

Abstract
The Epstein Barr virus (EBV) genome persists in infected host cells as a chromatinized episome and is subject to chromatin-mediated regulation. Binding of the host insulator protein CTCF to the EBV genome has an established role in maintaining viral latency type, and in other herpesviruses, loss of CTCF binding at specific regions correlates with viral reactivation. Here, we demonstrate that binding of PARP1, an important cofactor of CTCF, at the BZLF1 lytic switch promoter restricts EBV reactivation. Knockdown of PARP1 in the Akata-EBV cell line significantly increases viral copy number and lytic protein expression. Interestingly, CTCF knockdown has no effect on viral reactivation, and CTCF binding across the EBV genome is largely unchanged following reactivation. Moreover, EBV reactivation attenuates PARP activity, and Zta expression alone is sufficient to decrease PARP activity. Here we demonstrate a restrictive function of PARP1 in EBV lytic reactivation.
Keywords: Epstein Barr virus, Lytic reactivation, BZLF1, Zta, PARP1, CTCF
1. Introduction
Epstein Barr virus (EBV) is a gammaherpesvirus that is highly proficient in establishing immunoevasive latent infections. Upon infection of B cells, the EBV genome is circularized as a minichromosome and chromatinized by host proteins to establish a restricted latent gene expression program in which only a small percentage of viral genes are expressed. During latency, the virus is subject to low-level spontaneous reactivation (Phan et al., 2016); however, in order to generate a productive lytic infection, the virus must overcome tight regulation to initiate complete reactivation. During latency, the EBV genome is completely chromatinized and subject to suppressive chromatin marks and DNA methylation (Hammerschmidt, 2015; Tempera et al., 2010). Significant efforts have explored changes in the chromatin landscape during reactivation, although it appears these changes are often cell line-dependent and likely consequential, not causal, to reactivation (Flower et al., 2011; Ramasubramanyan et al., 2012; Murata and Tsurumi, 2013). The molecular mechanisms driving the process of reactivation are understood primarily in the context of transcriptional regulation of the BZLF1 promoter (Zp). Numerous factors that bind to Zp have been identified as either positive or negative regulators of BZLF1 transcription, as well as a few cellular factors that contribute to lytic reactivation (McKenzie and El-Guindy, 2015). Understanding the molecular mechanisms dictating the latent to lytic switch may offer therapeutic insight for oncolytic therapy in latency-associated tumors. Further, if these mechanisms are conserved among herpesviruses, we could identify therapeutic targets to prevent lytic outbreaks in infected individuals.
CTCF is a host insulator protein that demarcates chromatin boundaries and mediates three-dimensional chromatin loops in mammalian cells (Phillips and Corces, 2009). CTCF is frequently targeted by DNA viruses to manipulate viral gene expression (Pentland and Parish, 2015). Indeed, chromatin immunoprecipitation (ChIP) studies have identified a number of CTCF binding sites throughout the EBV genome (Chau et al., 2006; Day et al., 2007; Holdorf et al., 2011; Arvey et al., 2013). CTCF binding at latency promoters Qp and Cp prevents the spread of repressive chromatin to Qp and mediates latency type-specific promoter usage by forming alternative chromatin loops (Tempera et al., 2010, 2011). A prominent CTCF binding site is located proximal to the lytic promoter, Zp, but has yet to be characterized regarding its potential role in lytic reactivation. In HSV-1, CTCF occupancy is distinct during latent infection and reactivation; CTCF is lost from specific regions of the genome following HSV-1 reactivation (Ertel et al., 2012). Mixed reports in KSHV suggest that CTCF restricts lytic reactivation, at least in some cell lines (Chen et al., 2012; Li et al., 2014).
PARP1 is a host enzyme that post-translationally modifies proteins through the transfer of ADP-ribose from NAD+ onto acceptor proteins. PARP1 is well characterized in its roles in DNA damage and apoptosis, however, in recent years, PARP1 has also been implicated in chromatin modification, transcriptional regulation, and inflammation (Wei and Yu, 2016; Martin et al., 2015; Schiewer and Knudsen, 2014; Rosado et al., 2013; Laudisi et al., 2011). Evolutionary analyses have identified other PARP family proteins that likely have evolved a role in host-virus conflicts (Daugherty et al., 2014). Indeed, a body of evidence suggests that PARP1 may serve as a stress sensor—a function that would be especially relevant in regulating herpesvirus lytic reactivation (Mattiussi et al., 2007; Luo and Kraus, 2012). Reports in KSHV have shown that PARP1 is selectively downregulated by a lytic protein, and restricts KSHV lytic replication through various mechanisms, including poly(ADP-ribosyl)ation (PARylation) of the immediate early protein, Rta (Cheong et al., 2015; Gwack et al., 2003).
CTCF is also PARylated by PARP1, subsequently altering its insulator function (Yu et al., 2004; Farrar et al., 2011, Ong et al., 2013). Based on the established role of CTCF in EBV latency, the functional interactions of PARP1 and CTCF, and because CTCF and PARP1 are both implicated as viral restriction factors in other herpesviruses, we hypothesized that PARP1 might regulate CTCF binding in the EBV genome to control lytic reactivation. In this study, we specifically aimed to explore whether PARP1 regulates CTCF binding at the BZLF1 promoter.
2. Materials and methods
2.1. Cell culture and treatment
Cell lines were maintained in a humidified atmosphere containing 5% CO2 at 37 °C. B cells lines were cultured in suspension in RPMI 1640 supplemented with fetal bovine serum at a concentration of either 10% (Kem I, Akata) or 15% (lymphoblastoid cell lines [LCL], Kem III). HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS. All cell media were supplemented with 1% penicillin/streptomycin. Olaparib (Selleck Chemical) was dissolved in DMSO and diluted in the appropriate cell medium for treatment. B cells were treated with 2.5 μM olaparib for 24 h. HEK293 cells were treated with 100 μM N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG; Pfaltz & Bauer) for 10 min immediately prior to harvesting. Akata-EBV cells were treated with anti-human IgG (Sigma-Aldrich) at 10 μg/mL for 24 h to induce lytic reactivation.
2.2. Chromatin immunoprecipitation and quantitative PCR
Chromatin immunoprecipitation (ChIP) assays were performed according to the Upstate Biotechnology Inc. protocol as described previously, with minor modifications (Martin et al., 2016). Briefly, cells were fixed in 1% formaldehyde for 15 min, and DNA was sonicated using a sonic dismembrator (Fisher Scientific) to generate 200–500-bp fragments. Chromatin was immunoprecipitated with polyclonal antibodies to PARP1 (Active Motif) and CTCF (Millipore), and monoclonal antibody to Zta (Santa Cruz). Protein A/G magnetic beads (Pierce) were used for immunoprecipitation with monoclonal antibody. Realtime PCR was performed with a master mix containing 1X Maxima SYBR Green, 0.25 μM primers probing the CTCF binding site at the BZLF1 promoter, and 1/50 of the ChIP DNA per well. Primers are available upon request. Quantitative PCR reactions were carried out in triplicate using the ABI StepOnePlus PCR system. Data were analyzed by the ΔΔCT method relative to DNA input and normalized to the IgG control.
2.3. Chromatin immunoprecipitation and sequencing
We compared CTCF occupancy on the EBV genome during latency and reactivation in Akata-Zta, a cell line that harbors the Akata episome as well as a plasmid with a doxycycline-inducible bidirectional promoter that produces both BZLF1 and LNGFR (Ramasubramanyan et al., 2015). Cells were maintained in RPMI 1640 with 25 mM HEPES and 2 g/L NaHCO3 supplemented with 10% (v/v) Tet System Approved fetal bovine serum (Clontech) in 5% CO2 at 37 °C. Log-phase cultures were pretreated with 200 μM acyclovir (Sigma-Aldrich) for 1 h before induction with 500 ng/mL doxycycline (Sigma-Aldrich). Acyclovir prevents lytic replication of linear genomes that generates a heterogenous population of EBV DNA. After 1 day, cells were magnetically sorted using LNGFR Microbeads and LS columns (Miltenyi Biotec). Immunoprecipitation, library construction, sequencing, and bioinformatics were performed as previously described with minor modifications (Holdorf et al., 2011). Chromatin was purified using anti-CTCF (Millipore) from 2×107 cells per condition. ~200–400 bp amplicons of libraries were selected for with Agencourt RNAClean XP beads (Nugen). 50 bp sequence reads were mapped to an index containing both the human hg19 and the EBV reference (Genbank ID: NC_007605.1) genomes using Bowtie version 0.12.8 (Langmead et al., 2009) allowing up to 2 mismatches and 1 alignment. Peaks were visualized after normalization to the total number of mapped reads. Each occupancy profile was determined from a data set that yielded ~20 million mapped sequences.
2.4. CTCF binding site prediction and electrophoretic mobility shift assay
CTCF binding site prediction software was used to identify potential CTCF binding motifs in the BZLF1 promoter region (Ziebarth et al., 2013). Electrophoretic mobility shift assay (EMSA) was carried out using DNA oligonucleotides designed with the motif identified and a mutant oligonucleotide carrying point mutations within the motif. Reactions were carried out at room temperature for 30 min in Buffer A (25 mM Tris-HCl pH 7.5, 1 mM DTT, 10 mM NaCl, 0.01% NP-40, 0.1 mM ZnCl2, 0.1 mg/mL BSA, 10% glycerol) containing 0.2 μM CTCF and 5 nM radiolabeled WT or mutated dsDNA. Purified CTCF protein was a kind gift from Paul M. Lieberman (Chau et al., 2006). Competitor dsDNA was titrated in at 0.005–100 nM. Reactions were resolved in 5% non-denaturing polyacrylamide gels and exposed to a phosphor imaging screen. Oligonucleotide sequences for the motifs and competitor DNA are listed in Supplementary Table 1.
2.5. Western blot analysis
Cell lysates were prepared in radioimmunoprecipitate assay (RIPA) lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA; Millipore) supplemented with 1X protease inhibitor cocktail (Thermo Scientific). Protein extracts were obtained by centrifugation at 3000×g for 10 min at 4 °C. For nuclear fractions, nuclear soluble and chromatin-bound fractions were extracted from 2×106 cells using the Subcellular Protein Fractionation Kit for Cultured Cells kit (Invitrogen) according to manufacturer’s instructions. Protein concentration was measured using a bicinchoninic (BCA) protein assay (Pierce). Lysates were boiled with 1X Laemmli sample buffer (Bio-Rad) containing 1.25% β-mercaptoethanol (Sigma-Aldrich). Proteins were resolved by gel electrophoresis on a 4–20% polyacrylamide gradient Mini-Protean TGX precast gel (Bio-Rad) and transferred to an Immobilon-P membrane (Millipore). Membranes were blocked in 5% milk in PBS-T for 1 h at room temperature, and incubated overnight at 4 °C with primary antibodies against CTCF (Millipore), actin (Sigma-Aldrich), Zta (Santa-Cruz), PAR (Trevigen), FLAG (Abcam), gp125 (Abcam), PARP1 (Active Motif), Lamin B1 (Abcam), and H3 (Abcam).
2.6. shRNA-mediated knockdown
Plasmids encoding short hairpin RNAs (shRNA) specific to PARP1 and CTCF were purchased as glycerol stocks from Sigma-Aldrich. Two unique shRNA constructs were used for each target to ensure specificity. A non-targeting scrambled shRNA was a gift from David Sabatini (Addgene; 1864). All shRNA hairpin sequences are listed in Supplemental Table 1. Lentiviral particles were generated by transfecting HEK293T cells with the packaging plasmid psPAX2, the envelope plasmid pMD2.G, and shScramble, shCTCF, or shPARP1 plasmids, according the Addgene protocol. psPAX2 and pMD2.G plasmids were a gift from Didier Trono (Addgene; 12260, 12259). Supernatant containing lentivirus was collected at 48- and 72-h post-transfection, and filtered through a 0.45 μM filter. Akata-EBV cells were transduced by incubating with filtered media as described previously (Raver et al., 2013) and after 48 h, were selected with 1 μg/mL puromycin for a total of 48 h. 72 h post-transduction, Akata-EBV cells were treated with anti-IgG to induce lytic reactivation. Knockdown efficiency was assessed by western blot.
2.7. Viral copy number quantification
DNA was extracted from 1×106 cells using the GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol and quantified by NanoDrop (Thermo Scientific). For qPCR, 100 ng DNA was loaded per well in a master mix containing 1X Maxima SYBR Green (Fisher) and forward and reverse primers for the EBV DS region or a genomic control region (IDT). Primers are listed in Supplementary Table 1. PCR was carried out on an ABI StepOnePlus PCR system. Data were analyzed by the ΔΔCT method relative to the genomic control and normalized to the untreated control.
2.8. Determination of cellular PAR levels
Cellular poly[ADP-ribose] (PAR) levels were quantified using the PARP in vivo Pharmacodynamic Assay 2nd Generation (PDA II) kit (Trevigen) according to the manufacturer’s protocol. Briefly, 3×106 cells were lysed in the provided buffer, and protein concentration was determined with a bicinchoninic acid (BCA) protein assay (Pierce). Cell extracts were added to the provided antibody capture plate, incubated overnight at 4 °C. Wells were washed four times with phosphate-buffered saline containing 0.05% Tween-20 (PBS-T) and incubated with the polyclonal PAR detection antibody at room temperature for 2 h. After washing four times with PBS-T, extracts were incubated with goat-anti-rabbit IgG-HRP antibody at room temperature for 1 h. Wells were washed four times with PBS-T before adding the PARP PeroxyGlow reagent. Luminescence was measured using a POLARstar Optima microplate reader (BMG Labtech).
2.9. Plasmids and transfections
Plasmids for PARP1 and CTCF shRNAs were purchased from Sigma-Aldrich as E. coli glycerol stocks. Upon receipt, Luria broth was inoculated with the stock, a culture was grown overnight at 37 °C shaking, and plasmids were extracted using the Pureyield Plasmid Midiprep System (Promega). The shScramble construct was a gift from David Sabatini (Addgene; 1864). psPAX2 and pMD2.G plasmids were gifts from Didier Trono (Addgene; 12260, 12259). Addgene plasmids were provided as bacterial stabs and prepped as described above. The BZLF1 promoter firefly luciferase construct (LucZ), pRL Renilla luciferase construct, and FLAG-Zta plasmid were all kind gifts from Paul Lieberman. LucZ was generated in the Lieberman lab by amplification of BZLF1 −400 to +40 as an NheI/HindIII fragment in the pGL3 vector (Promega). The wild-type His-PARP1 construct was a generous gift from John Pascal (Langelier et al., 2008). The FLAG-BAP control expression construct was purchased from Sigma-Aldrich. Transfections for lentiviral particle generation were carried out using FuGENE 6 (Promega) according to the Addgene protocol. All other transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions for either 6- or 12-well format.
2.10. Luciferase assay
Luciferase assay was performed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. HEK293 Cells were transfected with pRL, LucZ, and constructs expressing either His-PARP1 or FLAG-Zta using Lipofectamine 2000 according to the manufacturer’s protocol. After 72 h, cells were lysed for 15 min at room temperature in the supplied Passive Lysis Buffer and transferred into a 96-well plate for luciferase assay. LARII and Stop & Glo Reagents were dispensed into wells via automated injectors. Luminescence was measured using the Promega GloMax Multi Detection System plate reader.
2.11. Statistical analysis
All experiments presented were conducted at least in triplicate to ensure reproducibility of results. The Prism statistical software package (GraphPad) was used to identify statistically significant differences between experimental conditions and control samples, using Student’s t-test as indicated in the figure legends.
3. Results
3.1. CTCF binds the BZLF1 promoter, but its binding is not affected by EBV lytic reactivation
Work in Kaposi’s sarcoma-associated herpesvirus has implicated CTCF as a restriction factor in lytic reactivation (Li et al., 2014). ChIP-seq studies in EBV-immortalized lymphoblastoid cell lines have identified numerous CTCF binding sites across the EBV genome (Holdorf et al., 2011; Arvey et al., 2013). Previous work from our lab and others has elucidated the role of CTCF binding in the regulation of EBV latent promoter selection and LMP1/2 expression (Tempera et al., 2011; Chen et al., 2014). Here, we ask whether the CTCF binding site proximal to the promoter of the immediate early gene BZLF1 is relevant to EBV lytic reactivation. EBV-positive B cells were subjected to chromatin immunoprecipitation with a CTCF antibody and probed for enrichment at the BZLF1 CTCF binding site (Fig. 1A). CTCF was enriched over IgG at the binding site in all cell lines examined, but was more heavily enriched in Type III latent cells.
Fig. 1.
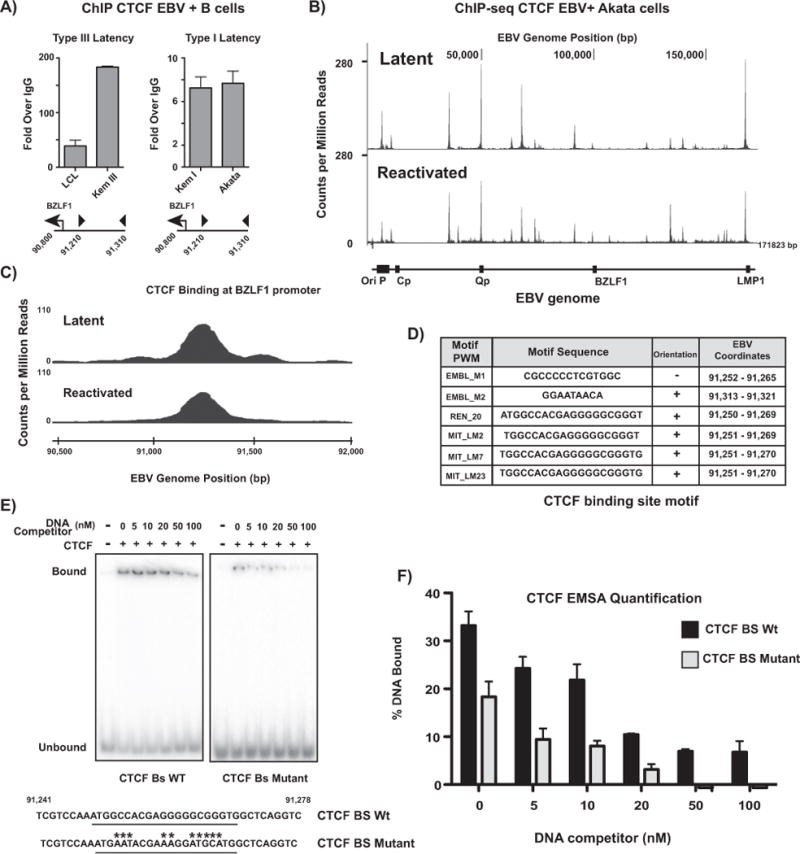
CTCF binds the BZLF1 promoter, but its binding is not affected by EBV lytic reactivation. (A) Chromatin immunoprecipitation (ChIP) for CTCF in type I and type III latent EBV-infected B cells at the BZLF1 promoter. qPCR data are presented as fold over IgG. Results are representative of three independent experiments, and show mean ± standard deviation. (B) ChIP-seq for CTCF in Akata-EBV cells carrying doxycycline-inducible BZLF1. CTCF binding is shown as counts per million reads normalized to total DNA across the EBV genome in latent and doxycycline-reactivated cells. (C) ChIP-seq experiment from (B) zoomed to the CTCF binding site at the BZLF1 promoter. (D) Table showing potential CTCF binding motifs in the BZLF1 promoter region CTCF binding site identified by ChIP-seq. Binding motifs were identified using CTCF binding site prediction algorithms (http://insulatordb.uthsc.edu/). (E) Electrophoretic mobility shift assay showing CTCF protein binding to both wild-type and mutant BZLF1 promoter CTCF binding site DNA. CTCF binding motif is underlined in the genome sequence. Point mutations are designated by asterisks. Gels are representative of three independent experiments. (F) Quantification of CTCF protein bound to wild-type and mutated BZLF1 CTCF binding motif DNA from EMSA in (E). Graph represents the % DNA bound to CTCF in the gel. DNA bound to protein is shown as mean ± standard deviation of gel quantification of three independent experiments.
In herpes simplex virus-1 (HSV-1), CTCF occupancy on the HSV-1 genome is significantly decreased at a number of CTCF binding sites following sodium butyrate-driven viral reactivation (Ertel et al., 2012). We next asked whether lytic reactivation of the virus might alter CTCF binding across the EBV genome. We performed ChIP-seq for CTCF in Akata-EBV cells containing a construct with a doxycycline-inducible BZLF1 promoter (Ramasubramanyan et al., 2015). Cells were treated with doxycycline for 24 h and sorted with magnetic beads to obtain a pure population of reactivated cells (Fig. 1B). CTCF binding was largely unchanged following lytic reactivation, including at the BZLF1 promoter (Fig. 1C), with one notable exception: lytic reactivation enriches for CTCF binding at the leftward OriLyt, a binding site not previously identified in the literature. Similar results were obtained in Mutu cells activated with anti-IgG/IgM (data not shown). We wanted to identify the consensus sequence associated with CTCF binding at the BZLF1 promoter. To identify the specific sequence element bound by CTCF near the BZLF1 promoter, we examined the region for candidate CTCF binding sites using a prediction algorithm (Ziebarth et al., 2013; Fig. 1D). To validate CTCF binding to this motif, we performed an EMSA, using the wild-type binding sequence identified and a mutant sequence (Fig. 1E). Quantification of the percent DNA bound to CTCF indicates that approximately 40% less mutant DNA sequence binds CTCF than the identified consensus sequence (Fig. 1F). Together, these data demonstrate that CTCF binding is unchanged at all but one novel CTCF binding site following viral reactivation.
3.2. CTCF binding at the BZLF1 promoter CTCF binding site is not involved in EBV lytic reactivation
To test whether CTCF restricts EBV lytic reactivation, we knocked down CTCF in Akata EBV cells. To avoid the confounding epigenetic effects of chemical inducers such as sodium butyrate and 5-azacytidine, we used anti-IgG to induce lytic reactivation in Akata EBV cells. CTCF knockdown was achieved by infecting Akata EBV cells with lentivirus containing shRNAs targeting CTCF. Protein knockdown was validated by western blot (Fig. 2A) and ChIP assays were performed to ensure that CTCF knockdown effectively removed CTCF from the BZLF1 CTCF binding site (Fig. 2B). Despite nearly complete CTCF knockdown, there was no effect on EBV lytic reactivation as measured by viral copy number (Fig. 2C) and Zta western blot (Fig. 2D). Collectively, these data suggest that CTCF binding proximal to the BZLF1 promoter is not relevant for EBV lytic reactivation.
Fig. 2.
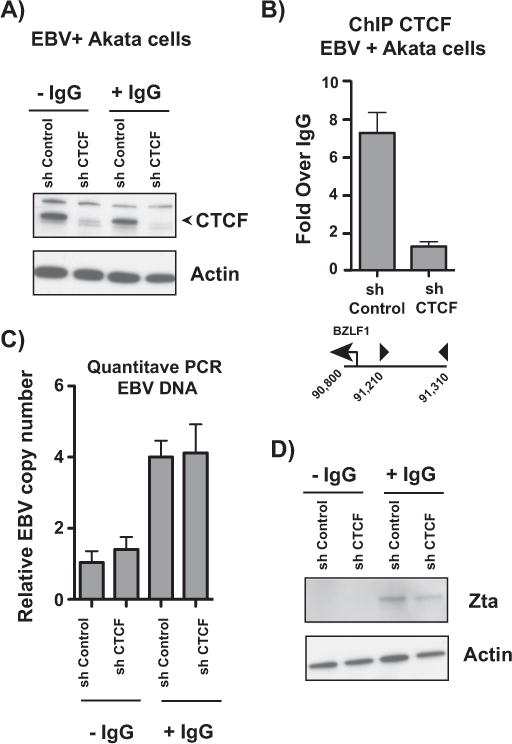
CTCF binding at the BZLF1 promoter CTCF binding site is not involved in EBV lytic reactivation. (A) Western blot showing CTCF knockdown in Akata-EBV cells. Actin served as a loading control. Cells were infected with lentivirus containing either non-targeting or CTCF shRNAs and induced with anti-IgG. Blot is representative of three independent knockdown experiments. (B) ChIP-qPCR for CTCF at the BZLF1 promoter in Akata-EBV cells infected with shControl or shCTCF lentivirus. Results are representative of three independent experiments, and show mean ± standard deviation. (C) qPCR showing EBV copy number in Akata-EBV cells under latent and anti-IgG-induced conditions after CTCF knockdown. Data are shown as EBV copy number relative to a host genomic control. Results are representative of three independent experiments, and show mean ± standard deviation. (D) Cells were treated as in (A) and protein lystates were subjected to western blot, probing for the viral protein Zta. Actin served as a loading control.
3.3. PARP1 colocalizes with CTCF at the BZLF1 CTCF binding site
CTCF has emerged as an important cofactor of the host protein PARP1 (Farrar et al., 2011; Ong et al., 2013; Nalabothula et al., 2015; Zhao et al., 2015). We hypothesized that PARP1 and CTCF may cooperate at the BZLF1 promoter to collectively restrict EBV lytic reactivation. We performed ChIP assays to assess PARP1 binding at the BZLF1 CTCF binding site in a panel of EBV-infected B cells (Fig. 3A). PARP1 binds the site at similar levels in all cell lines examined except for Kem I, in which PARP1 is more heavily enriched. Less PARP1 binding in these cell lines could explain why type III latent cells have greater baseline lytic expression, and why Akata EBV cells reactivate easily in vitro. In Akata EBV cells, PARP1 binding increases at the BZLF1 CTCF binding site upon reactivation with anti-IgG (Fig. 3B). This finding could indicate that the host selectively enriches for PARP1 during reactivation to restrict further activity from the promoter. In this way, PARP1 may act as a barrier against complete viral reactivation.
Fig. 3.
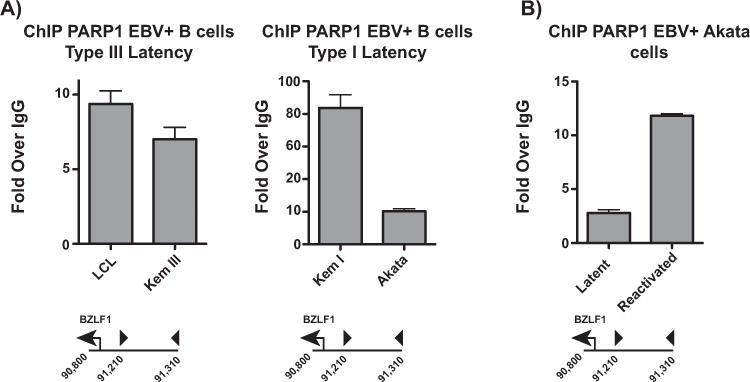
PARP1 colocalizes with CTCF at the BZLF1 CTCF binding site. (A) ChIP-qPCR for PARP1 in type I and type III latent EBV-infected B cells at the BZLF1 promoter. qPCR data are presented as fold over IgG. Results are representative of three independent experiments, and show mean ± standard deviation. (B) ChIP-qPCR for PARP1 in Akata-EBV cells during latency and after anti-IgG-induced reactivation. qPCR data are presented as fold over IgG. Results are representative of three independent experiments, and show mean ± standard deviation.
3.4. EBV lytic reactivation attenuates PARP activity through Zta
To further investigate the involvement of PARP in EBV lytic reactivation, both EBV-negative and EBV-positive Akata B cells were treated with anti-IgG to reactivate the virus over a 72-h time course. PARP activity was measured using a commercially available PAR ELISA (Fig. 4A) and PARylation of PARP1 was observed by western blot as a read-out of cellular PARP activity (Fig. 4B). In both cell lines, PAR levels increased dramatically at 6 h. However, in comparison to the EBV-negative cells, in the EBV-positive Akata cells, PARP activity was attenuated by approximately 2-fold between six and 24 h. In our system, this decrease in PARP activity is concurrent with peak expression of immediate-early proteins, specifically Zta (Fig. 4B). Thus, we hypothesized that Zta itself may be responsible for decreasing PARP activity. HEK293 cells were transfected with FLAG-tagged Zta and treated with MNNG to activate PARP to mimic the activation of PARP concurrent with Zta expression in B cells (Fig. 4D). PARP activity was measured by ELISA, and is shown here as relative PAR levels (Fig. 4C). Relative to a FLAG-tagged control protein, Zta decreased PARP activity in MNNG-activated cells by approximately 20% (p=0.0238), demonstrating that Zta expression alone is sufficient to decrease PARP activity.
Fig. 4.
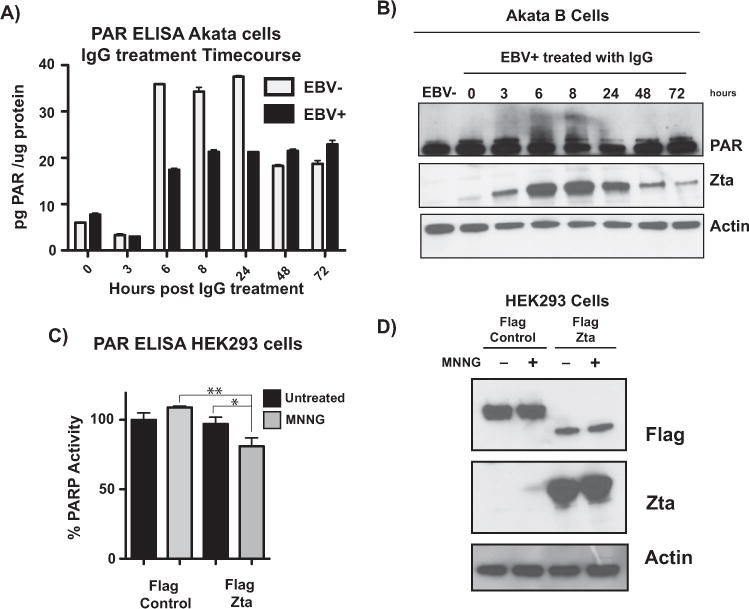
EBV lytic reactivation attenuates PARP activity through Zta. (A) ELISA measuring PAR levels in EBV-negative and -positive Akata cells. Cells were treated with anti-IgG over a 72-h time course, and protein lysates were subjected to ELISA. Data are quantified as pg of PAR per μg of protein, and show mean ± standard deviation; graph is representative of three independent experiments. (B) Western blot for protein lysates from (A) probing for PAR and Zta; actin served as a loading control. PAR western blot shows PARylated PARP1 as a readout of cellular PARP activity. (C) ELISA measuring PAR levels in HEK293 cells. Cells were transfected with FLAG-control or FLAG-Zta vectors and treated with MNNG to activate PARP. Data are shown as % PARP activity relative to the untreated control. Statistical significance is indicated by asterisks (**, p < 0.05; *, p < 0.1) (D) Western blot with lysates from (C), probing for FLAG and Zta to confirm transfection; actin served as loading control.
3.5. PARP inhibition with olaparib modestly affects Epstein Barr virus lytic reactivation
Since Zta expression inhibits PARP activity, we predicted that pharmacological inhibition of PARP activity would be advantageous for the virus, and enhance lytic reactivation. To test this hypothesis, Akata EBV cells were activated with anti-IgG and treated with 2.5 μM olaparib for 24 h to inhibit PARP1 activity (Fig. 5A). qPCR quantification of EBV copy number revealed that EBV lytic replication was only modestly increased (p=0.0172) after anti-IgG treatment (Fig. 5B). RNA was extracted from Akata EBV cells treated with anti-IgG after 0, 8, and 24 h. At 8 h, BZLF1 expression was significantly enhanced by olaparib treatment; this difference in expression was, however resolved by 24 h (Fig. 5C). We predict that olaparib’s ability to trap PARP1 to the DNA may contribute to inhibiting further transcription from the BZLF1 promoter (Shen et al., 2015). Western blot analysis of lytic proteins revealed no obvious differences in lytic protein expression with PARP inhibition (Fig. 5D).
Fig. 5.
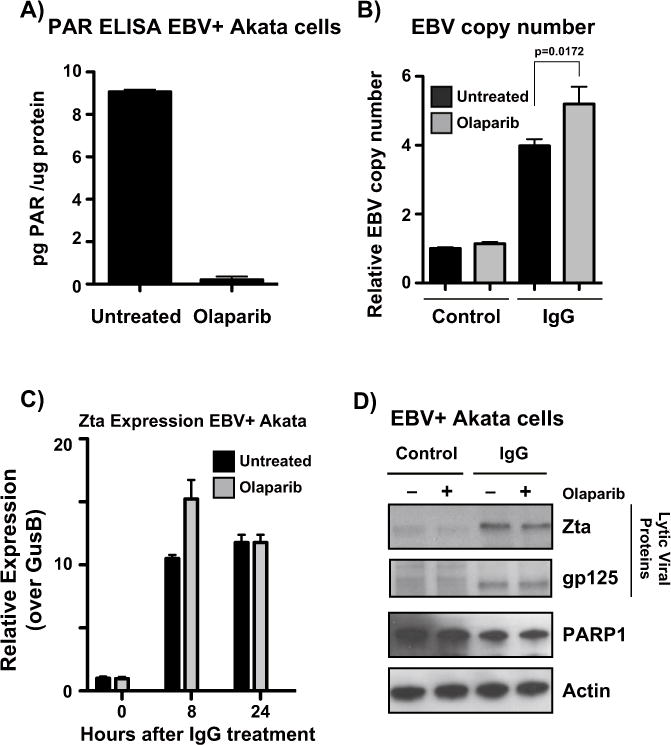
PARP inhibition with olaparib modestly affects Epstein Barr virus lytic reactivation. (A) ELISA measuring PAR levels in Akata-EBV cells. Cells were treated with 2.5 μM olaparib for 24 h to inhibit PARP activity. Data are shown as pg of PAR per μg of protein. (B) qPCR showing EBV copy number in Akata-EBV cells under latent and anti-IgG-induced conditions after olaparib treatment. Data are shown as EBV copy number relative to a host genomic control. Results are representative of three independent experiments, and show mean ± standard deviation. (C) qRT-PCR showing relative expression of BZLF1 in Akata-EBV cells. Cells were treated with 2.5 μM olaparib for 24 h and IgG for 0, 8, or 24 h. Graph is representative of three independent experiments, and shows mean ± standard deviation. (D) Western blot for protein lysates from (B); blot was probed for viral lytic proteins Zta and gp125, as well as PARP1, and actin as a loading control.
3.6. PARP1 knockdown enhances Epstein Barr virus lytic reactivation
Recent work in the PARP field has revealed new functions for PARP1 beyond its enzymatic activity (Zhao et al., 2015; Nalabothula et al., 2015). We hypothesized that in the context of EBV lytic reactivation, PARP1 binding may be more relevant than its activity. To understand the functional role of PARP1 in EBV lytic reactivation, Akata EBV cells were infected with lentivirus containing shRNAs for PARP1 and activated with anti-IgG (Fig. 6A). PARP1 knockdown alone was sufficient to drive EBV lytic replication, and significantly increased EBV copy number in the context of anti-IgG activation (Fig. 6B). Western blot analyses of Zta and the late lytic protein gp125 show that PARP1 knockdown enhances EBV lytic reactivation (Fig. 6C).
Fig. 6.
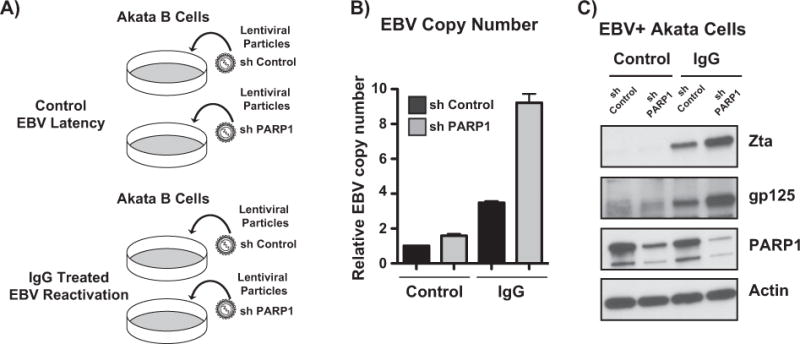
PARP1 knockdown enhances Epstein Barr virus lytic reactivation. (A) Akata-EBV cells were infected with lentivirus particles carrying non-targeting or PARP1 shRNAs. Cells were selected with puromycin, and treated with anti-IgG for 24 h to induce EBV reactivation. (B) qPCR showing EBV copy number in Akata-EBV cells under latent and anti-IgG-induced conditions after PARP1 knockdown. Data are shown as EBV copy number relative to a host genomic control. Results are representative of three independent experiments, and show mean ± standard deviation. (C) Western blot for lysates from experiment shown in (A). Blot was probed for PARP1 to confirm knockdown, and viral proteins Zta and gp125; actin served as a loading control.
3.7. PARP inhibition and PARP1 knockdown eliminate CTCF binding from the BZLF1 promoter in EBV-positive B cells
Our ChIP-seq data show that CTCF binding is not significantly altered at the CTCF binding site at the BZLF1 promoter after lytic reactivation. However, colocalization of CTCF and PARP1 prompted us to ask whether there exists some functional interplay between the two proteins. We reactivated Akata EBV cells with anti-IgG and performed CTCF ChIP assays at the promoter with either PARP inhibition or PARP1 knockdown. With both PARP inhibition and PARP1 knockdown, CTCF was removed from the binding site at the BZLF1 promoter (Fig. 7A,B). Since our data show that PARP1 knockdown, but not PARP inhibition significantly increases BZLF1 expression, CTCF is likely removed from the site as an indirect consequence of the loss of PARP activity. Olaparib treatment in type I Akata EBV and type III LCL cells also eliminates CTCF binding from the BZLF1 promoter (Fig. 7C,D). Since olaparib effectively phenocopies CTCF knockdown at the BZLF1 CTCF binding site, we treated a panel of type I and type III latent cells with olaparib to assess the effects of CTCF at the BZLF1 promoter on EBV reactivation. EBV copy number is unaffected by the removal of CTCF from the BZLF1 promoter in both type I and type III cell lines (Fig. 7E). These data further solidify the notion that the presence of CTCF at this binding site is not relevant for EBV lytic expression per se.
Fig. 7.
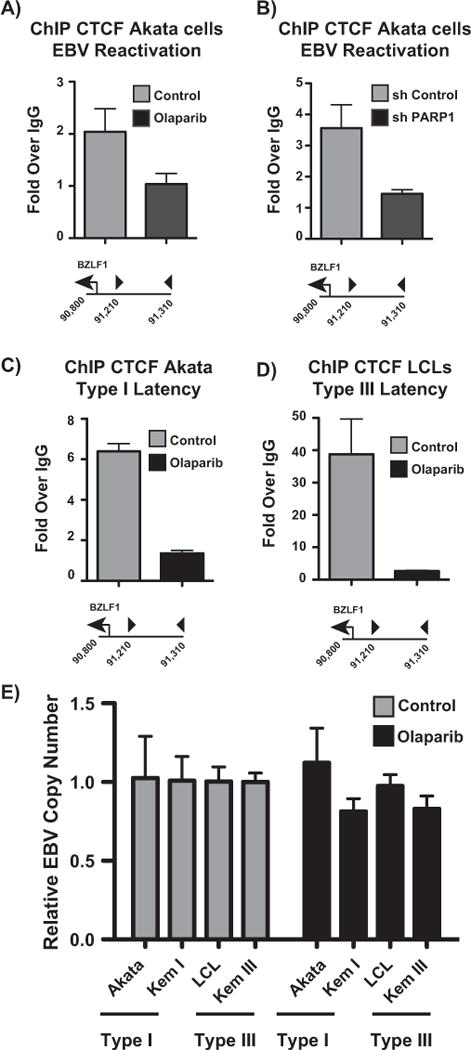
PARP inhibition and PARP1 knockdown eliminate CTCF binding from the BZLF1 promoter in EBV-positive B cells. (A) ChIP-qPCR for CTCF at the BZLF1 promoter in reactivated Akata-EBV cells treated with 2.5 μM olaparib for 24 h. (B) ChIP-qPCR for CTCF at the BZLF1 promoter in reactivated Akata-EBV cells infected with lentivirus containing shControl or shPARP1. (C) ChIP-qPCR for CTCF at the BZLF1 promoter in type I latent Akata-EBV cells with and without olaparib treatment. (D) ChIP-qPCR for CTCF at the BZLF1 promoter in type III latent LCLs with and without olaparib treatment. (E) qPCR showing EBV copy number in a panel of type I and III latent EBV-positive B cell lines. Cells were treated with 2.5 μM olaparib for 24 h. Data are shown as EBV copy number relative to a host genomic control. Results in (A–E) are representative of three independent experiments, and show mean ± standard deviation.
3.8. PARP inhibition and PARP1 knockdown increase Zta binding to the BZLF1 promoter
We activated Akata EBV cells with anti-IgG, with and without olaparib treatment, and performed cellular fractionation to obtain nuclear-free and chromatin-bound protein fractions (Fig. 8A). Western blot for Zta demonstrates that when PARP is inhibited, Zta becomes more enriched in the chromatin-bound fraction, while PARP1 localization is maintained. Enrichment of Zta in the chromatin-bound fraction suggests that the transactivator function of Zta may be enhanced in response to PARP inhibition. In order to determine whether Zta is functioning as a transactivator specifically at the BZLF1 promoter, we assessed Zta binding at the BZLF1 CTCF binding site by ChIP. Indeed, both PARP inhibition (Fig. 8B) and PARP1 knockdown (Fig. 8C) enrich for Zta binding at the BZLF1 promoter, indicating that PARP1 likely acts to prevent Zta transactivator function at the BZLF1 CTCF binding site. Finally, to assess the role of PARP1 and Zta on transcription of the BZLF1 promoter, we performed a luciferase assay. HEK293 cells were transfected with a pGL3 vector expressing luciferase under control of the BZLF1 promoter (LucZ) in addition to either FLAG-tagged Zta or His-tagged PARP1 (Fig. 8E). Zta expression alone is sufficient to drive luciferase expression, while PARP1 expression drives only low-level luciferase expression (Fig. 8D), relative to LucZ alone. Although we do observe slightly increased luciferase activity with PARP1 over the LucZ control, it is possible that this is due to a global increase in transcription with PARP1 overexpression (Krishnakumar and Kraus, 2010; Martin et al., 2015). Overexpression of PARP1, or even Zta alone, is likely not sufficient to drive robust expression from the tightly regulated BZLF1 promoter, especially in uninfected HEK293 cells. These data suggest that there are likely additional factors, both positive and negative, required to turn the BZLF1 promoter to a completely “on” state in B cells.
Fig. 8.
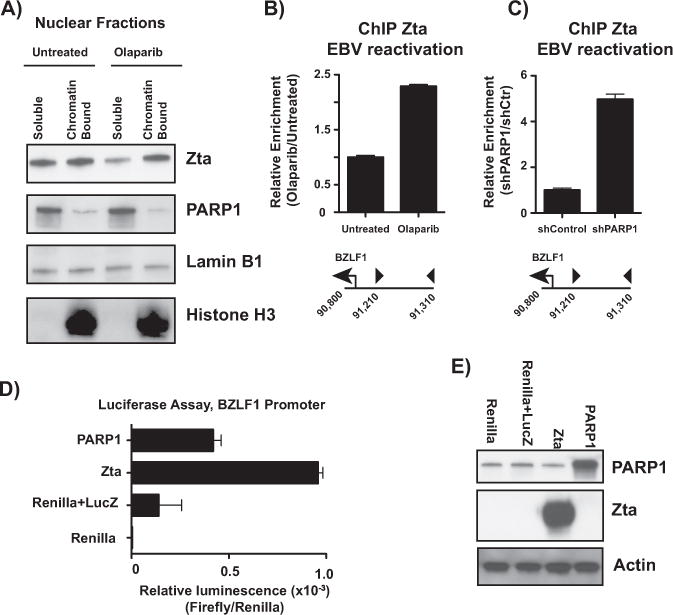
PARP inhibition and PARP1 knockdown increase Zta binding to the BZLF1 promoter. (A) Nuclear soluble and chromatin-bound fractions were extracted from Akata-EBV cells treated with and without olaparib. Fractions were subjected to western blot for Zta, PARP1, Lamin B1 as a nuclear control, and histone H3 as a chromatin fraction control. (B) ChIP-qPCR for Zta in Akata-EBV cells treated with and without olaparib at the BZLF1 promoter. Data are shown as relative enrichment over the untreated control. Data are representative of three independent experiments, and show mean ± standard deviation. (C) ChIP-qPCR for Zta in Akata-EBV cells infected with lentivirus carrying shRNA for PARP1 at the BZLF1 promoter. Data are shown as relative enrichment over the non-targeting control. Data are representative of three independent experiments, and show mean ± standard deviation. (D) HEK293 cells were transfected with constructs expressing Renilla luciferase, firefly luciferase under control of the BZLF1 promoter (LucZ), plus constructs expressing Zta and PARP1. Data are shown as relative luminescence over Renilla luciferase, and show mean ± standard deviation. (E) Western blot of HEK293 lysates from transfection in (D). Blot was probed for PARP1 and Zta to confirm overexpression; actin served as a loading control.
4. Discussion
Taken together, these data support a model in which PARP1 acts as a barrier to productive lytic reactivation. During latency, CTCF and PARP1 are colocalized at the BZLF1 promoter, restricting transcription. Spontaneous reactivation during latency may result in increased PARP1 enrichment at the site, subsequently blocking Zta binding, and ultimately resulting in abortive reactivation (Fig. 9A). Indeed, this may reflect what we observe in anti-IgG induction in cell lines as well as with PARP inhibition—modest, but unsustainable reactivation. Indeed, BZLF1 expression is transiently enhanced by PARP inhibition after anti-IgG induction, indicating that PARP activity may serve as an early barrier to restrict BZLF1 transcription. Early attenuation of PARP activity by EBV during reactivation may enhance transcription from the BZLF1 promoter, but, if PARP1 is still bound to the promoter, and there is an insufficient amount of Zta produced to overcome PARP1 binding, the promoter is not able to switch to a fully “on” state. Additionally, PARylation of Zta may further inhibit the transactivator function of the protein, similar to what has been observed in KSHV (Gwack et al., 2003). During a true reactivation event, Zta would be expressed at levels sufficient to inhibit and downregulate PARP1 and release PARP1 binding from the site, allowing robust BZLF1 expression and complete viral reactivation (Fig. 9B). The mechanisms dictating the interaction between PARP1 and Zta during latency and throughout the process of reactivation are unclear, but there are likely additional factors, both positive and negative, involved. Our data suggest that both PARP catalytic activity and PARP1 binding to the BZLF1 promoter contribute uniquely to the restrictive nature of PARP1. Nonetheless, our data clearly demonstrate that PARP1 restricts EBV lytic reactivation.
Fig. 9.
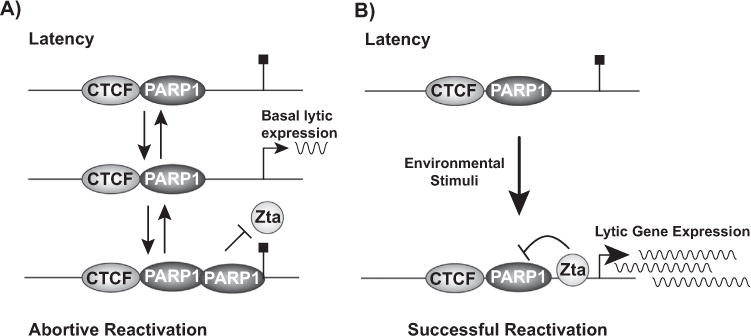
PARP1 binds to the BZLF1 promoter to restrict Epstein Barr Virus lytic reactivation. (A) During latency, CTCF and PARP1 are bound to the BZLF1 promoter to prevent transcription. During a spontaneous, abortive reactivation event, PARP1 is enriched at the BZLF1 promoter. Attenuation of PARP activity by the virus permits transient BZLF1 expression, but PARP enrichment by the host at the promoter ultimately prevents Zta from binding to the promoter, halting BZLF1 transcription. (B) Upon proper stimulation, such as environmental or stress stimuli, Zta is expressed at levels sufficient to inhibit PARP activity and eliminate PARP1 binding from the BZLF1 promoter. When PARP1 is removed from the promoter, Zta binds, and transcription is significantly enhanced to drive robust BZLF1 expression and lytic reactivation.
Antiviral properties of PARP1 have been reported in other gammaherpesviruses. In KSHV, PARP1 interacts with and PARylates Rta to repress Rta-mediated transactivation (Gwack et al., 2003). A recent study identified a KSHV protein that interacts with PARP1, targeting it for proteasomal degradation, and thus reducing levels of inhibitory PARylated-Rta (Cheong et al., 2015). Here, we identify a similar restrictive function for PARP1 in EBV. Mechanistically, our data suggest that binding of PARP1 to the BZLF1 promoter restricts transcription. However, Zta’s ability to repress PARP activity suggests multilayered regulation in respect to both host and virus. For example, it is possible that Zta, like KSHV Rta, is PARylated by PARP1 to regulate its transactivator function; this post-translational mechanism could account for distinct roles for PARP activity and PARP1 binding to the EBV genome in lytic reactivation. Accordingly, we intend to investigate in the future the mechanisms by which PARP1 and Zta manipulate each other throughout the course of lytic reactivation.
Interestingly, a previous study used the PARP1 inhibitor 3-ABA to conclude that PARP inhibition impairs EBV lytic cycle progression (Mattiussi et al., 2007). This apparent discrepancy is likely due to numerous differences in technical approaches. First, 3-aminobenzamide (3-ABA) is a potent inhibitor of apoptosis. In our system, cells treated with olaparib eventually die by apoptosis (data not shown). Thus, the results of the previous study could be attributed primarily to the role of PARP1 in apoptosis, rather than its activity and the subsequent effects on EBV lytic progression, i.e. when apoptosis does not progress, neither does the lytic cycle. Second, the conclusions in this paper are drawn primarily from experiments in Raji cells, in which EBV cannot complete the lytic cycle. In experiments from Jijoye cells, 3-ABA significantly increases the percent of EA-positive cells relative to untreated cells after 24-h induction. Lastly, in their experiments, cells were induced with a combination of phorbol ester, sodium butyrate, and TGF-β2. Because these drugs have broad epigenetic effects, and the underlying mechanisms of lytic reactivation are at least in part epigenetic, we opted for B cell receptor crosslinking for induction. Despite these technical differences, we can conclude that PARP1 certainly plays a distinct role in EBV reactivation.
Although lytic EBV infection is not typically associated with EBV-associated tumors, there is speculation that spontaneous lytic activation may contribute to tumorigenesis (Hong et al., 2005). One such study demonstrated that a “superlytic” virus expressing high levels of BZLF1 could induce lymphomas in humanized mice (Ma et al., 2012). It is thus possible that enhanced expression of BZLF1 might suppress PARP activity, subjecting cells to increased DNA damage and tumorigenic mutations. In addition, the lymphomas in the mice exhibited abortive lytic infection, in which expression of the early lytic protein BMRF1 was mutually exclusive with that of LMP1 in cells. Previously, we identified a positive association between the latency protein LMP1 and PAR levels, in which LMP1 activates PARylation through PARP1 (Martin et al., 2016). Although LMP1 is upregulated during lytic reactivation, paradoxically, LMP1 suppresses lytic induction and progression through a number of unique mechanisms (Chang et al., 2004; Adler et al., 2002; Prince et al., 2003). Our data and others imply a mechanism by which Zta and LMP1 antagonistically manipulate PARP1, at least during the early stages of reactivation. This antagonistic relationship could involve a feedback mechanism in which LMP1 is upregulated by Rta, triggering PARP activity shortly after lytic induction; upon sufficient Zta expression, LMP1 is downregulated, effectively attenuating PARP activity for permissive viral production.
CTCF binding is critical for EBV latency, but has been largely unexplored in the context of lytic reactivation. The Tsurumi lab noted that a recombinant virus carrying point mutations in the CTCF binding site flanking the BZLF1 promoter exhibited no notable differences in BZLF1 expression from wild-type virus (Murata and Tsurumi, 2013). Although these negative data were not presented in the paper, our data are consistent with this observation, that CTCF binding plays little to no role in lytic reactivation in EBV. This is in stark contrast with reports in alphaherpesviruses, in which CTCF occupancy is lost in the LAT, ICP0, and ICP4 regions of the HSV-1 genome following reactivation (Ertel et al., 2012). In the closely related gammaherpesvirus KSHV, reports are mixed regarding the role of CTCF in reactivation; the Lieberman lab observed no effect on KSHV reactivation following CTCF knockdown, whereas the Swaminathan lab reported a massive increase in KSHV virion production (Chen et al., 2012; Li et al., 2014). EBV is unique from other herpesviruses in that it can switch between various latency types in addition to lytic. Accordingly, it is likely that the distinct gene expression programs in EBV are regulated with greater complexity than in other herpesviruses. It is thus possible that in EBV, CTCF is primarily involved in switching between latency types, rather than the lytic switch, as observed in HSV-1 and KSHV.
During latency, CTCF binds at specific sites to mediate latency-type-specific chromatin loops (Tempera et al., 2011). We predict that CTCF binding across the EBV genome functions primarily to maintain chromatin structure and organization. Although CTCF binding is inherently dynamic, cells containing recombinant viruses with mutated CTCF binding sites require a period of months before changes in latency type are observed (Tempera et al., 2010; Hughes et al., 2012). It seems that, at least in cell lines, CTCF binding and CTCF-mediated chromatin loops in EBV are important for stable, long-term changes. This may be explained by the fact that CTCF binding is itself epigenetically regulated, but also dictates epigenetic changes through its insulator function. PARylation of CTCF alters CTCF binding and CTCF-mediated chromatin loops (Ong et al., 2013; Farrar et al., 2010). Early on in this study, we hypothesized that changes in PARP1 activity would drive lytic reactivation by altering CTCF binding. Although our studies did show that PARP1 alters CTCF binding, these changes were not sufficient to drive reactivation. Thus, we conclude that it may be more pertinent to address whether changes in PARP1 activity might alter CTCF binding and subsequent changes in latency. In this study, we show that both PARP inhibition and PARP1 knockdown result in the loss of CTCF binding at the BZLF1 promoter. Even though the loss of CTCF binding at this specific site is irrelevant for lytic reactivation, it is worth investigating whether PARP1 mediates CTCF binding across the EBV genome in the context of latency. LMP1-mediated activation of PARP1 in type III latency may specifically alter CTCF binding and the chromatin landscape of the EBV genome.
5. Conclusions
This study establishes a role for PARP1 in restricting EBV lytic reactivation, and further reveals that CTCF binding to the EBV genome is largely irrelevant to reactivation (Figs. 1 and 2). Furthermore, we show that EBV suppresses PARP activity in the early stages of viral reactivation, and that Zta expression alone is sufficient to decrease PARP activity (Fig. 4). We predicted that restricting PARP1 activity would be advantageous for the virus; however, PARP1 inhibition is insufficient to enhance complete reactivation of the virus (Fig. 5). Knockdown of PARP1 significantly enhances lytic reactivation, suggesting that PARP1 binding restricts viral reactivation (Fig. 6). Indeed, EBV reactivation is enhanced only when PARP1 is absent from the BZLF1 promoter (data not shown). We propose a model in which PARP1’s enzymatic activity and PARP1 binding play distinct roles in initiating EBV lytic reactivation, but ultimately, PARP1 binding at the BZLF1 promoter acts as a strong barrier to robust BZLF1 expression. Here, we demonstrate that PARP1 binds to the BZLF1 promoter to restrict EBV lytic reactivation, and that CTCF, although important in reactivation of other herpesviruses, is dispensable for EBV lytic reactivation.
Supplementary Material
Acknowledgments
We thank Jeffery T. Sample (Pennsylvania State University) and Paul M. Lieberman (The Wistar Institute) for providing cell lines, as well as the aforementioned individuals for providing various reagents. We also thank Xuan Mo and Rachael Werner for technical assistance. This work was supported by a National Institutes of Health grant to I.T. from the National Institute of Allergy and Infectious Diseases (R00AI099153). Study design, data collection and interpretation, and the publication of the work were not influenced by our funding source.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.virol.2017.04.006.
References
- Adler B, Schaadt E, Kempkes B, Zimber-Strobl U, Baier B, Bornkamm GW. Control of Epstein-Barr virus reactivation by activated CD40 and viral latent membrane protein 1. Proc Natl Acad Sci USA. 2002;99(1):437–442. doi: 10.1073/pnas.221439999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvey A, Tempera I, Lieberman P. Interpreting the Epstein-Barr virus (EBV) epigenome using high-throughput data. Viruses. 2013;5(4):1042–4254. doi: 10.3390/v5041042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Lee HH, Chang SS, Hsu TY, Wang PW, Chang YS, et al. Induction of Epstein-Barr virus latent membrane protein 1 by a lytic transactivator Rta. J Virol. 2004;78(23):13028–13036. doi: 10.1128/JVI.78.23.13028-13036.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau CM, Zhang XY, McMahon SB, Lieberman PM. Regulation of Epstein-Barr virus latency type by the chromatin boundary factor CTCF. J Virol. 2006;80(12):5723–5732. doi: 10.1128/JVI.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Wikramasinghe P, Showe L, Lieberman PM. Cohesins repress kaposi’s sarcoma-associated herpesvirus immediate early gene transcription during latency. J Virol. 2012;86(17):9454–9464. doi: 10.1128/JVI.00787-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Martin KA, Lu F, Lupey LN, Mueller JM, Lieberman PM, Tempera I. Epigenetic deregulation of the LMP1/LMP2 locus of Epstein-Barr virus by mutation of a single CTCF-cohesin binding site. J Virol. 2014;88(3):1703–1713. doi: 10.1128/JVI.02209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong WC, Park JH, Kang HR, Song MJ. Downregulation of Poly(ADP-Ribose) polymerase 1 by a viral processivity factor facilitates lytic replication of gammaherpesvirus. J Virol. 2015;89(18):9676–9682. doi: 10.1128/JVI.00559-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Young JM, Kerns JA, Malik HS. Rapid evolution of PARP genes suggests a broad role for ADP-ribosylation in host-virus conflicts. PLoS Genet. 2014;10(5):e1004403. doi: 10.1371/journal.pgen.1004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day L, Chau CM, Nebozhyn M, Rennekamp AJ, Showe M, Lieberman PM. Chromatin profiling of Epstein-Barr virus latency control region. J Virol. 2007;81(12):6389–6401. doi: 10.1128/JVI.02172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel MK, Cammarata AL, Hron RJ, Neumann DM. CTCF occupation of the herpes simplex virus 1 genome is disrupted at early times postreactivation in a transcription-dependent manner. J Virol. 2012;86(23):12741–12759. doi: 10.1128/JVI.01655-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar D, Rai S, Chernukhin I, Jagodic M, Ito Y, Yammine S, et al. Mutational analysis of the poly(ADP-ribosyl)ation sites of the transcription factor CTCF provides an insight into the mechanism of its regulation by poly(ADP-ribosyl) ation. Mol Cell Biol. 2010;30(5):1199–1216. doi: 10.1128/MCB.00827-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar D, Chernukhin I, Klenova E. Poly(ADP-ribose) Polymerase. Vol. 780. Humana Press; Totowa, NJ: 2011. Generation of Poly(ADP-ribosyl)ation deficient mutants of the transcription factor, CTCF; pp. 293–312. 〈. 〉. [DOI] [PubMed] [Google Scholar]
- Flower K, Thomas D, Heather J, Ramasubramanyan S, Jones S, Sinclair AJ. Epigenetic control of viral life-cycle by a DNA-methylation dependent transcription factor. PLoS One. 2011;6(10):e25922. doi: 10.1371/journal.pone.0025922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y, Nakamura H, Lee SH, Souvlis J, Yustein JT, Gygi S, et al. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol Cell Biol. 2003;23(22):8282–8294. doi: 10.1128/MCB.23.22.8282-8294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt W. The epigenetic life cycle of Epstein-Barr virus. Curr Top Microbiol Immunol. 2015;390(Pt1):103–117. doi: 10.1007/978-3-319-22822-8_6. [DOI] [PubMed] [Google Scholar]
- Holdorf MM, Cooper SB, Yamamoto KR, Miranda JL. Occupancy of chromatin organizers in the Epstein–Barr virus genome. Virology. 2011;415(1):1–5. doi: 10.1016/j.virol.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol. 2005;79(22):13993–14003. doi: 10.1128/JVI.79.22.13993-14003.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DJ, Marendy EM, Dickerson CA, Yetming KD, Sample CE, Sample JT. Contributions of CTCF and DNA methyltransferases DNMT1 and DNMT3B to Epstein-Barr virus restricted latency. J Virol. 2012;86(2):1034–1045. doi: 10.1128/JVI.05923-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell. 2010;39(5):736–749. doi: 10.1016/j.molcel.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier MF, Servent KM, Rogers EE, Pascal JM. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J Biol Chem. 2008;283(7):4105–4114. doi: 10.1074/jbc.M708558200. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudisi F, Sambucci M, Pioli C. Poly (ADP-Ribose) polymerase-1 (PARP-1) as immune regulator. Endocr Metab Immune Disord Drug Targets. 2011;11(4):326–333. doi: 10.2174/187153011797881184. [DOI] [PubMed] [Google Scholar]
- Li DJ, Verma D, Mosbruger T, Swaminathan S. CTCF and Rad21 Act as host cell restriction factors for Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog. 2014;10(1):e1003880. doi: 10.1371/journal.ppat.1003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26(5):417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma SD, Yu X, Mertz JE, Gumperz JE, Reinheim E, Zhou Y, et al. An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J Virol. 2012;86(15):7976–7987. doi: 10.1128/JVI.00770-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KA, Lupey LN, Tempera I. Epstein-Barr virus oncoprotein LMP1 mediates epigenetic changes in host gene expression through PARP1. J Virol. 2016;90(19):8520–8530. doi: 10.1128/JVI.01180-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KA, Cesaroni M, Denny MF, Lupey LN, Tempera I. Global transcriptome Analysis reveals that Poly(ADP-Ribose) polymerase 1 regulates gene expression through EZH2. Mol Cell Biol. 2015;35(23):3934–3944. doi: 10.1128/MCB.00635-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiussi S, Tempera I, Matusali G, Mearini G, Lenti L, Fratarcangeli S, et al. Inhibition of Poly(ADP-ribose)polymerase impairs Epstein Barr Virus lytic cycle progression. Infect Agents Cancer. 2007;2(1):18. doi: 10.1186/1750-9378-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie J, El-Guindy A. Epstein Barr Virus. Vol. 2. Vol. 391. Springer International Publishing; Cham: 2015. Epstein-Barr virus lytic cycle reactivation; pp. 237–261. 〈. 〉. [DOI] [PubMed] [Google Scholar]
- Murata T, Tsurumi T. Epigenetic modification of the Epstein-Barr virus BZLF1 promoter regulates viral reactivation from latency. Front Genet. 2013;4 doi: 10.3389/fgene.2013.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalabothula N, Al-jumaily T, Eteleeb AM, Flight RM, Xiaorong S, Moseley H, et al. Genome-wide profiling of PARP1 reveals an interplay with gene regulatory regions and DNA methylation. PLoS One. 2015;10(8):e0135410. doi: 10.1371/journal.pone.0135410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Van Bortle K, Ramos E, Corces VG. Poly(ADP-ribosyl)ation regulates insulator function and intrachromosomal interactions in Drosophila. Cell. 2013;155(1):148–159. doi: 10.1016/j.cell.2013.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentland I, Parish J. Targeting CTCF to control virus gene expression: a common theme amongst diverse DNA viruses. Viruses. 2015;7(7):3574–3585. doi: 10.3390/v7072791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan AT, Fernandez SG, Somberg JJ, Keck KM, Miranda JL. Epstein-Barr virus latency type and spontaneous reactivation predict lytic induction levels. Biochem Biophys Res Commun. 2016;474(1):71–75. doi: 10.1016/j.bbrc.2016.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince S, Keating S, Fielding C, Brennan P, Floettmann E, Rowe M. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycle induction and progress via different mechanisms. J Virol. 2003;77(8):5000–5007. doi: 10.1128/JVI.77.8.5000-5007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasubramanyan S, Osborn K, Flower K, Sinclair AJ. DynaMic chromatin environment of key lytic cycle regulatory regions of the Epstein-barr virus genome. J Virol. 2012;86(3):1809–1819. doi: 10.1128/JVI.06334-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasubramanyan S, Osborn K, Al-Mohammad R, Naranjo Perez-Fernandez IB, Zuo J, Balan N, et al. Epstein-Barr virus transcription factor Zta acts through distal regulatory elements to directly control cellular gene expression. Nucleic Acids Res. 2015;43(7):3563–3577. doi: 10.1093/nar/gkv212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raver RM, Panfil AR, Hagemeier SR, Kenney SC. The B-cell-specific transcription factor and master regulator Pax5 promotes Epstein-Barr virus latency by negatively regulating the viral immediate early protein BZLF1. J Virol. 2013;87(14):8053–8063. doi: 10.1128/JVI.00546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado MM, Bennici E, Novelli F, Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology. 2013;139(4):428–437. doi: 10.1111/imm.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiewer MJ, Knudsen KE. Transcriptional roles of PARP1 in cancer. Mol Cancer Res: MCR. 2014;12(8):1069–1080. doi: 10.1158/1541-7786.MCR-13-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Aoyahi-Scharber M, Wang B. Trapping poly(ADP-ribose) polymerase. J Pharmacol Exp Ther. 2015;353(3):446–457. doi: 10.1124/jpet.114.222448. [DOI] [PubMed] [Google Scholar]
- Tempera I, Klichinsky M, Lieberman PM. EBV latency types adopt alternative chromatin conformations. PLoS Pathog. 2011;7(7):e1002180. doi: 10.1371/journal.ppat.1002180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempera I, Wiedmer A, Dheekollu J, Lieberman PM. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 2010;6(8):e1001048. doi: 10.1371/journal.ppat.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Yu X. Functions of PARylation in DNA damage repair pathways. Genom Proteom Bioinform. 2016;14(3):131–139. doi: 10.1016/j.gpb.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Ginjala V, Pant V, Chernukhin I, Whitehead J, Docquier F, et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat Genet. 2004;36(10):1105–1110. doi: 10.1038/ng1426. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sifakis EG, Sumida N, Millán-Ariño L, Scholz BA, Svensson JP, et al. PARP1- and CTCF-mediated interactions between active and repressed chromatin at the lamina promote oscillating transcription. Mol Cell. 2015;59(6):984–997. doi: 10.1016/j.molcel.2015.07.019. [DOI] [PubMed] [Google Scholar]
- Ziebarth JD, Bhattacharya A, Cui Y. CTCFBSDB 2.0: a database for CTCF-binding sites and genome organization. Nucleic Acids Res. 2013;41(D1):D188–D194. doi: 10.1093/nar/gks1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.