

Abstract
Melanocytic tumors originating in the central nervous system (MT-CNS) are rare tumors that generally have a favorable prognosis, however malignant tumors do occur. Pathogenetically MT-CNS are not well characterized. Similar to uveal melanoma and blue nevi, they frequently harbor activating GNAQ or GNA11 mutations. Rare NRAS mutations have also been reported. Other mutations have not yet been described. We analyzed 19 MT-CNS, 7 uveal melanomas and 19 cutaneous melanomas using a targeted next generation sequencing approach analyzing 29 genes known to be frequently mutated in other melanocytic tumors (in particular uveal and cutaneous melanomas). In concordance with previous studies, cutaneous melanoma samples showed frequent NRAS or BRAF mutations, as well as mutations in other genes (e.g. NF1, RAC1, PIK3CA, ARID1A). Metastasized uveal melanomas exhibited mutations in GNAQ, GNA11 and BAP1. In contrast, MT-CNS almost exclusively demonstrated mutations in GNAQ (71 %) or GNA11 (12 %). Interestingly both GNA11 mutations identified were detected in MT-CNS diagnosed as intermediate grade melanocytomas which also recurred. One of these recurrent cases also harbored an inactivating BAP1 mutation and was found to have lost one copy of chromosome 3. Our findings show that while MT-CNS do have GNAQ or GNA11 mutations, they rarely harbor other recurrent mutations found in uveal or cutaneous melanomas. Considering chromosome 3 and BAP1 loss are robust markers of poor prognosis in uveal melanoma, it will prove interesting to determine whether these genomic alterations are also of prognostic significance in MT-CNS.
Keywords: Melanocytoma, BAP1, GNAQ, GNA11
Introduction
Primary melanocytic tumors of the CNS (MT-CNS) arise from the leptomeninges and histologically represent a spectrum of lesions ranging from well-differentiated melanocytomas to malignant melanomas. Melanocytomas are the most common MT-CNS and are frequently located in the posterior fossa or upper spinal cord [1]. Melanocytomas generally show benign clinical behavior, with the majority of patients being cured by complete tumor excision. However, recurrences are not infrequent [2]. Overtly malignant MT-CNS, generally designated primary CNS melanomas, are extremely rare.
In patients in whom a proliferation of melanocytes in the central nervous system is identified, the possibility of melanoma metastasis (from a cutaneous, mucosal or uveal primary tumor) must be excluded before a primary CNS tumor can be diagnosed, as the former occurs considerably more frequently than MT-CNS.
Recent years have brought about a wealth of information regarding the genetic alterations in various melanocytic tumors. In cutaneous melanoma, in addition to previously known mutations such as BRAF and NRAS, recurrent mutations in a multitude of other genes have been identified (e.g. NF1, RAC1, ARID2, PPP6C, MAP2K1) [3, 4]. In uveal melanoma, activating mutations in GNAQ and GNA11 were identified [5], as well as mutations in BAP1 [6], SF3B1 [7] and EIF1AX [8]. Inactivating BAP1 mutations are associated with poor prognosis, whereas SF3B1 and EIF1AX mutations primarily occur in tumors which do not metastasize.
In MT-CNS, the occurrence of activating GNAQ and GNA11 mutations has been well documented [9–11]. The common occurrence of these mutations in uveal melanoma and their rarity in cutaneous melanoma points toward a pathogenetic relationship of MT-CNS with uveal melanomas. This is further supported by the finding that TERT promoter mutations, frequent events in cutaneous melanoma [12, 13], are rare in uveal melanoma [14], and are not present in CNS melanocytomas [11]. In rare, mainly pediatric MT-CNS cases, NRAS mutations have been reported [15, 16]. The presence of other recurrent mutations in MT-CNS is currently unknown.
Recent work by Küsters-Vandevelde et al. [17]. demonstrated GNAQ and GNA11 mutations in CNS melanocytomas and found at least one tumor to have copy number alterations similar to uveal melanoma (loss of chromosome 3 and gains of chromosome 8q) [18, 19]. Another melanocytoma was found to have a gain of chromosome 6, an alteration that is frequent in both uveal and cutaneous melanomas [19–21]. A detailed study by Koelsche et al. [22] analyzed copy number alterations, methylation profiles and individual activating gene mutations in melanocytomas, schwannomas and melanomas. Compared to the other tumor types, melanocytomas showed a distinct DNA methylation profile. Chromosome 6p gains and chromosome 3 losses were found to be mutually exclusive and were observed in 6/18 (33 %) and 3/18 (17 %) melanocytoma samples, respectively.
The aim of our study was to analyze the occurrence of gene mutations known to be frequent in cutaneous or uveal melanoma in a cohort of MT-CNS using a next generation targeted sequencing approach.
Materials and methods
Sample selection
Samples of MT-CNS were retrieved from the Institute of Neuropathology, Essen, Germany, the Institute of Neuropathology, Bonn, Germany, as well as the Institute of Pathology, Catholic University in Rome. None of the melanocytoma patients had a clinical history of a synchronous uveal, cutaneous or other melanoma. Uveal and cutaneous melanoma samples used as control tissue samples were retrieved from the Department of Dermatology, Essen, Germany. Tumor tissue was identified and DNA of sufficient quality for genetic analysis was isolated after macrodissection. Eight samples (indicated in Table 1), where specific gene mutations (GNAQ and GNA11) had been screened by pyrosequencing, were previously reported [15]. The study was performed in accordance with the guidelines of the Ethics committee of the Faculty of Medicine of the University Duisburg-Essen.
Table 1. List of identified mutations.
| Pat. | Sample | Diagnosis | Type | BRAF | NRAS | GNAQ | GNA11 | BAP1 | Other |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 MT-CNS | Melanocytoma | P | WT | WT | WT | WT | WT | |
| 2 | 2 MT-CNS | Melanocytoma | P | WT | WT | Q209L | WT | WT | |
| 3 | 3 MT-CNS+ | Melanocytoma | P | WT | WT | Q209P | WT | WT | |
| 4 | 4 MT-CNS+ | Melanocytoma | P | WT | WT | Q209L | WT | WT | |
| 5 | 5 MT-CNS | Melanocytoma | P | WT | WT | WT | WT | WT | TP53 H214 fs, ARID1A S614A |
| 6 | 6 MT-CNS+ | IG Melanocytoma | P | WT | WT | Q209L | WT | WT | SMARCA4 P197S |
| 7 | 7 MT-CNS+ | IG Melanocytoma | P | WT | WT | Q209L | WT | WT | |
| 8 | 8 MT-CNS | IG Melanocytoma | P | WT | WT | Q209P | WT | WT | WT1 A343S |
| 9 | 9 MT-CNS | IG Melanocytoma | P | WT | WT | Q209L | WT | WT | |
| 10 | 10 MT-CNS | IG Melanocytoma | P | WT | WT | Q209L | WT | WT | GNAQ V206I |
| 11 | 11 MT-CNS | IG Melanocytoma | P | WT | WT | Q209L | WT | WT | |
| 12 | 12 MT-CNS | IG Melanocytoma | P | WT | WT | Q209L | WT | WT | |
| 13 | 13 MT-CNS | IG Melanocytoma | P | WT | WT | WT | WT | WT | SMARCA4 R167Q |
| 14 | 14 MT-CNS | IG Melanocytoma | P | WT | WT | Q209L | WT | WT | |
| 15 | 15 MT-CNS+ | IG Melanocytoma | P | WT | WT | Q209P | WT | WT | |
| 15 | 16 MT-CNS+ | IG Melanocytoma | R | WT | WT | Q209P | WT | WT | |
| 16 | 17 MT-CNS+ | IG Melanocytoma | P | WT | WT | WT | Q209L | R60* | |
| 16 | 18 MT-CNS+ | IG Melanocytoma | R | WT | WT | WT | Q209L | R60* | |
| 17 | 19 MT-CNS | IG Melanocytoma | R | WT | WT | WT | Q209L | WT | |
| 1 | 1 UM | Uveal melanoma | M | WT | WT | WT | Q209L | WT | TERT G804S |
| 2 | 2 UM | Uveal melanoma | M | WT | WT | WT | WT | R385* | FBXW7 R505C, TP53 R248Q, R273C |
| 3 | 3 UM | Uveal melanoma | M | WT | WT | WT | Q209L | G115 fs | |
| 4 | 4 UM | Uveal melanoma | M | WT | WT | Q209P | WT | WT | |
| 5 | 5 UM | Uveal melanoma | M | WT | WT | Q209P | WT | WT | |
| 6 | 6 UM | Uveal melanoma | M | WT | WT | WT | Q209L | I210 fs | BAP1 A648del# |
| 7 | 7 UM | Uveal melanoma | M | WT | WT | WT | Q209L | L97P | |
| 1 | 1 CM | Cut. melanoma | P | V600E | WT | WT | WT | WT | PTEN A328 fs*15, ARID1A R1721*# |
| 2 | 2 CM | Cut. melanoma | P | V600K | WT | WT | WT | WT | |
| 3 | 3 CM | Cut. melanoma | P | WT | Q61K | WT | WT | WT | RAC1 P29S, CTNNB1 S33F, PIK3CA P377L |
| 4 | 4 CM | Cut. melanoma | M | WT | Q61K | WT | WT | WT | RAC1 P29S, ARID1A Q588*, WT1 R366C# |
| 5 | 5 CM | Cut. melanoma | M | V600E | WT | WT | WT | WT | CTNNB1 S45P |
| 6 | 6 CM | Cut. melanoma | M | T599_V600insT | WT | WT | WT | WT | ARID1A G423Q, ARID2 S297F |
| 7 | 7 CM | Cut. melanoma | P | V600K | WT | WT | WT | WT | MAP2K1 P124L |
| 8 | 8 CM | Cut. melanoma | P | N581S | WT | WT | WT | WT | NF1 T923 fs, ARID1A R1202Q, KIT F469L |
| 9 | 9 CM | Cut. melanoma | P | P239L | WT | WT | WT | WT | TP53 G245C, ARID1A S574F, SF3B1 P355S |
| 10 | 10 CM | Cut. melanoma | CL | WT | Q61K | WT | WT | WT | KIT V569A, NF1 R440* |
| 11 | 11 CM | Cut. melanoma | CL | V600E | WT | WT | WT | WT | MITF L117F |
| 12 | 12 CM | Cut. melanoma | M | V600E | WT | WT | WT | WT | IDH1 V178I |
| 13 | 13 CM | Cut. melanoma | P | V600E | WT | WT | WT | WT | WT1 G334R |
| 14 | 14 CM | Cut. melanoma | M | V600E | WT | WT | WT | WT | |
| 15 | 15 CM | Cut. melanoma | P | WT | WT | WT | WT | WT | PIK3CA G109del, NF1 H553 fs |
| 16 | 16 CM | Cut. melanoma | P | WT | Q61R | WT | WT | WT | NF1 L2416P |
| 17 | 17 CM | Cut. melanoma | M | WT | WT | WT | WT | WT | KIT K642E, SF3B1 R625H |
| 18 | 18 CM | Cut. melanoma | P | V600E | WT | WT | WT | WT | |
| 19 | 19 CM | Cut. melanoma | P | WT | Q61K | WT | WT | WT | ARID1A R1906Q |
Italics—mutations known or assumed to be activating; bold—loss of function mutations; normal—missense mutation (frequently with unknown functional consequences); Pat patient; MT-CNS melanocytic tumor of the central nervous system; UM uveal melanoma; CM cutaneous melanoma; IG intermediate grade; Cut. cutaneous; fs frame shift;
stop codon (nonsense mutation); ins insertion; del deletion; P primary tumor; R recurrence; CL cell line
All mutations listed in the table were found to have an allelic frequency of at least 15 %
More detailed information and additional mutations are presented in Supplemental Table 3
Samples were previously analyzed for GNAQ and GNA11 mutations by pyrosequencing [15]
Histopathology and immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) tissue samples of 19 MT-CNS were obtained. For histopathologic examination, 2 μm-thin sections were routinely stained with hematoxylin-and-eosin. Additional immunhistochemical (IHC) stainings for S100 (1:5000, Dako, Glostrup, Danmark; Z0311), melanocytic markers Melan A (1:100, Dako, Glostrup, Danmark; M7196) and HMB45 (1:200, Dako, Glostrup, Danmark; M0634), BAP1 (described below), and the proliferation marker Ki67/MIB1 (1:200, Zytomed, Berlin, Germany; MSK0810) were performed.
Diagnoses were made based on criteria described by Brat et al. [23]. Melanocytomas are well-differentiated MT-CNS with no or very low mitotic activity (0–1 mitoses per 10 HPF) devoid of CNS infiltration. Ki67/MIB1-staining is ≤2 %. Intermediate grade melanocytomas are characterized by increased mitotic activity and microscopic CNS invasion, but are not sufficiently anaplastic to warrant the designation of malignant melanoma. Ki67/MIB1 staining ranges from 1 to 4 %.
All histologic and IHC sections were reviewed by at least two histopathologists (JvdN, KGG, MG, TP). The clinico-pathologic details are summarized in Supplemental Table 1.
BAP1 immunohistochemistry
The BAP1 antibody used was rabbit polyclonal raised against a synthetic peptide corresponding to amino acids 430–729 of the BAP1 molecule (clone C-4, Santa Cruz Biotechnology Inc.). For immunohistochemical examination, 5 μm-thin sections were cut from representative FFPE tissue samples from each tumor. For antigen retrieval, the sections were transferred into a jar containing EDTA buffer solution (pH 8.0) at 90 °C for 52 min. The primary antibody to detect BAP-1 (diluted 1:50 at 36 °C for 24 min) was used in combination with a highly sensitive and specific polymer detection system applying the chromogen permanent red, resulting in an orange-red reaction product (Ultra view universal alkaline phosphatase detection kit, Ventana®). The sections were counterstained with haematoxylin for 5 min. All stainings were performed by means of Ventana® Benchmark XT Autostainer. Tumors were scored as positive or negative according to nuclear staining of BAP1.
DNA isolation
10 μm-thick sections of FFPE tissue were deparaffinized according to the following protocol: 2 steps of 5 min xylene, 5 min 100 % ethanol, 5 min 95 % ethanol, 5 min 70 % ethanol, 5 min 50 % ethanol, rinsing in water. After drying, tumor tissue was manually macrodissected from the sections. Genomic DNA was isolated using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Targeted sequencing
A custom amplicon-based sequencing panel covering 29 genes (Supplemental Table 2) known to be recurrently mutated in cutaneous and uveal melanoma was designed and prepared applying the GeneRead Library Prep Kit from QIAGEN® according to the manufacturer's instructions. For adapter ligation and barcoding of individual samples, the NEBNext Ultra DNA Library Prep Mastermix Set and NEBNext Multiplex Oligos for Illumina from New England Biolabs were applied. Twelve samples were sequenced in parallel on an Illumina MiSeq next generation sequencer.
Sequencing analysis was performed applying the CLC Cancer Research Workbench from QIAGEN®. In brief, the following steps were applied. The workflow in CLC included adapter trimming and read pair merging before mapping to the human reference genome (hg19). Insertions and deletions as well as single nucleotide variant detection, local realignment and primer trimming followed. Additional information was then obtained regarding potential mutation type, known single nucleotide polymorphisms and conservation scores by cross-referencing varying databases (COSMIC, ClinVar, dbSNP, 1000 Genomes Project, HAPMAP and PhastCons-Conservation_scores_hg19). After the CLC processing, resulting csv files were analyzed manually. Mutations affecting the protein coding portion of the gene were considered if predicted to result in non-synonymous amino acid changes. To eliminate questionable low frequency background mutations calls, not uncommon in our experience with FFPE amplicon sequencing approaches [24], mutations were reported if the overall coverage of the mutation site was ≥30 reads, ≥15 reads reported the mutated variant and the frequency of mutated reads was ≥15 %.
Sanger-sequencing was performed for EIF1AX exon 1 applying the primers F-CCTCCAGCACCTACTTGGTC and R-CTGGGTGACCTGCAATCTAC as previously described [21].
Detailed listing of sequence analysis settings
The following analysis modules were applied sequentially in CLC Cancer Research Workbench using the notated settings: 1. Trim sequences (trimming barcode primers from NEB [Trim_NEBNextUltra_Adapter_List]); 2. Merge overlapping pairs (settings: mismatch cost: 2; minimum score: 8, gap cost: 3, maximum unaligned end mismatches: 0); 3. Map reads to reference (alignment to the human reference genome hg19, adjusted settings: mismatch cost: linear gap cost selected, insertion cost: 3, deletion cost: 3 length fraction: 0.5, similarity fraction: 0.8, auto-detect paired distances selected); 4. Local realignment (option realign unaligned ends activated, multi-pass realignment set to 2); 5. InDels and structural variants (analysis for both insertions and deletions was applied allowing 3 maximum mismatches (unaligned end breakpoints) and requiring 2 minimum reads); 6. Trim primers of mapped reads (selecting file for primer sequences obtained from QIAGEN); 7. Low frequency variant detection (significance set to 1 %, minimum coverage to 10, minimum count to 2); 8. Configure QC for targeted sequencing (bed file of targeted region, minimum coverage of 30, options ignore non-specific matches and ignore broken pairs activated.); 9. Remove false positives (settings: minimum frequency of 2 %, minimum forward/reverse read balance of 0.05, minimum average base quality of 20.0); 10. Remove variants outside targeted regions (bed file of targeted genes) 11. Add information from overlapping genes (by cross-referencing ensemble_v74genes and ensemble_v74_mRNA); 12. Add exon number (obtained cross-referencing ensemble_v74_mRNA); 13. Add information about amino acid changes (applying ensemble v74CDS, hg18 and v74mRNA, and the standard genetic code for translation); 14. Add information from COSMIC (cross-referencing COSMIC); 15. Add information from ClinVar (cross-referencing to Clinvar_20131203); 16. Add information from common dbSNP (cross-referencing to dbSNP); Add information from 1000 genomes project (cross-referencing to 1000 genomes phase 1; EUR, AMR, AFR); Add information from HapMap (cross-referencing to HAPMAP phase 3; CHD, CHB, ASW, GIH, CEU, LWK, MEX, MKK, TSI, HCB, YRI, JPT); 19. Add conservation scores (cross-referencing to PhastCons-Conservation_scores_ hg19).
Copy number analysis
Array-based comparative genomic hybridization (CGH) was performed to analyze DNA copy number alterations (CNAs) applying Agilent® 180 K CGH arrays. Methods for hybridization and analysis have been described previously [21, 25–28].
Results
Patients and histopathological characteristics
The MT-CNS samples came from 17 patients aged 16–79 years, including 9 females (41–79 years) and 8 males (16–79 years). These tumors included 5 melanocytomas, and 14 intermediate grade melanocytomas, of which 3 were recurrent samples. Clinical pathological information is presented in Supplemental Table 1. In one case, MT-CNS 19, only the recurrent sample was available for analysis.
Targeted next generation sequencing
The sequencing of all samples with the 29 gene assay identified recurrent activating Q209 mutations in GNAQ and GNA11 in the MT-CNS (Table 1; Fig. 1). Mutations in GNAQ were identified in 13 samples from 12 patients (12 of 17 = 71 %). Mutations in GNA11 were found in three samples from two patients (2 of 17 = 12 %). An inactivating BAP1 mutation leading to the formation of a stop codon at residue 60 (R60*) was identified in two samples of the same patient (the primary tumor and a recurrence, shown in Fig. 2). Other recurrent mutations were not identified in the MT-CNS samples. As controls, 7 uveal melanomas and 19 cutaneous melanomas were sequenced. The uveal melanoma samples harbored recurrent GNAQ (n = 2, 29 %) and GNA11 mutations (n = 4, 57 %). BAP1 mutations were identified in 4 samples (57 %), of which 3 (75 %) were clearly inactivating, leading to loss of the functional protein (Supplemental Fig. 1). The cutaneous samples showed a range of mutations. BRAF V600 mutations were detected in 10 of 19 (53 %) samples, including 7 V600E, 2 V600K and 1 T599_V600insT alterations. Another two samples carried N581S and P239L mutations of unclear functional significance. NRAS mutations were found mutually exclusively with BRAF mutations in 5 (26 %) samples and included 4 Q61K and 1 Q61R alterations. Other rarer mutations were identified, including NF1, PTEN, ARID1A, RAC1, KIT, SF3B1 and other mutations (see Table 1; Supplemental Table 3 reporting mutations with an allelic frequency ≥15 %). In 14 of the MT-CNS samples, exon 1 of the gene EIF1AX, which was not covered in the next gen sequencing panel, was sequenced by Sanger-sequencing, in search of recurrent mutations which are frequently seen in primary uveal melanoma samples; no EIF1AX mutations were identified.
Fig. 1.
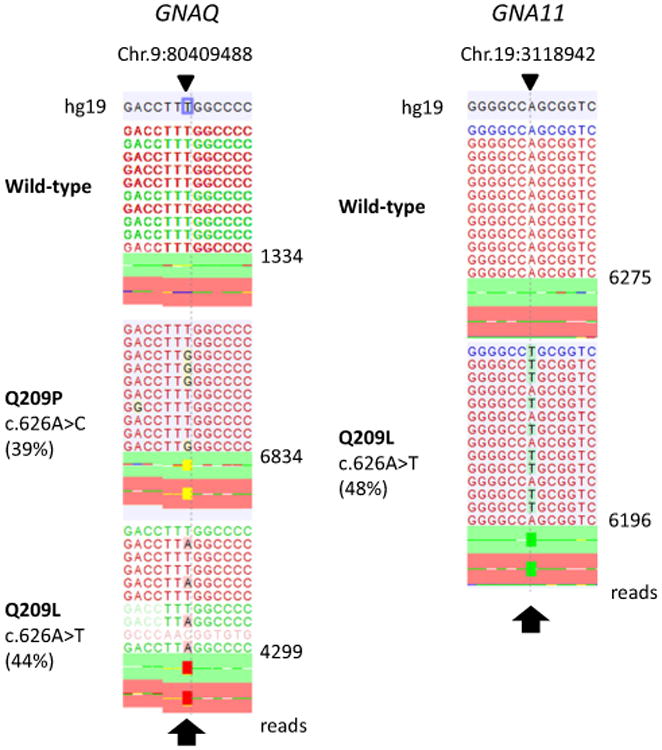
Recurrent GNAQ and GNA11 mutations in melanocytic tumors of the central nervous system. Plot of aligned sequencing reads in the location where recurrent mutations were found in the genes GNAQ (left) and GNA11 (right). Wild-type sequences are shown on the top. For GNAQ, a c.616A > C p.Q209P mutation is shown in the middle (sample 15) and a c.626A > T p.Q209L mutation (sample 12) on the bottom. For GNA11, the c.626A > T p.Q209L mutation (sample 17) is shown, observed in two melanocytic tumors of the central nervous system. The arrows highlight the mutation site
Fig. 2.
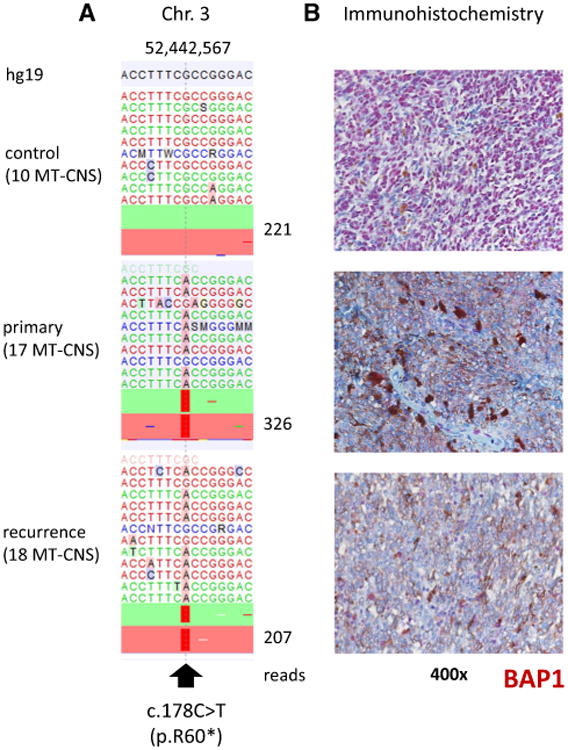
Inactivating BAP1 mutation resulting in BAP1 protein loss in a melanocytic tumor of the central nervous system. a Plot of aligned sequencing reads in the location where a BAP1 c.178C > T, p.R60* mutation was identified. The primary and recurrent tumor (sample 17; 18, respectively) harboring this mutation are shown in the middle and bottom plot. The number of sequence reads is notated on the right. Mutations were found in 88 and 75 % of reads from the primary and recurrent tumor (sample 17; 18, respectively). b Corresponding BAP1 immunohistochemistry; the top picture, with a strong nuclear BAP1 staining, represents the situation found in most samples. The two lower pictures from the primary and recurrent tumor (sample 17; 18, respectively) show complete loss of BAP1 expression (only background staining remains)
BAP1 immunohistochemistry
Expression of BAP1 protein by tumors was analyzed by IHC in all MT-CNS cases. Almost all cases showed a convincing nuclear staining, showing BAP1 protein primarily located in the nucleus. In two samples, the primary and recurrent tumor from the same patient harboring the BAP1 R60* mutations, BAP1 IHC showed absent nuclear expression (Fig. 2).
Copy number alterations
Genome wide chromosomal copy number analysis was performed on both samples identified as having inactivating BAP1 c.178C > T, p.R60* mutations as well as two other MT-CNS (also intermediate-grade melanocytoma) samples, both harboring GNAQ c.626A > T, p.Q209L mutations. Whereas no or minimal changes (gain of 6p and loss of parts of 16q) were observed in the BAP1-wild-type MT-CNS (Fig. 3), several alterations were seen in the BAP1-mutant tumors. These included losses of Chr. 1p, gains of Chr. 8 and most importantly a complete loss of one copy of Chr. 3.
Fig. 3.
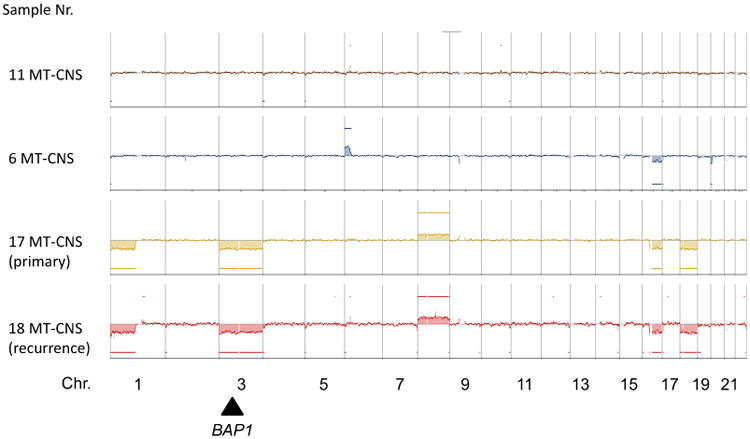
Copy number alterations of selected melanocytic tumors of the central nervous system. Shown are whole genome DNA copy number profiles obtained by CGH (comparative genomic hybridization). The primary and recurrent tumor (sample 17; 18, respectively) from the central nervous system melanocytic tumor case harboring a BAP1 R60* nonsense mutation are shown on the bottom. The approximate location of BAP1 on Chr. 3 is designated by the black arrow
Discussion
In our study, a larger cohort of MT-CNS was screened for mutations in a range of genes known to be recurrently mutated in other melanocytic tumors, in particular cutaneous and uveal melanoma. Other than the known mutations in GNAQ and GNA11, none of the other genes analyzed were found to harbor recurrent mutations in MT-CNS from different patients. These results indicate that MT-CNS have a distinct genetic mutation profile, differing from both uveal and cutaneous melanoma.
In the uveal melanoma samples analyzed, 4 out of 7 (57 %) had mutations in BAP1. Three of these (75 %) were clearly inactivating, being nonsense or frameshift mutations resulting in a nonfunctional protein (Supplemental Fig. 1). This frequency is similar to previously reported results [6]. Potentially events inactivating BAP1, such as promoter methylation or homozygous deletions, not detected by our sequencing approach, may have taken place in some of the remaining samples. As all samples available for analysis in the study were metastases, it was not surprising that other mutations such as SF3B1 and EIF1AX, which are known to be associated with a good prognosis, were not observed [7, 8].
The mutations identified in cutaneous melanomas reflect those described in previous studies [3, 4] with activating BRAF and NRAS mutations in 53 and 26 % of samples, respectively. Additionally, our screen identified a number of other recently described mutations such as two hotspot R29 RAC1 mutations [4], 3 inactivating NF1 mutations, 2 inactivating ARID1A mutations and 2 KIT mutations. One KIT mutation, K642E, is clearly activating and was identified in an NRAS and BRAF wild-type sample [29]. The described mutations detected in cutaneous melanoma samples, were not identified in MT-CNS.
The high mutation frequency of GNAQ and GNA11 mutations detected in MT-CNS is intriguing. As we previously reported [15], in contrast to uveal melanomas, MT-CNS samples much more commonly harbor GNAQ mutations than GNA11 mutations. Concordant with our previous study [11], our targeted next generation sequencing in the current study identified 71 % GNAQ and 12 % GNA11 mutations. Küsters-Vandevelde et al. also recently reported more frequent GNAQ mutations (37 %) than GNA11 mutations (10 %) in MT-CNS. Blue nevi, benign melanocytic proliferations of the skin, also show a similar distribution with 55 % GNAQ and 7 % GNA11 mutations reported in one study [5]. The distribution of mutations in primary uveal melanoma samples is more evenly distributed, however still shows slightly more GNAQ (45–47 %) than GNA11 (32–44 %) mutations [5, 30]. In contrast, a higher frequency of GNA11 (57–60 %) to GNAQ (20–22 %) mutations has been reported in uveal melanoma metastases [5, 31]. The shift in mutation frequencies from GNAQ to GNA11 from benign to increasingly malignant tumors may indicate GNA11 mutations are associated with a more malignant phenotype in this entity.
Interestingly, both of the two mutant GNA11 cases (three samples from two patients) we observed in MT-CNS were rated intermediate grade melanocytomas. Furthermore, both mutant GNA11 MT-CNS cases were tumors that recurred. One of these cases also harbored an inactivating BAP1 mutation. Küsters-Vandevelde et al. also reported that all melanocytomas in their cohort with GNA11 mutations were of intermediate grade [17]. If future studies with larger case numbers report similar findings to our study, this could signify GNA11 mutations are also associated with a more aggressive phenotype in MT-CNS.
Considering the similar occurrence of GNAQ and GNA11 mutations in a high frequency of MT-CNS and uveal melanomas, it would seem likely that MT-CNS could potentially also harbor mutations in other genes known to be relevant in uveal melanoma, in particular the recently identified mutations in SF3B1, EIF1AX and BAP1 [6–8]. Mutations in these genes are found in most uveal melanoma samples and with rare exceptions are mutually exclusive. BAP1 mutations are associated with metastasis and poor prognosis [6], whereas SF3B1 and EIF1AX mutations primarily occur in tumors with a favorable prognosis [8]. As MT-CNS are mostly benign tumors with a favorable prognosis, it would seem likely that they could also harbor SF3B1 or EIF1AX mutations. However, in our study, no such mutations were present. One case (of 17 = 6 %) did harbor an inactivating BAP1 mutation. These results suggest that mutations in uveal melanoma genes other than GNAQ and GNA11 are rare in MT-CNS.
Although only identified in one MT-CNS case (1 of 17 = 6 %), the case with BAP1 inactivation and Chr. 3 loss is intriguing. Other recent studies have shown that losses of Chr. 3 in MT-CNS are rare, however they do occur [17, 22]. Similar to our findings, the chromosomal alterations in these cases are highly reminiscent of uveal melanomas with a poor prognosis [19, 32]. As such, screening for inactivating BAP1 mutations and/or Chr. 3 loss could represent a relevant prognostic marker in MT-CNS. In our MT-CNS case harboring both a loss of Chr. 3 and an inactivating BAP1 mutation, both patient history and MRI scans showed no sign of uveal melanoma. Unfortunately, detailed patient follow-up data was not available, but the tumors recurrence alone could indicate more aggressive behavior. As BAP1 loss was clearly demonstrated by IHC, genetic screening may not be necessary. Additional studies with greater case numbers are needed to determine the full prognostic value of BAP1 and Chr. 3 status in MT-CNS.
Our study screened for the presence of mutations in many genes known to be recurrently mutated in cutaneous and uveal melanomas. The results show that MT-CNS are genetically distinct from cutaneous melanomas. Although MT-CNS share frequent GNAQ and GNA11 mutations with uveal melanomas, mutations in other uveal melanoma genes were very rare. This argues that MT-CNS are genetically related but still distinct from uveal melanomas. It stands to reason that MT-CNS must harbor additional, as yet unidentified gene mutations warranting whole-exome or whole-genome studies. The identification of novel gene mutations unique to MT-CNS would be a valuable diagnostic tool to help distinguish MT-CNS from metastases of melanocytic tumors from other sites.
Supplementary Material
Acknowledgments
We would like to thank Marion Schwamborn and Nicola Bielefeld for their excellent technical support. Assistance from staff of Melanoma Institute Australia and Royal Prince Alfred Hospital and funding support from the National Health and Medical Research Council (of the Commonwealth Government of Australia) and the Cancer Institute New South Wales is also gratefully acknowledged. Support from the NIH Cancer Center Support Grant P30 CA008748 is gratefully acknowledged.
Funding: This study was not funded by any type of grant or sponsor. There was no external influence on the decision to publish or content of the manuscript.
Lisa Zimmer has honoraria from Roche, Bristol-Meyers Squibb, and Amgen, and travel support from Merck Sharp and Dohme and Bristol-Meyers Squibb. Bastian Schilling has received honoraria from Roche and travel support as well research funding from Bristol-Myers Squibb. Dirk Schadendorf is on the advisory board or has received honoraria from Roche, Genentech, Novartis, Amgen, GlaxoSmithKline, Bristol-Myers Squibb, Boeh-ringer Ingelheim, and Merck Sharp and Dohme.
Footnotes
Electronic supplementary material: The online version of this article (doi:10.1007/s11060-015-2052-2) contains supplementary material, which is available to authorized users.
Compliance with ethical standards: Conflict of interests: All other authors have nothing to declare.
References
- 1.Goldgeier MH, Klein LE, Klein-Angerer S, Moellmann G, Nordlund JJ. The distribution of melanocytes in the leptomeninges of the human brain. J Invest Dermatol. 1984;82:235–238. doi: 10.1111/1523-1747.ep12260111. [DOI] [PubMed] [Google Scholar]
- 2.Wadasadawala T, Trivedi S, Gupta T, Epari S, Jalali R. The diagnostic dilemma of primary central nervous system melanoma. J Clin Neurosci Off J Neurosurg Soc Australia. 2010;17:1014–1017. doi: 10.1016/j.jocn.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 3.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, Sznol M, Kluger HM, Brash DE, Stern DF, Materin MA, Lo RS, Mane S, Ma S, Kidd KK, Hayward NK, Lifton RP, Schlessinger J, Boggon TJ, Halaban R. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, Heguy A, Dolgalev I, Khanin R, Busam K, Speicher MR, O'Brien J, Bastian BC. Mutations in GNA11 in uveal melanoma. New Engl J Med. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013 doi: 10.1038/ng.2523. doi:10. 1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, Lohmann DR, Zeschnigk M. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933–936. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Küsters-Vandevelde HV, Klaasen A, Küsters B, Groenen PJ, van Engen-van Grunsven IA, van Dijk MR, Reifenberger G, Wesseling P, Blokx WA. Activating mutations of the GNAQ gene: a frequent event in primary melanocytic neoplasms of the central nervous system. Acta Neuropathol. 2010;119:317–323. doi: 10.1007/s00401-009-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murali R, Wiesner T, Rosenblum MK, Bastian BC. GNAQ and GNA11 mutations in melanocytomas of the central nervous system. Acta Neuropathol. 2012;123:457–459. doi: 10.1007/s00401-012-0948-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gessi M, van de Nes J, Griewank K, Barresi V, Buckland ME, Kirfel J, Caltabiano R, Hammes J, Lauriola L, Pietsch T, Waha A. Absence of TERT promoter mutations in primary melanocytic tumors of the central nervous system. Neuropathol Appl Neurobiol. 2014 doi: 10.1111/nan.12138. [DOI] [PubMed] [Google Scholar]
- 12.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 13.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griewank KG, Murali R, Schilling B, Scholz S, Sucker A, Song M, Susskind D, Grabellus F, Zimmer L, Hillen U, Steuhl KP, Schadendorf D, Westekemper H, Zeschnigk M. TERT promoter mutations in ocular melanoma distinguish between conjunctival and uveal tumours. Br J Cancer. 2013;109:497–501. doi: 10.1038/bjc.2013.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gessi M, Hammes J, Lauriola L, Dorner E, Kirfel J, Kristiansen G, Muehlen A, Denkhaus D, Waha A, Pietsch T. GNA11 and N-RAS mutations: alternatives for MAPK pathway activating GNAQ mutations in primary melanocytic tumours of the central nervous system. Neuropathol Appl Neurobiol. 2013;39:417–425. doi: 10.1111/j.1365-2990.2012.01288.x. [DOI] [PubMed] [Google Scholar]
- 16.Küsters-Vandevelde HV, Küsters B, van Engen-van Grunsven AC, Groenen PJ, Wesseling P, Blokx WA. Primary melanocytic tumors of the central nervous system: a review with focus on molecular aspects. Brain Pathol. 2015;25:209–226. doi: 10.1111/bpa.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Küsters-Vandevelde HV, van Engen-van Grunsven IA, Coupland SE, Lake SL, Rijntjes J, Pfundt R, Küsters B, Wesseling P, Blokx WA, Groenen PJ. Mutations in G protein encoding genes and chromosomal alterations in primary leptomeningeal melanocytic neoplasms. POR, Pathology oncology research. 2014 doi: 10.1007/s12253-014-9841-3. doi:10. 1007/s12253-014-9841-3. [DOI] [PubMed] [Google Scholar]
- 18.Thomas S, Putter C, Weber S, Bornfeld N, Lohmann DR, Zeschnigk M. Prognostic significance of chromosome 3 alterations determined by microsatellite analysis in uveal melanoma: a long-term follow-up study. Br J Cancer. 2012;106:1171–1176. doi: 10.1038/bjc.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehlers JP, Worley L, Onken MD, Harbour JW. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14:115–122. doi: 10.1158/1078-0432.CCR-07-1825. [DOI] [PubMed] [Google Scholar]
- 20.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. New Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 21.Griewank K, Westekemper H, Murali R, Mach M, Schilling B, Wiesner T, Schimming T, Livingstone E, Sucker A, Grabellus F, Metz C, Susskind D, Hillen U, Speicher MR, Woodman SE, Steuhl KP, Schadendorf D. Conjunctival melanomas harbor BRAF and NRAS mutations and copy number changes similar to cutaneous and mucosal melanomas. Clin Cancer Res Off J Am Assoc Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-13-0163. [DOI] [PubMed] [Google Scholar]
- 22.Koelsche C, Hovestadt V, Jones DT, Capper D, Sturm D, Sahm F, Schrimpf D, Adeberg S, Bohmer K, Hagenlocher C, Mechtersheimer G, Kohlhof P, Muhleisen H, Beschorner R, Hartmann C, Braczynski AK, Mittelbronn M, Buslei R, Becker A, Grote A, Urbach H, Staszewski O, Prinz M, Hewer E, Pfister SM, von Deimling A, Reuss DE. Melanotic tumors of the nervous system are characterized by distinct mutational, chromosomal and epigenomic profiles. Brain Pathol. 2015;25:202–208. doi: 10.1111/bpa.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brat DJ, Giannini C, Scheithauer BW, Burger PC. Primary melanocytic neoplasms of the central nervous systems. Am J Surg Pathol. 1999;23:745–754. doi: 10.1097/00000478-199907000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Murali R, Chandramohan R, Moller I, Scholz SL, Berger M, Huberman K, Viale A, Pirun M, Socci ND, Bouvier N, Bauer S, Artl M, Schilling B, Schimming T, Sucker A, Schwindenhammer B, Grabellus F, Speicher MR, Schaller J, Hillen U, Schadendorf D, Mentzel T, Cheng DT, Wiesner T, Griewank KG. Targeted massively parallel sequencing of angiosarcomas reveals frequent activation of the mitogen activated protein kinase pathway. Oncotarget. 2015;6:36041–36052. doi: 10.18632/oncotarget.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, Windpassinger C, Wackernagel W, Loy S, Wolf I, Viale A, Lash AE, Pirun M, Socci ND, Rutten A, Palmedo G, Abramson D, Offit K, Ott A, Becker JC, Cerroni L, Kutzner H, Bastian BC, Speicher MR. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, Du J, Kau T, Thomas RK, Shah K, Soto H, Perner S, Prensner J, Debiasi RM, Demichelis F, Hatton C, Rubin MA, Garraway LA, Nelson SF, Liau L, Mischel PS, Cloughesy TF, Meyerson M, Golub TA, Lander ES, Mellinghoff IK, Sellers WR. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. Journal of clinical oncology. Off J Am Soc Clin Oncol. 2006;24:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 30.Daniels AB, Lee JE, MacConaill LE, Palescandolo E, Van Hummelen P, Adams SM, DeAngelis MM, Hahn WC, Gragoudas ES, Harbour JW, Garraway LA, Kim IK. High throughput mass spectrometry-based mutation profiling of primary uveal melanoma. Invest Ophthalmol Vis Sci. 2012;53:6991–6996. doi: 10.1167/iovs.12-10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griewank KG, van de Nes J, Schilling B, Moll I, Sucker A, Kakavand H, Haydu LE, Asher M, Zimmer L, Hillen U, Thompson JF, Scolyer RA, Schadendorf D, Murali R. Genetic and clinico-pathologic analysis of metastatic uveal melanoma. Modern pathology. Off J United States Can Acad Pathol. 2014;27:175–183. doi: 10.1038/modpathol.2013.138. [DOI] [PubMed] [Google Scholar]
- 32.Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jöckel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222–1225. doi: 10.1016/s0140-6736(96)90736-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.