Sasaki M, Mori Y. Phase I study of palbociclib, a cyclin-dependent kinase 4/6 inhibitor, in Japanese patients. Cancer Sci. 2016;107:755–763. doi: 10.1111/cas.12932. [DOI] [PMC free article] [PubMed] [Google Scholar]