

Graphical Abstract
A family of Shear-thinning Hydrogels for Injectable Encapsulation and Long-term Delivery (SHIELD) has been designed and synthesized with controlled in situ stiffening properties to regulate the stem cell secretome. We demonstrate that SHIELD with an intermediate stiffness (200 – 400 Pa) could significantly promote the angiogenic potential of human adipose-derived stem cells (hASCs).
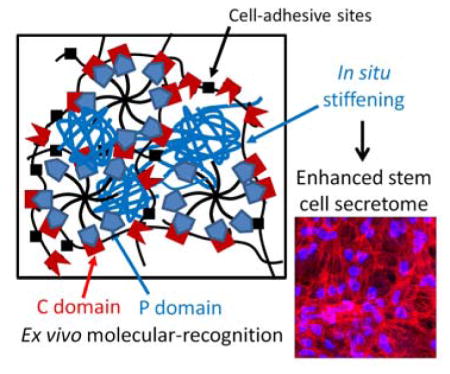
Keywords: Injectable hydrogels, Stem cell secretome, Molecular recognition, Angiogenic function, Reinforcing network
Tissue ischemia, including stroke, ischemic heart diseases, and peripheral arterial diseases, is the leading cause of human morbidity and mortality globally.[1] The transplantation of autologous cells into the ischemic tissue of patients has emerged as a novel therapeutic strategy to induce angiogenesis and blood reperfusion.[1] Adipose-derived stem cells (ASC) and mesenchymal stem cells (MSCs) are among the most promising cell types for treatment, with over 20 registered clinical trials.[2, 3] While the mechanism behind the therapeutic efficacy of the cells is somewhat contentious, the secretion of paracrine and trophic factors has been shown to play a dominant role.[4] Alternatively, researchers have been directly delivering growth factors to the site of injury;[5] however, stem cells serve as a long-term reservoir of a myriad of important growth factors critical for tissue regeneration. To improve this stem cell secretome, research efforts have explored multiple strategies, including cytokine or hypoxic preconditioning and genetic manipulations.[6] A relatively unexplored strategy has been modulation of the cellular microenvironment to improve the stem cell secretome. While several studies have evaluated the role of the cellular microenvironment in guiding stem cell differentiation,[7] much less attention has been given to monitoring of the secretome. Furthermore, the studies to date have been performed on 2D substrates or have used materials with limited clinical potential.[8, 9, 10] Nonetheless, these reports support the hypothesis that cellular microenvironment can dramatically impact the stem cell secretome. For example, in vitro 2D experiments have shown that increased substrate stiffness could significantly increase the interleukin-8 (IL-8) expression of MSCs cultured on polyacrylamide gels.[10] In addition, conditioned media from MSCs attached to a 2D polyacrylamide gel was found to enhance tubulogenesis of human microvascular endothelial cells (hMVECs).[8] Furthermore, ASCs encapsulated within 3D polyethylene glycol/hyaluronic acid (PEG/HA) hydrogels showed significantly higher secretion of vascular endothelial growth factor (VEGF) and placenta-derived growth factor (PDGF) compared to cells cultured on top of 2D hydrogels.[11] Based on these observations, we hypothesized that tuning the stiffness of 3D injectable hydrogels may be used to modulate the secretion of pro-angiogenic factors by ASCs.
To evaluate this hypothesis, we formulated a series of gels with shear moduli (G′) spanning ~10 – 1000 Pa. We previously reported the design of an injectable hydrogel (G′ ~10–100 Pa) that utilized two different physical crosslinking mechanisms to protect ASCs during the injection process and to enhance cell transplantation efficiency in vivo.[12–14] The viability and retention of transplanted ASCs at the injection site were found to be significantly enhanced up to two weeks post-injection compared to delivery of cells in saline or single-network hydrogel controls.[12] In this study, we further expanded this family of dual-network hydrogels to extend the range of achievable stiffness. Specifically, we created a series of injectable hydrogels termed SHIELD (Shear-thinning Hydrogels for Injectable Encapsulation and Long-term Delivery) with two stages of gelation: 1) weak gelation ex situ between a star-shaped peptide-polyethylene glycol (PEG) copolymer assembling with an engineered recombinant protein (C7); and 2) stronger gelation in situ via a thermal phase transition of a thermoresponsive component to form a reinforcing network (Figure 1a). The in situ mechanical properties were tuned by varying the molecular weight (MW) and weight percentage (wt%) of the thermoresponsive polymer poly(N-isopropylacrylamide) (PNIPAM). The MW of PNIPAM was precisely controlled using Reversible Addition-Fragmentation chain Transfer (RAFT) polymerization. Human ASCs were cultured within these hydrogels and assessed for proliferation, spreading, and pro-angiogenic factor secretion at both the genetic and protein levels. Finally, the biofunctionality of the secreted factors was quantified using hMVECs.
Figure 1.

Schematic and mechanical properties of Shear-thinning Hydrogel for Injectable Encapsulation and Long-term Delivery (SHIELD). (a) Schematic of SHIELD ex vivo and in situ network formation. (b) Shear storage moduli (G′) of all SHIELD formulations at 25 and 37 °C at 1 Hz. * p < 0.05, n ≥ 3. (c) Stress relaxation and the time constant of relaxation (λ) for all SHIELD formulations.
To induce distinct stiffening properties in situ, PNIPAM thiols with two different MWs, 11 and 30 kDa, were synthesized via RAFT polymerization to achieve uniform chain lengths (polydispersity indices of 1.1 for both samples). These two PNIPAM thiols were conjugated to one or two arms of an 8-arm PEG (Table in Figure 1). The remaining arms of the PEG-PNIPAM copolymer were conjugated with the P domain peptide (Figure 1a). The resulting peptide-PEG-PNIPAM was mixed with C7 engineered protein. All formulations had a final 10 wt% polymer composition and a C:P domain ratio of 1:1. Within seconds, all mixtures formed a weak, physical network ex vivo due to specific and reversible peptide binding between C and P domains. Further increasing the temperature to 37 °C led to the formation of a reinforcing network of the peptide-PEG-PNIPAM copolymer with controlled stiffness. Shear moduli (G′) of various SHIELD formulations were determined using dynamic oscillatory rheology (Figure 1b). At room temperature, all SHIELD formulations have G′ ~10 – 50 Pa, which enables easy hand injection. Furthermore, this ex vivo stiffness range is hypothesized to enhance transplanted cell viability, as previous studies demonstrated that shear-thinning hydrogels with G′ less than 50 Pa could protect cells from membrane damage during the syringe injection process.[15] At body temperature, the stiffness of SHIELD-0 with 0 wt% thermoresponsive PNIPAM remained constant as expected. In clear contrast, the G′ of SHIELD-1 (with 1 wt% PNIPAM) increased ten-fold to ~100 Pa due to the formation of the secondary PNIPAM network. As the PNIPAM wt% was further increased, this thermo-stiffening effect was significantly increased with G′ up to ~1,000 Pa for SHIELD-4 (Figure 1b). Stress relaxation behavior was also altered by the secondary PNIPAM network. Upon application of 100% strain, the time constant of relaxation (λ) increased from 5 s for SHIELD-0 to 19 s for SHIELD-4 (Figure 1c). This range of λ mimics the two relaxation modes of muscle, which exhibits a fast λ varying from 0.03 s to 8.4 s and a slow λ varying from 2.2 s to 93.8 s.[16] Several reports have recently demonstrated that in addition to initial substrate stiffness, stress relaxation behavior may influence many cellular functions.[17] Therefore, for new biomaterials formulations, it is critically important to characterize both the initial substrate stiffness as well as time-dependent rheological behavior.
Human ASCs (hASCs) possess enormous potential for multiple regenerative medicine therapies and are isolated through voluntary lipoaspiration of adult fat tissue to circumvent procurement and ethical concerns.[2, 18] hASC proliferation within a 3D hydrogel was evaluated by encapsulating cells within SHIELD and ejecting through a 28-gauge needle into a circular mold (4 mm diameter) at a flow rate of 1 mL/min. The hASCs within SHIELD were then brought to physiological temperature to induce formation of the secondary reinforcing network and analyzed for cell proliferation at several time points up to 2 weeks (Figure 2a). hASCs were found to remain proliferative within hydrogels from SHIELD-0 to SHIELD-2.5. In these hydrogels, the cells exhibited well-spread cytoskeletal morphologies with distinct actin filament networks (Figure 2a,b). Quantification of cell number suggests that cell proliferation within SHIELD-1.25 and SHIELD-2.5 is significantly higher than other SHIELD formulations. Interestingly, SHIELD-4 did not support cell proliferation and spreading, possibly due to the presence of a dense secondary PNIPAM network.
Figure 2.

hASC proliferation and angiogenic function within SHIELD. (a) Cell number within SHIELD at days 1, 3, 7, and 14 post-injection. (b) Confocal 3D projection images of hASCs cultured within SHIELD stained with DAPI (blue) for cell nuclei and rhodamine phalloidin (red) for F-actin cytoskeleton at day 14 post-injection. (c) Relative mRNA expression of hASCs within SHIELD at day 14 post-injection. (d) Relative growth factor concentration of collected media conditioned by hASCs within SHIELD at day 14 post-injection. * p < 0.05, n = 4.
A key role of hASCs in functional recovery has been hypothesized to be the secretion of paracrine factors that promote endogenous cell function. Therefore, we next evaluated the level of gene expression and secreted paracrine factors that are known to influence angiogenesis. We selected several important angiogenic factors including angiopoietin (ANG), fibroblast growth factor-2 (FGF-2), hepatocyte growth factor (HGF), PDGF, VEGF-A, and VEGF-C. Real-time PCR results indicate that the gene expression levels of these factors at days 7 and 14 post-injection are modulated by different SHIELD formulations, with the intermediate SHIELD-1.25 and SHIELD-2.5 hydrogels supporting significantly higher gene expression (Figure 2c, S2, Supporting Information). This finding is consistent with previous 2D experiments using MSCs cultured on polyacrylamide gels that demonstrated that matrix mechanical properties could influence the stem cell secretome.[8] To confirm the translation of gene expression to protein expression, semi-quantitative ELISA-based microarrays were conducted to measure the secretion of these angiogenic factors over 14 days post-injection (Figure 2d). We found that the secretion of these factors is consistent with gene expression data. Using quantitative VEGF-ELISA analysis, we also found that the amount of VEGF secreted from hASCs within SHIELD followed the same trends (Figure S3, Supporting Information), indicating that SHIELD with appropriate secondary network formation could promote the angiogenic potential of hASCs.
To evaluate whether the enhanced secretome of hASCs could lead to better angiogenic function, the bioactivity of the secreted factors was quantified using hMVECs. Conditioned media were prepared using hASCs cultured within the two best performing SHIELD formulations (SHIELD-1.25 and SHIELD-2.5) and SHIELD-0 for comparison. In addition, two positive control groups used media supplemented wtih VEGF at 5 or 100 ng/mL, while basal media only served as a negative control. This hASC-conditioned media was then delivered to hMVECs within a Matrigel sandwich to evaluate the effects on network formation. This protocol is a common in vitro measure of angiogenic-like behavior.[19] As expected, the hMVECs treated with unconditioned basal media showed minimal network formation, while hMVECs treated with a high concentration of VEGF (100 ng/mL) exhibited branching and looping morphologies (Figure 3a). The conditioned media prepared using hASCs within all SHIELD formulations supported formation of significantly more network junctions and loops than basal media, confirming bioactivity of the secreted factors (Figure 3b,c,d). When comparing among the different SHIELD formulations, hASC-conditioned media from SHIELD-1.25 and SHIELD-2.5 resulted in significantly improved network formation over SHIELD-0. Interestingly, these two SHIELD formulations also supported better network formation than the basal media supplemented with 5 ng/mL VEGF. Based on our VEGF-ELISA analysis (Figure S3, Supporting Information), we estimate that the SHIELD-1.25 and SHIELD-2.5 conditioned media contained less than 5 ng/mL VEGF. Thus, taken together, these data indicate that the stem cell secretome could offer more effective treatment due to the presence of a myriad of growth factors compared to the VEGF alone. These data are consistent with the gene and protein expression data, suggesting that enhanced secretome led to better angiogenic function of hMVECs. These data are also consistent with the literature that suggests a cocktail of growth factors is required for effective network formation.[20]
Figure 3.

(a) Confocal 3D projection images of hMVECs within Matrigel sandwiches stained with DAPI (blue) for cell nuclei and rhodamine phalloidin (red) for F-actin cytoskeleton after 2 days exposure to various media treatments. Quantification of (b) number of junctions, (c) total network length, and (d) loop perimeter per image (733 μm × 733 μm). * p < 0.05, n = 5.
We have demonstrated that an injectable hydrogel with controlled in situ network formation can be used to encapsulate hASCs within a 3D microenvironment and regulate their secretome, which has been shown to play an important role in tissue regeneration. The enhanced hASC secretome may serve as a long-term reservoir of crucial trophic factors to promote host cell migration, proliferation, differentiation, angiogenesis, and immunomodulation to foster tissue regeneration.[6, 21] As an example, the cell secretome offers an array of potential therapeutic mechanisms for cardiovascular repair, including tissue preservation, neovascularization, cardiac remodeling, anti-inflammatory responses, and potential endogenous regeneration.[6] Here we have identified two hydrogel formulations, SHIELD-1.25 and SHIELD-2.5, which support cell survival, proliferation, and pro-angiogenic secretion for at least two weeks. Therefore, SHIELD has great potential to be used as a cell delivery vehicle that provides a dynamic, physical microenvironment to support long-term cell survival and angiogenic function. Ongoing work is exploring the application of SHIELD in cell transplantation using different tissue ischemia disease models, including peripheral arterial disease and myocardial infarction. Specifically, to demonstrate SHIELD injectability into cardiac tissue, a preliminary study was performed to evaluate material delivery and retention in healthy rats (n = 3 per treatment group, Fig. S6a, Supporting Information). Even the stiffest SHIELD formulation tested, SHIELD-2.5, was successfully and safely delivered to the myocardium with a 28-gauge needle. Consistent with our previous results,[12, 13, 22] SHIELD stiffness had an inverse correlation with biodegradation rate. At 16 days post-transplantation, the retention of SHIELD-0 was 40% compared to that of SHIELD-2.5 (Fig. S6b, Supporting Information). Current work is underway to evaluate the potential in vivo functionality of these materials to facilitate cell transplantation for the treatment of ischemic cardiomyopathy.
In summary, we have developed a series of injectable, physically-crosslinked hydrogels with controlled stiffening properties to regulate the stem cell secretome in situ. We demonstrated that SHIELD with an intermediate stiffness range (200 – 400 Pa) could significantly promote angiogenic potential of hASCs, thereby enhancing therapeutic efficacy and minimizing the number of transplanted cells required for cell-based regenerative medicine therapies.
Experimental Section
Material synthesis
8-arm polyethylene glycol vinyl sulfone (8-arm PEG-VS) with nominal molecular weights of 20,000 g/mol were purchased from Nanocs (Boston, MA). P domain peptide (EYPPYPPPPYPSGC, 1563 g/mol) was purchased through custom peptide synthesis from Genscript Corp (Piscataway, NJ). All other chemicals were purchased from Sigma-Aldrich (Milwaukee, WI) unless otherwise noted. PNIPAM endcapped with a thiol group (PNIPAM-SH) was synthesized using Reversible Addition-Fragmentation chain Transfer (RAFT) polymerization via two steps.[23] In the first step, N-isopropylacrylamide, methyl-2-(n-butyltrithiocarbonyl) propanoate as a RAFT agent, and azobisisobutyronitrile (AIBN) as an initiator were added to anhydrous N,N-dimethylformamide (DMF). The mixture was deoxygenated, and the reaction was carried out at 70 °C for 5 h. Then the PNIPAM-chain transfer agent (PNIPAM-CTA) polymer was purified by precipitation in diethyl ether and dried under vacuum. The second step of aminolysis of PNIPAM-CTA was conducted by adding PNIPAM-CTA, dimethylphenyl phosphine (DMPP), and n-hexylamine to tetrahydrofuran (THF). The mixture was stirred at ambient temperature for 2 h. PNIPAM-SH was precipitated in diethyl ether and dried under vacuum. Two molecular weights of PNIPAM-SH were synthesized and characterized by gel permeation chromatography (GPC) at room temperature in tetrahydrofuran (THF) as the eluent at a flow rate of 1.0 mL/min using a Viscotek chromatograph and a Viscotek S3580 refractive index detector (Houston, TX) and standard monodisperse polystyrenes for calibration. The two PNIPAM-SH have weight-average molecular weights (Mw) of 11,400 and 30,500 g/mol and polydispersity indices (PDI) of 1.1 (Figure S4, Supporting Information). The amount of thiol end-group was also quantified using Ellman’s reagent (ThermoFisher). A Michael-type addition of the two PNIPAM-SH to 8-arm PEG-VS were conducted at a thiol:VS ratio of 1:8 or 1:4 in the presence of triethanolamine buffer (pH 8.0) and tris(2-carboxyethyl)phosphine (TCEP) at 25 °C for 4 h. The rest of the unreacted arms of PEG-VS were further reacted with excess P domain peptide for 24 h. Peptide-PEG-PNIPAM copolymers were dialyzed (Molecular Weight Cut-Off = 30,000 g/mol) against deionized water to remove unreacted peptide and then lyophilized. For comparison, peptide-PEG copolymer was synthesized by reacting 8-arm PEG-VS with excess P domain peptide and purified as described above. The chemical structure was confirmed by 1H NMR spectrometry, acquired on a Varian Inova 500 MHz NMR spectrometer using deuterium oxide as a solvent (Figure S5, Supporting Information). 1H NMR (500 MHz, D2O) for peptide-PEG-PNIPAM: δ = 7.0, 6.7 (tyrosine aromatic protons), 3.6 (PEG backbone protons), 1.8, 1.4 (PNIPAM backbone protons), 1.0 (s, -C(CH3)2); peptide-PEG: δ = 7.0, 6.7 (tyrosine aromatic protons), 3.6 (PEG backbone protons).
The C7 recombinant protein polymer was cloned, synthesized, and purified as reported previously.[17] Briefly, the DNA sequence encoding the C7 linear protein block copolymer was cloned into the pET-15b vector (Novagen) and transformed into the BL21(DE3)pLysS Escherichia coli host strain (Life Technologies). The protein was expressed following isopropyl β-D-1-thiogalactopyranoside (IPTG) induction, purified by affinity chromatography via the specific binding of N-terminal polyhistidine tag to Ni-nitrilotriacetic acid resin (Qiagen), dialyzed against phosphate-buffered saline (PBS), and concentrated by diafiltration across Amicon Ultracel filter units (Millipore).
Hydrogel preparation
Each WW domain in C7 was treated as one C unit, and each pendant P domain peptide group in the peptide-PEG-PNIPAM copolymer was treated as one P unit. All SHIELD formulations were designed to have a final C:P ratio of 1:1 and a 10% w/v of total polymer in PBS. Weight percentage of the PNIPAM component was used to name five SHIELD formulations from SHIELD-0 to SHIELD-4, with 0 wt% to 4 wt% PNIPAM moiety, respectively (Figure 1a). SHIELD-0 was formed by mixing C7 and peptide-PEG copolymer. SHIELD-1, SHIELD-2.5, and SHIELD-4 were formed by mixing C7 with the appropriate peptide-PEG-PNIPAM copolymer (see table in Figure 1a). SHIELD-1.25 was formed by mixing C7 with a blend of peptide-PEG and peptide-PEG-PNIPAM at a 1:1 ratio.
Rheological characterization
Dynamic oscillatory rheology experiments were performed on a stress-controlled rheometer (AR-G2, TA Instrument) using a 20-mm diameter cone-plate geometry (n ≥ 3). Samples were loaded immediately onto the rheometer after mixing and a humidity chamber was secured in place to prevent dehydration. Frequency sweeps from 0.1 – 20 Hz at 25 °C and 37 °C were performed at 5% constant strain to obtain storage moduli (G′) and loss moduli (G″). Stress relaxation experiments were performed at 100% strain at 37 °C. Time constant of relaxation (λ) was calculated based on a standard Maxwell model for viscoelastic fluids.
In vitro cell proliferation within 3D hydrogels
hASCs were obtained from de-identified human lipoaspirate from the flank and thigh regions by suction assisted liposuction. All tissue donors responded to an Informed Consent approved by the Stanford Institutional Review Board. hASCs were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 IU/ml penicillin/streptomycin at 37 °C and 5% atmospheric CO2. Cells were expanded and passaged by trypsinization for subsequent use. In vitro proliferation experiments were performed with 30-μl gel volume containing 5 x 104 cells. Cell suspension (5 μl) was first mixed with C7 (10% w/v in PBS) before further mixing with the peptide-PEG-PNIPAM copolymer solution (20% w/v in PBS). The volumes of C7 and peptide-PEG-PNIPAM copolymer solution were adjusted to achieve a final C:P ratio of 1:1 at a total cell-laden hydrogel concentration of 10% w/v. The final mixing step with peptide-PEG-PNIPAM was performed in the barrel of a 1-mL insulin syringe fitted with a 28 G needle for cell injection. The mixture was allowed to gel for 5 min before injecting into a circular silicone mold (diameter = 4 mm, height = 2.5 mm) within a 24-well plate using a syringe pump (SP220I; World Precision Instruments) at a flow rate of 1 mL/min. Cell number was determined using PrestoBlue assay (ThermoFisher) at days 1, 3, 7, and 14 post-injection (n = 5), according to manufacturer’s instructions. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 solution in PBS, and stained with rhodamine phalloidin (1:300, Life Technologies) and 4′,6-diamidino-2-phenylindole (DAPI, 1 μg/mL, Life Technologies). Images were collected using a confocal microscope (Leica SPE) by creating z-stacks of greater than 200-μm depth with 2.4-μm intervals between slices in the middle of the hydrogel and then compressing into a maximum projection.
Angiogenic gene expression and growth factor secretion
hASCs were injected into molds and cultured within 30-μl gel volume of various SHIELD formulations containing 5 x 105 cells for 7 and 14 days. Total RNA was isolated using a TRIzol® Reagent (Ambion) with a Phase Lock Gel (5 PRIME) according to the manufacturer’s protocol. The amount of the total RNA from each sample was quantified using a Nanodrop1000 spectrophotometer (Thermo Scientific). Reverse transcription of isolated RNA was then performed using a T100 Thermal Cycler (Bio-Rad) according to the manufacturer’s protocol. Using serial dilutions of cDNA, qPCR was performed for several angiogenic genes including ANG, FGF-2, HGF, PDGF, VEGFA, and VEGFC (see Table S1 for primer sequences). Amplification reactions were performed in a total volume of 15 μl PCR mixture containing 7.5 μl Fast SYBR Green Master Mix™ (Applied Biosystems), 0.45 μM of each PCR primer, 1.6 μl dH2O and 5 μl cDNA samples. After initial denaturation at 95 °C for 10 s, targets were amplified using 40 cycles of 95 °C, 10 s, and then 60 °C, 30 s (StepOnePlusTM Real-Time PCR Machine, Applied Biosystems). All samples were analyzed in triplicate. Relative mRNA level was determined by the 2−ΔΔCt method, with 18S used as the endogenous control. The quantification of angiogenic growth factor secretion was conducted using the conditioned hASC media collected every 3–4 days for 14 days post-injection. The growth factor concentration of the total cell media was quantified using ELISA-based Human Angiogenesis Antibody Array and the Quantikine Human VEGF Immunoassay (R&D systems) according to the manufacturer’s protocol.
In vitro functional assay
Conditioned basal media was prepared by culturing 5 x 105 hASCs within 30-μl gel volume of various SHIELD formulations in EBM-2 basal media (Lonza) for two days. In parallel, human microvascular endothelial cells (hMVECs) were cultured in complete growth media (EGM-2 BulletKit, Lonza). Bioactivity of the hASC-conditioned medium was assessed using a traditional Matrigel sandwich assay.[19, 24] In brief, hMVECs (1 x 104 cells) were seeded on top of growth factor reduced Matrigel (BD Biosciences, 200 μl in a 48-well), allowed to attach for 4 h, and covered with another layer of Matrigel (200 μl) to create a Matrigel sandwich. The Matrigel sandwich was immersed in unconditioned basal media or hASC-conditioned media. Following incubation at 37 °C for two days, the Matrigel sandwich was fixed in 4% paraformaldehyde and 2% glutaraldehyde, and permeabilized with 0.25% Triton X-100 solution in PBS. The cells were stained with rhodamine phalloidin and DAPI, and analyzed using confocal microscropy as described above. Number of junctions, total tubule length, and loop perimeter were quantified for each image using ImageJ software.[19, 24]
Intramyocardial delivery of SHIELD
All experiments followed protocols approved by the Stanford Administrative Panel on Laboratory Animal Care. NIH guidelines for the care and use of laboratory animals (8th edition, revised 2011) were observed. For in vivo delivery of SHIELD, male adult Wistar rats (Charles River Laboratories) were anesthetized with isoflurane and a left fourth interspace thoracotomy was performed.[25] Fifty μL of SHIELD conjugated with a Cyanine5.5 near-infrared dye (Lumiprobe) was epicardially injected into the anterolateral wall of the left ventricle. SHIELD retention was quantified at 16 days post-injection from explanted hearts (n = 3 per treatment group).
Statistical analysis
All data are presented as mean ± standard deviation. Statistical comparisons were performed by one-way analysis of variance (ANOVA) with Tukey post-hoc test. Values were considered to be significantly different when the p value was <0.05.
Supplementary Material
Acknowledgments
We acknowledge support from NIH (F32HL128094-01 to L.C. and R21-EB020235 to S.C.H) and Stanford Neuroscience Institute (Interdisciplinary Scholar Awards to L.C.), Stanford Bio-X (IIP-7-75), CIRM (RT3-07948), and Coulter Foundation (CP-2014-4). The authors thank Xiangyi Zhang and Prof. Robert Waymouth for use of GPC, and Andreina Parisi Amon, Allison Nauta, Benjamin Levi, and Prof. Michael Longaker for hASC isolation.
Footnotes
Supporting Information is available from the Wiley Online Library.
Contributor Information
Dr. Lei Cai, Department of Materials Science and Engineering, Stanford Neuroscience Institute, Stanford University, Stanford, CA 94305, USA
Ruby E. Dewi, Department of Materials Science and Engineering, Stanford University, Stanford, CA 94305, USA
Andrew B. Goldstone, Department of Cardiothoracic Surgery, Department of Bioengineering, Stanford University, Stanford, CA 94305, USA
Jeffrey E. Cohen, Department of Cardiothoracic Surgery, Department of Bioengineering, Stanford University, Stanford, CA 94305, USA
Amanda N. Steele, Department of Cardiothoracic Surgery, Department of Bioengineering, Stanford University, Stanford, CA 94305, USA
Prof. Y. Joseph Woo, Department of Cardiothoracic Surgery, Department of Bioengineering, Stanford University, Stanford, CA 94305, USA
Prof. Sarah C. Heilshorn, Department of Materials Science and Engineering, Department of Bioengineering, Stanford University, Stanford, CA 94305, USA
References
- 1.Le Huu A, Paul A, Xu L, Prakash S, Shum-Tim D. Ther Deliv. 2013;4:503. doi: 10.4155/tde.13.13. [DOI] [PubMed] [Google Scholar]; Blanco M, Castillo J. Nature reviews Neurology. 2013;9:68. doi: 10.1038/nrneurol.2012.274. [DOI] [PubMed] [Google Scholar]; Annex BH. Nat Rev Cardiol. 2013;10:387. doi: 10.1038/nrcardio.2013.70. [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Qin F, Ge M, Shu Q, Xu J. J Cardiovasc Transl Res. 2014;7:651. doi: 10.1007/s12265-014-9585-1. [DOI] [PubMed] [Google Scholar]
- 3.Kim N, Cho SG. The Korean journal of internal medicine. 2013;28:387. doi: 10.3904/kjim.2013.28.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ascheim DD, Gelijns AC, Goldstein D, Moye LA, Smedira N, Lee S, Klodell CT, Szady A, Parides MK, Jeffries NO, Skerrett D, Taylor DA, Rame JE, Milano C, Rogers JG, Lynch J, Dewey T, Eichhorn E, Sun B, Feldman D, Simari R, O’Gara PT, Taddei-Peters WC, Miller MA, Naka Y, Bagiella E, Rose EA, Woo YJ. Circulation. 2014;129:2287. doi: 10.1161/CIRCULATIONAHA.113.007412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beeson W, Woods E, Agha R. Facial plastic surgery : FPS. 2011;27:378. doi: 10.1055/s-0031-1283056. [DOI] [PubMed] [Google Scholar]; Wei X, Yang X, Han Z-p, Qu F-f, Shao L, Shi Y-f. Acta Pharmacol Sin. 2013;34:747. doi: 10.1038/aps.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee K, Silva EA, Mooney DJ. Journal of the Royal Society, Interface / the Royal Society. 2011;8:153. doi: 10.1098/rsif.2010.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]; Awada HK, Johnson NR, Wang Y. J Control Release. 2015;207:7. doi: 10.1016/j.jconrel.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shah NJ, Hyder MN, Quadir MA, Dorval Courchesne N-M, Seeherman HJ, Nevins M, Spector M, Hammond PT. Proceedings of the National Academy of Sciences. 2014;111:12847. doi: 10.1073/pnas.1408035111. [DOI] [PMC free article] [PubMed] [Google Scholar]; Martino MM, Briquez PS, Maruyama K, Hubbell JA. Adv Drug Deliver Rev. 2015;94:41. doi: 10.1016/j.addr.2015.04.007. [DOI] [PubMed] [Google Scholar]; Kim IL, Pfeifer CG, Fisher MB, Saxena V, Meloni GR, Kwon MY, Kim M, Steinberg DR, Mauck RL, Burdick JA. Tissue Engineering Part A. 2015;21:2680. doi: 10.1089/ten.tea.2015.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranganath SH, Levy O, Inamdar MS, Karp JM. Cell Stem Cell. 2012;10:244. doi: 10.1016/j.stem.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schraufstatter IU, Discipio RG, Khaldoyanidi S. Frontiers in bioscience. 2011;16:2271. doi: 10.2741/3853. [DOI] [PubMed] [Google Scholar]; Shudo Y, Cohen JE, Goldstone AB, MacArthur JW, Patel J, Edwards BB, Hopkins MS, Steele AN, Joubert LM, Miyagawa S, Sawa Y, Woo YJ. Cytotherapy. 18:510. doi: 10.1016/j.jcyt.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdeen AA, Weiss JB, Lee J, Kilian KA. Tissue Eng Part A. 2014;20:2737. doi: 10.1089/ten.tea.2013.0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright B, Hopkinson A, Leyland M, Connon CJ. PLoS One. 2013;8:e70860. doi: 10.1371/journal.pone.0070860. [DOI] [PMC free article] [PubMed] [Google Scholar]; Barcan GA, Zhang X, Waymouth RM. Journal of the American Chemical Society. 2015;137:5650. doi: 10.1021/jacs.5b02161. [DOI] [PubMed] [Google Scholar]; Chan JMW, Zhang X, Brennan MK, Sardon H, Engler AC, Fox CH, Frank CW, Waymouth RM, Hedrick JL. Journal of Chemical Education. 2015;92:708. [Google Scholar]; Zhang X, Waymouth RM. ACS Macro Letters. 2014;3:1024. doi: 10.1021/mz500525n. [DOI] [PubMed] [Google Scholar]
- 10.Seib FP, Prewitz M, Werner C, Bornhauser M. Biochemical and biophysical research communications. 2009;389:663. doi: 10.1016/j.bbrc.2009.09.051. [DOI] [PubMed] [Google Scholar]
- 11.Hassan W, Dong Y, Wang W. Stem cell research & therapy. 2013;4:32. doi: 10.1186/scrt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai L, Dewi RE, Heilshorn SC. Advanced Functional Materials. 2015;25:1344. doi: 10.1002/adfm.201403631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parisi-Amon A, Mulyasasmita W, Chung C, Heilshorn SC. Adv Healthc Mater. 2013;2:428. doi: 10.1002/adhm.201200293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong Po Foo CT, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. Proc Natl Acad Sci U S A. 2009;106:22067. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguado BA, Mulyasasmita W, Su J, Lampe KJ, Heilshorn SC. Tissue Eng Part A. 2012;18:806. doi: 10.1089/ten.tea.2011.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbott BC, Lowy J. Proceedings of the Royal Society of London. Series B, Biological sciences. 1956;146:281. doi: 10.1098/rspb.1957.0011. [DOI] [PubMed] [Google Scholar]
- 17.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Adv Mater. 2014;26:865. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chaudhuri O, Gu L, Darnell M, Klumpers D, Bencherif SA, Weaver JC, Huebsch N, Mooney DJ. Nat Commun. 2015:6. doi: 10.1038/ncomms7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nauta A, Seidel C, Deveza L, Montoro D, Grova M, Ko SH, Hyun J, Gurtner GC, Longaker MT, Yang F. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:445. doi: 10.1038/mt.2012.234. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mizuno H, Tobita M, Uysal AC. Stem Cells. 2012;30:804. doi: 10.1002/stem.1076. [DOI] [PubMed] [Google Scholar]
- 19.Staton CA, Reed MW, Brown NJ. International journal of experimental pathology. 2009;90:195. doi: 10.1111/j.1365-2613.2008.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Nat Biotech. 2001;19:1029. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]; Di Santo S, Yang Z, Wyler von Ballmoos M, Voelzmann J, Diehm N, Baumgartner I, Kalka C. PLoS ONE. 2009;4:e5643. doi: 10.1371/journal.pone.0005643. [DOI] [PMC free article] [PubMed] [Google Scholar]; Di Santo S, Seiler S, Fuchs AL, Staudigl J, Widmer HR. PLoS ONE. 2014;9:e95731. doi: 10.1371/journal.pone.0095731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaewsuwan S, Song SY, Kim JH, Sung JH. Expert opinion on biological therapy. 2012;12:1575. doi: 10.1517/14712598.2012.721763. [DOI] [PubMed] [Google Scholar]
- 22.Mulyasasmita W, Cai L, Hori Y, Heilshorn SC. Tissue Eng Part A. 2014 doi: 10.1089/ten.tea.2013.0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho TH, Levere M, Soutif JC, Montembault V, Pascual S, Fontaine L. Polymer Chemistry. 2011;2:1258. [Google Scholar]; Li M, De P, Li H, Sumerlin BS. Polymer Chemistry. 2010;1:854. [Google Scholar]
- 24.Francescone RA, 3rd, Faibish M, Shao R. J Vis Exp. 2011 doi: 10.3791/3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atluri P, Miller JS, Emery RJ, Hung G, Trubelja A, Cohen JE, Lloyd K, Han J, Gaffey AC, MacArthur JW, Chen CS, Woo YJ. The Journal of thoracic and cardiovascular surgery. 2014;148:1090. doi: 10.1016/j.jtcvs.2014.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cohen JE, Purcell BP, MacArthur JW, Jr, Mu A, Shudo Y, Patel JB, Brusalis CM, Trubelja A, Fairman AS, Edwards BB, Davis MS, Hung G, Hiesinger W, Atluri P, Margulies KB, Burdick JA, Woo YJ. Circulation Heart failure. 2014;7:619. doi: 10.1161/CIRCHEARTFAILURE.113.001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.