

Abstract
Background:
Opioids are critical for managing cancer pain, but may provide inadequate relief and/or unacceptable side effects in some cases.
Objective:
To assess the analgesic efficacy of adjunctive Sativex (Δ9-tetrahydrocannabinol (27 mg/mL): cannabidiol (25 mg/mL)) in advanced cancer patients with chronic pain unalleviated by optimized opioid therapy.
Methods:
This report describes two phase 3, double-blind, randomized, placebo-controlled trials. Eligible patients had advanced cancer and average pain numerical rating scale (NRS) scores ≥4 and ≤8 at baseline, despite optimized opioid therapy. In Study-1, patients were randomized to Sativex or placebo, and then self-titrated study medications over a 2-week period per effect and tolerability, followed by a 3-week treatment period. In Study-2, all patients self-titrated Sativex over a 2-week period. Patients with a ≥15% improvement from baseline in pain score were then randomized 1:1 to Sativex or placebo, followed by 5-week treatment period (randomized withdrawal design).
Results:
The primary efficacy endpoint (percent improvement (Study-1) and mean change (Study-2) in average daily pain NRS scores) was not met in either study. Post hoc analyses of the primary endpoints identified statistically favourable treatment effect for Sativex in US patients <65 years (median treatment difference: 8.8; 95% confidence interval (CI): 0.00–17.95; p = 0.040) that was not observed in patients <65 years from the rest of the world (median treatment difference: 0.2; 95% CI: −5.00 to 7.74; p = 0.794). Treatment effect in favour of Sativex was observed on quality-of-life questionnaires, despite the fact that similar effects were not observed on NRS score. The safety profile of Sativex was consistent with earlier studies, and no evidence of abuse or misuse was identified.
Conclusions:
Sativex did not demonstrate superiority to placebo in reducing self-reported pain NRS scores in advanced cancer patients with chronic pain unalleviated by optimized opioid therapy, although further exploration of differences between United States and patients from the rest of the world is warranted.
Keywords: Pain, advanced cancer pain, cannabinoids, opioids, numerical rating scale, randomized control trial
Introduction
Chronic pain is highly prevalent in patients with advanced cancers. Thus, among the more than 30 million cancer patients worldwide, cancer-related pain is estimated to affect 24–60% of patients undergoing active treatment and 62–90% of those with incurable late-stage disease.1,2 This pain harms the functional capacity and psychological well-being of patients, while imposing substantial burdens on the overall healthcare system.3–5 Opioids, the most important tools for managing moderate to severe cancer pain, play central and indispensable roles in the cancer-pain treatment ladder as advocated by the World Health Organization.6,7 Nonetheless, opioid therapy is highly associated with significant and unacceptable side effects and may provide inadequate relief in some cancer patients, even following dose adjustment or in combination with standard adjuvant analgesics.8 As a result, too many patients spend their last weeks, months or even years in discomfort and disability.9 Thus, a substantial unmet need exists for new analgesics that effectively supplement opioids in patients with significant cancer pain not wholly alleviated by opioids.
Cannabinoids (CBs) have been identified as potential adjuvant analgesics in the setting of cancer pain.10 CBs are synthesized in small glands or trichomes on the flowers and main fan leaves of late-stage cannabis plants.11 These trichomes exude a resin containing a specific mixture of compounds unique to each genetic strain of plant (chemotype). Sativex is an oral mucosal spray formulated from Cannabis sativa L extracts that contains two potentially therapeutic CBs, Δ9-tetrahydrocannabidiol (THC) and cannabidiol (CBD), in an approximate 1:1 ratio.12 The extract also contains smaller amounts of other compounds, including minor CBs, terpenoids, flavonoids and sterols.13
Two prior randomized double-blind phase 2/3 studies demonstrated that Sativex had encouraging analgesic effects in advanced cancer patients with pain not fully alleviated by opioid therapy.14–16 Both studies enrolled patients with baseline scores ≥4 on a 0- to 10-point average daily pain numerical rating scale (NRS), despite ongoing treatment with opioids. In one of the studies, participants were randomized to receive either low-dose Sativex (1–4 sprays/day; n = 91), medium-dose Sativex (6–10 sprays/day; n = 88), high-dose Sativex (11–16 sprays/day; n = 90), or matching placebo (n = 91).16 The primary efficacy endpoint, that is, 30% response rate on an average daily pain NRS, was similar for Sativex and placebo (treatment effect, p = 0.59). However, a secondary continuous responder analysis of average daily pain demonstrated that the proportion of patients reporting analgesia was greater for Sativex than placebo overall (p = 0.035), specifically in the low-dose (p = 0.008) and medium-dose (p = 0.039) groups. In the low-dose group, results were similar for mean average pain (p = 0.006), mean worst pain (p = 0.011), and mean sleep disruption (p = 0.003). High-dose Sativex was not effective in the trial and demonstrated poor tolerability, indicating that 11 or more sprays per day provides an unsatisfactory risk/benefit profile in most patients.
In the other reported study, patients with advanced cancer and inadequate pain-relief despite optimized opioid care were randomized to 2 weeks of treatment with Sativex (n = 60), THC alone (n = 58) or placebo (n = 59).14 The primary analysis, change from baseline in average daily pain NRS score, favoured Sativex over placebo (−1.37 vs. −0.69; p = 0.024), while the THC group lacked a statistically significant benefit in the same comparison (−1.01 vs. −0.69; p = 0.204). Twice as many patients in the Sativex group exhibited a 30% decrease on the pain NRS, considered a clinically relevant response,17 compared to patients in the placebo group (23 (43%) vs. 12 (21%); odds ratio (OR) = 2.81; p = 0.006). By comparison, the numbers of patients with clinically significant responses in the THC and placebo groups were similar (12 (23%) vs. 12 (21%); OR = 1.10; p = 0.28). In another open-label extension study,15 39 patients who completed the 2-week titration continued on Sativex (n = 39). Sativex treatment was well tolerated, with no evidence of loss of effect for the relief of cancer pain with long-term use and did not seek to increase their dose. Median treatment duration was 25 days (range, 2–529 days). Change from baseline in mean Brief Pain Inventory-Short Form scores for the ‘pain severity’ and ‘worst pain’ domains improved at each visit in the Sativex treatment group. Similarly, the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-C30 scores showed improvements from baseline scores in the domains of insomnia, pain and fatigue.
Given these encouraging results and the enormous need for new analgesics in this setting, it was of great interest to determine the effectiveness of Sativex in advanced cancer patients suffering from cancer-related pain unalleviated by opioids. To do so, three phase 3 clinical studies were conducted, using pain NRS scores as the primary endpoint. The studies also assessed the effects of Sativex on opioid consumption, sleep disruption and patient quality of life. Due to the significant amount of data generated across the programme, the current report describes combined results from only two of the three trials. An additional report describing results from the third trial will be published elsewhere.
Methods
Ethics
The studies described in this report were in compliance with International Conference on Harmonization Good Clinical Practice guidelines for conducting, recording and reporting clinical trials, as well as for archiving essential documents. Consistent with ethical principles for the protection of human research subjects, no trial procedures were performed on trial candidates until written consent had been obtained. The informed consent form, protocol and amendments for the study were submitted to and approved by the institutional review board or independent ethics committee for each respective trial site or country.
Study designs
Both trials described in this report were phase 3, double-blind, multicenter, randomized, placebo-controlled trials. Study 1 (ClinicalTrials.gov identifier: NCT01361607) had a classic randomized clinical trial (RCT) design in which patients were randomized prior to receiving study therapy. Study 2 (NCT01424566) had an enriched enrolment with randomized withdrawal (EERW) design chosen to detect clinically significant analgesic effect (responders group), in which patients were titrated with active treatment and only randomized if satisfactory effect was achieved; consequently, patients who could not tolerate or respond to active treatment were excluded from final efficacy analyses. Both trials were in accordance with the Initiative on Methods, Measurement and Pain Assessment in Clinical Trials II (IMMPACT II), which recommends appropriate methodologies and core outcome measures in chronic pain clinical trials.17
Study 1
Study 1 was conducted in 399 patients at 101 centres in Belgium, Bulgaria, Czech Republic, Estonia, Germany, Hungary, Latvia, Lithuania, Poland, Romania, the United Kingdom and the United States. Eligible patients enrolled in the study had an advanced incurable stage of cancer, were ≥18 years of age, and had a clinical diagnosis of cancer-related pain that was unalleviated by an optimized maintenance dose of Step 3 opioid therapy. Opioid therapy was considered optimized if (1) a dose increase was clinically inappropriate due to opioid-related side effects or (2) further efficacy benefit was not expected at higher doses (for the second definition, patients had to be receiving ≥90 mg morphine equivalents/day, inclusive of maintenance and breakthrough opioids). The maintenance opioid was preferably a sustained-release formulation, but an around-the-clock immediate-release formulation was acceptable. To be eligible, patients also had to fulfil the following criteria on each of three consecutive days during the screening period: ≤4 opioid breakthrough analgesic episodes per day (averaged over the 3 days), a stable maintenance opioid therapy dose, average pain ≥ 4 and ≤8 on a 0–10 NRS and average pain scores on the NRS that did not change by more than 2 points from the beginning to end of screening (i.e. no more than a 2-point difference between the highest and lowest scores, with all scores remaining between 4 and 8). Key exclusion criteria in study 1 included baseline use of morphine at >500 mg morphine equivalents/day (inclusive of maintenance and breakthrough opioids), current use of more than one type of breakthrough opioid analgesic, planned clinical interventions that would affect pain, and any history of schizophrenia or substance abuse including recreational use of cannabis product.
Following 1:1 randomization to either Sativex oral mucosal spray (THC (27 mg/mL): CBD (25 mg/mL)) or matching placebo, eligible patients entered an initial titration period lasting up to 14 days (Figure 1(a)).
Figure 1.

(a) Study designs for study 1 and (b) study designs for study 2.
Treatment was initiated as a single spray in the evening of the first day of treatment and was gradually increased by one additional spray per day (15 minutes apart) according to a pre-specified dose escalation protocol (Supplementary Table 1) until patients experienced unacceptable side effects, received acceptable pain relief, or reached the maximum allowed daily dosage of 10 sprays per day (patients were advised to reach at least 3 sprays per day). Patients were advised to take the medication at home and to remain home until at least 3 hours after their first dose due to potential sleepiness and dizziness following the medication. Patients were advised to initiate treatment with a single evening spray, as the most common side effects of Sativex, especially during the early titration stage, are somnolence and dizziness. To avoid undesirable side effect during the day, patients were recommended to administer higher number of sprays in the evening hours, despite the fact that cancer patients typically experience more intense pain in the morning hours.
Following the 2-week Sativex or placebo titration period, patients continued study drug administration at the same dose (i.e. the same number of sprays per day) for an additional 3 weeks, for a total treatment period of 5 weeks. Whenever possible, stable doses of all other prescribed pain medications were to be continued during the study period. Two weeks after the end of treatment, patients were contacted by phone for follow-up safety evaluations.
Study 2
Study 2 was conducted in 65 centres in Australia, Bulgaria, Germany, Hungary, India, Israel, Italy, Lithuania, Poland, Romania, Spain, Taiwan and the United Kingdom. Eligibility criteria were identical to those in study 1. The most salient difference between the two trials was that study 2 had a two-part randomized withdrawal design (Figure 1(b)).
In Part A, all patients underwent Sativex titration during a single-blind treatment period lasting 10 days, followed by 4 days of therapy at the titrated dose. Patients who demonstrated an improvement of 15% or more on pain NRS score were advanced to Part B, where they were randomized 1:1 to Sativex or placebo in a double-blind fashion. Patients then received study treatments at their self-titrated doses for 5 weeks. Two weeks after the end of treatment, patients were contacted by phone for follow-up safety evaluations.
Efficacy outcomes
In both studies, primary and key secondary endpoints were derived from patient diary listings reported through an interactive voice response system.
Study 1
In study 1, all efficacy assessments occurred during screening, immediately before dosing on day 1, and 3 weeks (day 22) and 5 weeks (day 36) later. The primary endpoint was percent improvement from baseline to the end of treatment in average pain NRS score.
Secondary efficacy endpoints included mean change from baseline to the end of treatment in average pain NRS score, worst pain NRS score, sleep disruption NRS and maintenance, breakthrough, and total opioid use per day in morphine equivalents. Patients also completed the following questionnaires: Subject Global Impression of Change (SGIC), Patient Satisfaction Questionnaire (PSQ), Physician Global Impression of Change (PGIC) and a constipation NRS.
Study 2
Efficacy assessments in Part A (titration) were used to identify patients eligible for subsequent double-blind treatment and are not reported here. In Part B (double-blind treatment), efficacy assessments were conducted immediately prior to dosing on day 1, and 3 weeks (day 22) and 5 weeks (day 36) later. The primary endpoint was the mean change from the randomization baseline to the end of treatment in average pain NRS score. Other secondary efficacy endpoints were the same as in study 1, including questionnaires.
Safety analysis
Safety and tolerability was assessed in both studies by documenting the nature and severity of treatment-emergent adverse events (TEAEs) at every patient visit. Clinical laboratory tests and vital sign readings were also performed at every visit during the screening and treatment periods. Patients completed the Columbia Suicide Severity Rating Scale (C-SSRS) every visit during the treatment period.
Statistical analysis
All patients who were randomized and received at least one dose of study medication comprised the safety analysis set. All patients who were randomized, received at least one dose of study medication, and had at least one post-randomized efficacy endpoint comprised the intention-to-treat (ITT) analysis set. For study 1, 380 patients (190 per treatment arm, Sativex or placebo) provided approximately 90% power to show statistical significance in percent improvement from baseline to end of treatment in NRS average pain score between Sativex and placebo using a two-sided Wilcoxon rank sum test with 5% significance level, based on a mean difference of 25 and a common standard deviation (SD) of 70 in the rank data of the percent change from baseline in NRS average pain score and a dropout rate of 12%. For study 2, based on previous experience with Sativex in this population, it was estimated that approximately 540 patients would enter Part A and receive Sativex treatment in order to yield 216 patients to be randomized in Part B. An interim analysis was planned to be performed when half of the planned number of patients completed the study (including early termination). The O’Brien-Fleming group sequential boundaries were used to allocate alpha levels of 0.003 to the interim look and 0.049 to the final analysis. Assuming an SD of 1.8 point in change from baseline in 0–10 NRS average pain, 216 patients would provide approximately 90% power to detect a mean treatment difference of 0.8 points and preserve an overall nominal alpha level of 0.05 (2-sided) with the interim and final analyses.
Wilcoxon rank-sum test was conducted for percent improvement in average pain NRS score (from baseline to end of treatment in study 1, and from eligibility pre-treatment baseline to end of treatment in study 2). Analysis of covariance (ANCOVA) was applied on the primary and the key secondary efficacy endpoints, including percent improvement, average pain/worst pain/sleep disruption scores, with corresponding baseline value as a covariate and treatment group as a factor. The time-course of these four efficacy endpoints from week 1 through week 5 was also analysed using Mixed-Effect Model Repeat Measurement (MMRM) on the ITT analysis set in both studies.
For both study 1 and study 2, the primary endpoint and the key secondary endpoints were tested with their Type I error controlled by use of a hierarchical gate-keeping procedure.
In each study, p-values from Wilcoxon rank-sum tests on percent improvement and ANCOVA on average pain/worst pain/sleep disruption scores were used for the hierarchical gate-keeping procedure in the sequence of the primary endpoint and the key secondary endpoints. No adjustments for covariates were made for the analyses of the other secondary endpoints in both studies with analysis of variance (ANOVA), including PGIC, SGIC or PSQ, and daily total, maintenance, and breakthrough opioid dose. Subgroup analyses for region (United States and rest of world (ROW)) were performed for the primary and the key secondary endpoints.
Results
Patients
In study 1, 528 patients were screened for enrolment (Figure 2). Of these, 399 fulfilled eligibility criteria and were randomized to Sativex (n = 200) or placebo (n = 199). Two patients randomized to Sativex did not administer any study drug and were therefore included in the demographic analyses, but not in the safety and efficacy analyses. During the subsequent 5-week titration/treatment period, 64 (32.0%) patients in the Sativex group and 41 (20.6%) in the placebo group withdrew from the study. The most common reasons for discontinuation were an adverse event (38 (19.0%) vs. 29 (14.6%) in the Sativex and placebo groups, respectively) and withdrawal of consent (19 (9.5%) vs. 8 (4.0%)). Twenty patients (10%) died in the Sativex group and 25 (12.6%) died in the placebo group. In total, 136 (68.0%) patients completed the study on Sativex and 158 (79.4%) on placebo. Of the 397 randomized patients, 279 were recruited in Europe and 120 were recruited in the United States.
Figure 2.

CONSORT flow diagram for study 1.
ITT: intention-to-treat.
*Two patients in the Sativex group did not administer any IMP, so although these patients were included in the patient demographic data, they were excluded from the efficacy and safety analysis sets.
In study 2, 508 patients were screened for enrolment, and 406 of whom fulfilled eligibility criteria and entered the study (Figure 3).
Figure 3.

CONSORT Flow Diagram for study 2: (a) Patient disposition in the single-blind Sativex titration period. (b) Patient disposition in the double-blind, randomized, placebo-controlled treatment period.
*Two patients who entered Phase A did not administer any IMP; they were excluded from the efficacy and safety analysis sets and were not counted as discontinued
During the subsequent 2-week single-blind Sativex titration period, 198 (48.8%) withdrew from the study. The most common reasons for discontinuation were an adverse event (71 (17.5%)) and withdrawal of consent (16 (3.9%)). At the end of the titration period, 108 patients did not meet the pre-specified criteria for subsequent randomization, the most common issue (n = 78) being failure to demonstrate a 15% improvement in average pain NRS score during titration. The remaining 206 eligible patients were randomized to Sativex (n = 103) or placebo (n = 103). Over the ensuing 5-week double-blind treatment period, 25 (24.3%) of the Sativex patients and 15 (14.6%) of the placebo patients withdrew, most commonly due to an adverse event. Forty-two (10.3%) patients died during the titration phase, and 32 (15.5%) died during the randomized treatment period (23 (22.3%) vs. 9 (8.7%) in the Sativex and placebo groups, respectively). In total, 78 patients completed the study on Sativex and 88 on placebo. Among the 406 patients who enrolled in the trial, 388 were recruited in Europe and 18 were recruited in Taiwan.
Demographic and baseline characteristics of patients in study 1 and study 2 were well balanced (Table 1).
Table 1.
Demographics and baseline characteristics.
| Study 1 |
Study 2 |
||||
|---|---|---|---|---|---|
| Sativex (n = 200) | Placebo (n = 199) | Single-blind Sativex Titration |
Double-blind treatment |
||
| Sativex (n = 404) | Sativex (n = 103) | Placebo (n = 103) | |||
| Age, mean year (SD) | 60.0 (11.0) | 59.6 (11.0) | 61.2 (11.2) | 61.4 (10.9) | 61.6 (11.8) |
| Male, n (%) | 106 (53.0) | 97 (48.7) | 228 (56.4) | 63 (61.2) | 55 (53.4) |
| Race, n (%) | |||||
| White | 193 (96.5) | 180 (90.5) | 384 (95.0) | 98 (95.1) | 101 (98.1) |
| Black | 3 (1.5) | 8 (4.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Asian | 1 (0.5) | 2 (1.0) | 20 (5.0) | 2 (1.9) | 7 (3.4) |
| Other | 3 (1.5)a | 9 (4.5)a | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| BMI, mean kg/m2 (SD) | 25.2 (6.3) | 25.6 (6.4) | 24.2 (5.2) | 23.7 (5.1) | 24.1 (5.5) |
| Time since cancer diagnosis, mean year (SD) | 4.1 (4.2) | 3.5 (5.0) | 3.2 (4.3) | 3.1 (3.6) | 3.0 (3.3) |
| Type of cancer pain, n (%) | |||||
| Neuropathic | 27 (13.5) | 23 (11.6) | 41 (10.1) | 11 (10.7) | 12 (11.7) |
| Somatic | 9 (4.5) | 17 (8.5) | 29 (7.2) | 9 (8.7) | 6 (5.8) |
| Visceral | 21 (10.5) | 22 (11.1) | 39 (9.7) | 12 (11.7) | 8 (7.8) |
| Mixed | 111 (55.5) | 116 (58.3) | 220 (54.5) | 56 (54.4) | 54 (52.4) |
| Bone | 32 (16.0) | 21 (10.6) | 71 (17.6) | 14 (13.6) | 20 (19.4) |
| Other | 0 (0.0) | 0 (0.0) | 4 (1.0) | 1 (1.0) | 3 (2.9) |
| Pain duration, mean year (SD) | 2.0 (2.7) | 1.7 (2.1) | 1.2 (1.5) | 1.3 (1.6) | 1.6 (2.0) |
| Average pain NRS score, mean (SD)b | 5.7 (1.2) | 5.8 (1.1) | 5.6 (1.1) | 5.6 (1.1) | 5.6 (1.2) |
| Use of breakthrough opioid, n (%) | 129 (64.5) | 124 (62.3) | 271 (67.1) | 59 (57.3) | 73 (70.9) |
| Daily opioid use, mean morphine equivalents (SD) | |||||
| Maintenance | 170.4 (118.7) | 182.4 (124.3) | 187.6 (114.8) | 185.5 (123.7) | 175.3 (106.5) |
| Breakthrough | 28.8 (40.2) | 25.3 (38.1) | 30.6 (44.0) | 26.8 (36.1) | 34.0 (48.5) |
| Total | 199.2 (131.1) | 207.7 (135.4) | 218.2 (130.1) | 212.3 (136.4) | 209.4 (121.4) |
BMI: body mass index; NRS: numerical rating scale; SD: standard deviation.
Other included: Hispanic/Latin/Latino (1 Sativex and 6 placebo patients), Afro Caribbean (1 placebo patient), 2 Black/White (both placebo patients), 1 Multi-racial (a Sativex patient) and 1 Hawaiian Islander (a Sativex patient).
Mean value over the days starting with the first day of the 3-day eligibility period through to the day before the first dose of study medication.
Across studies and treatment groups, enrolees had an average pain duration of 1.2–2.0 years and an average pain NRS score of 5.6–5.8 out of 10 at pre-titration baseline (see Methods). Approximately 60–70% of patients required breakthrough opioid use to manage their cancer-related pain, which arose from a variety of underlying sources (neuropathic, somatic, visceral, mixed, bone and other). Mean total daily opioid use at baseline, encompassing both maintenance and breakthrough therapy, ranged from approximately 199–218 morphine equivalents per day across all treatment groups. The distribution and characteristics of cancers among the enrolled patients are presented in Supplementary Tables 2 and 3.
Study drug exposure
In study 1 , the average number of sprays administered per day during the first week of therapy was 3.7 in both treatment groups. Average daily dosing plateaued and remained stable for the remaining 4 weeks of the treatment period, with placebo-treated patients self-administering, on average, 1 spray more per day than Sativex-treated patients (7.4 vs. 6.3 sprays per day). Consistent with this, a greater number of patients in the placebo group took more than 6 sprays of study medication per day, on average, over the entire treatment period (119 (60.1%) vs. 84 (42.2%)).
In study 2, the average number of Sativex sprays administered per day was 3.6 during the first week and 6.4 during the second week of single-blind titration (Part A). The average number of daily sprays remained stable throughout the subsequent 5-week double-blind treatment period (Part B), with a mean daily dosing of 6.5 sprays per day and 6.3 sprays per day in the Sativex and placebo groups, respectively.
Primary endpoint analyses
The primary efficacy endpoint in study 1 was the percent improvement in average pain NRS score from baseline to end of treatment. Since the analysis was non-parametric in nature, the percent improvement was calculated as a median difference between groups, where a positive value indicated a treatment difference in favour of Sativex. At the end of treatment in study 1, patients in the Sativex group had a median percent improvement from baseline in average pain NRS score of 7.2% compared with 9.5% in the placebo group (median difference = −1.84%; confidence interval (CI): −6.19%, 1.50%; p = 0.274).
In a post hoc analysis of the primary endpoint in study 1, a difference was observed between the effects of Sativex in the United States versus those in the rest of the world. In the United States, the median difference between Sativex and placebo was 5.14 (p = 0.12), while the median difference in the rest of the world was −6.03 (p = 0.03). Of particular interest was the observation that a statistically favourable treatment effect for Sativex was observed in patients <65 years of age from the United States (median treatment difference = 8.8; 95% CI: 0.00, 17.95; p = 0.040), but was not observed in patients <65 years of age from the rest of the world (median treatment difference = 0.2; 95% CI: −5.00, 7.74; p = 0.794).
The primary efficacy endpoint in study 2 was the mean change in average pain NRS score from randomization baseline to end of treatment. Mean average pain scores increased from 3.2 to 3.7 across the double-blind treatment period in the Sativex group; the analogous values in the placebo group were 3.1 and 3.6, respectively. Thus, there was a worsening of equal severity in both treatment groups (estimated treatment effect −0.02; 95% CI: −0.42, 0.38; p = 0.917). Analysis of the primary endpoint by subgroup did not reveal any demographic groups in which Sativex treatment was significantly better than placebo for improving average pain NRS scores (note no US patients were enrolled in this study).
Secondary endpoints
In both study 1 and study 2, change from baseline to end of treatment for average pain NRS score (study 1), percent improvement score from baseline to end of treatment in average pain NRS during Part B (study 2), worst pain NRS score (both studies), and sleep disruption NRS score (both studies) showed no significant treatment differences between Sativex and placebo (Tables 2 and 3).
Table 2.
Summary of outcomes in study 1.
| Primary efficacy endpointa | Estimated treatment difference (p-value) | 95% CI |
|---|---|---|
| Median percent improvement in average pain NRS score | ||
| Wilcoxon rank-sum testb | −1.84 (0.274) | −6.19, 1.50 |
| ANCOVAc | −2.00 (0.454) | −7.27, 3.26 |
| MMRMd | 1.12 (0.709)♦ | −4.77, 7.01 |
| Secondary efficacy endpointsa,e | Estimated treatment effect (p-value) | 95% CI |
| Average pain NRS score | ||
| ANCOVAc | 0.12 (0.434) | −0.18, 0.42 |
| MMRMd | −0.06 (0.723)♦ | −0.39, 0.27 |
| Worst pain NRS score | ||
| ANCOVAc | 0.11 (0.496) | −0.21, 0.44 |
| MMRMd | −0.10 (0.600)♦ | −0.46, 0.27 |
| Sleep disruption NRS score | ||
| ANCOVAc | 0.06 (0.7322) | −0.28, 0.39 |
| MMRMd | −0.17 (0.3615)♦ | −0.53, 0.19 |
| Questionnaire Outcomesa,f | Estimated Treatment Effect (p-value)g | 95% CI |
| SGIC score | ||
| Week 3 | −0.24 (0.041)♦♠ | −0.46, −0.01 |
| Week 5 | −0.31 (0.022)♦♠ | −0.57, −0.04 |
| Last visit | −0.29 (0.022)♦♠ | −0.53, −0.04 |
| PGIC score | ||
| Week 3 | −0.20 (0.086)♦ | −0.42, 0.03 |
| Week 5 | −0.27 (0.037)♦♠ | −0.52, −0.02 |
| Last Visit | −0.16 (0.182)♦ | −0.40, 0.08 |
| PSQ score | ||
| Week 3 | −0.36 (0.018)♦♠ | −0.66, −0.06 |
| Week 5 | −0.21 (0.199)♦ | −0.52, 0.11 |
| Last visit | −0.03 (0.823)♦ | −0.32, 0.26 |
| Impact on opioid usea | Estimated treatment effect (p value)c | 95% CI |
| Daily total opioid doseh | −9.35 (0.053)♦ | −18.81, 0.12 |
| Daily maintenance opioid doseh | −3.63 (0.321)♦ | −10.80, 3.55 |
| Daily breakthrough opioid doseh | −4.17 (0.075)♦ | −8.76, 0.42 |
| Constipation NRS Score | 0.16 (0.514) | −0.32, 0.63 |
ANCOVA: analysis of covariance; CI: confidence interval; MMRM: Mixed-Effect Model Repeated Measure; NRS: numerical rating scale; PGIC: Physician Global Impression of Change; PSQ: Patient Satisfaction Questionnaire; SGIC: Subject Global Impression of Change; CI: confidence interval; ITT: intention-to-treat; ANOVA: analysis of variance.
Result is numerically in favour of Sativex.
Result is statistically in favour of Sativex.
Results are calculated from baseline to the end of treatment in the ITT population, unless otherwise designated.
Estimate of the median difference between Sativex and placebo, together with 95% CI, was calculated using the Hodges-Lehmann approach.
Treatment difference (last visit – baseline) and 95% CI are derived from ANCOVA model with treatment as factor and baseline value as covariate.
Treatment difference (week 5 – baseline) and 95% CI are derived from a MMRM with treatment, week and treatment by week interaction as fixed effects; the baseline value and baseline by week interaction as covariates; and week as the time variable for repeated measures.
The hierarchical testing procedure adopted to control for Type I error prevented formal statistical significance testing of the key secondary efficacy endpoints on the grounds that the primary endpoint analysis was negative; unadjusted p-values shown are for reference only.
No adjustment for multiplicity was included in analyses for the ‘other’ secondary endpoints; multiplicity issues should therefore be allowed for when interpreting the results.
Derived from an ANOVA model.
Opioid doses are expressed as an oral morphine equivalent in mg.
Estimated odds ratio (p-value) obtained from logistic regression, with treatment as a factor in the model.
Table 3.
Summary of Outcomes in Part B of study 2.
| Primary efficacy endpoint, baseline to EOTa | Estimated treatment difference (p-value) | CI |
|---|---|---|
| Average pain NRS score | ||
| ANCOVAb | −0.02 (0.917)♦ | −0.42, 0.38 |
| MMRMc | −0.30 (0.117)♦ | −0.67, 0.07 |
| Secondary efficacy endpointsa,d | Estimated treatment effect (p-value) | CI |
| Percent improvement in average pain NRS score | ||
| Wilcoxon rank-sum teste | −2.13 (0.558)♦ | −10.12, 5.11 |
| ANCOVAb | −1.23 (0.757)♦ | −9.05, 6.59 |
| MMRMc | 2.73 (0.485) | −4.96, 10.41 |
| Worst Pain NRS score | ||
| ANCOVAf | −0.32 (0.124)♦ | −0.73, 0.09 |
| MMRMg | −0.58 (0.004)♦♠ | −0.98, −0.19 |
| Sleep Disruption NRS score | ||
| ANCOVAf | −0.31 (0.089)♦ | −0.67, 0.05 |
| MMRMg | −0.40 (0.020)♦♠ | −0.74, −0.07 |
| Questionnaire outcomes, baseline to EOTa,h | Estimated treatment effect (p-value)i | CI |
| SGIC score | ||
| ANOVA | −0.14 (0.450)♦ | −0.51, 0.23 |
| PGIC score | ||
| ANOVA | 0.03 (0.888) | −0.35, 0.41 |
| PSQ score | ||
| ANOVA | −0.11 (0.538)♦ | −0.47, 0.25 |
| Impact on opioid use, post titration baseline to EOTa | Estimated treatment effect (p-value)f | CI |
| Daily total opioid dosej | −7.11 (0.405)♦ | −23.92, 9.69 |
| Daily maintenance opioid Dosej | −8.93 (0.104)♦ | −19.69, 1.84 |
| Daily breakthrough opioid dosej | 1.81 (0.769) | −10.34, 13.96 |
| Constipation NRS score | 0.36 (0.193) | −0.18, 0.89 |
ANCOVA: analysis of covariance; CI: confidence interval; EOT: end of treatment; MMRM: Mixed-Effect Model Repeated Measure; NRS: numerical rating scale; PGIC: Physician Global Impression of Change; PSQ: Patient Satisfaction Questionnaire; SGIC: Subject Global Impression of Change; ITT: intention-to-treat; ANOVA: analysis of variance.
Result is numerically in favour of Sativex.
Result is statistically in favour of Sativex.
Results are mean values in the ITT population, unless otherwise designated.
Treatment difference and 95% CI are derived from ANCOVA model with treatment as factor and post titration baseline value as covariate.
Treatment difference and 95% CI are derived from a MMRM with treatment, week and treatment by week interaction as fixed effects, post titration baseline value and baseline-by-week interaction as covariates and week as the time variable for repeated measures.
The hierarchical testing procedure adopted, to control for Type I error, prevented formal statistical significance testing of the key secondary efficacy endpoints on the grounds that the primary endpoint analysis was negative; unadjusted p-values shown are for reference only.
Estimate of the median difference between Sativex and placebo, together with 95% CI, was calculated using the Hodges-Lehmann estimates approach.
Treatment difference (EOT – post titration baseline (week 2)) and 95% CI are derived from ANCOVA model with treatment as factor and post titration baseline value as covariate.
Treatment difference (week 7 – post titration baseline (week 2)) and 95% CI are derived from a MMRM with treatment, week and treatment by week interaction as fixed effects; post titration baseline value and baseline-by-week interaction as covariates; and week as the time variable for repeated measures.
No adjustment for multiplicity was included in analyses for the ‘other’ secondary endpoints; multiplicity issues should therefore be allowed for when interpreting the results.
Derived from an ANOVA model.
Opioid doses are expressed as an oral morphine equivalent in mg.
In study 1, Sativex was associated with greater improvements in self-reported global impression of change based on SGIC score, as well as greater physician perceived improvements in ‘general functional abilities’ relative to placebo (Table 2). Across the treatment period, SGIC scores significantly favoured Sativex at week 3 (p = 0.041), week 5 (p = 0.022) and last visit (p = 0.022), whereas PGIC was significantly in favour of Sativex at week 5 (p = 0.037). In study 2, there were no differences between treatment groups for SGIC, PGIC or PSQ (Table 3).
Adjunctive Sativex did not significantly impact opioid use in either study, although total daily opioid dosage (last visit – baseline) exhibited a treatment effect in favour of Sativex that trended towards significance (p = 0.053) in study 1 (Table 2). However, it is important to note that, according to the protocol, all other medications prescribed for pain were to be continued during the study period at a stable dose (see Methods). No effect on constipation NRS score was observed in either study (Tables 2 and 3).
Safety
Overall, 68% of Sativex-treated patients reported at least one TEAE in study 1, compared with 64% in the placebo group (Table 4). The most commonly TEAEs (all-causalities) in the Sativex-treated group were neoplasm progression (32 patients (16.1%)), somnolence (24 patients (12.1%)) and nausea (19 patients (9.5%)). The incidence of each of these TEAEs was similar in the placebo-treated group, with the exception of somnolence (8 patients (4.0%)).
Table 4.
Treatment-emergent adverse events in ≥5% in study 1.
| Event, n (%) | Sativex (n = 199) | Placebo (n = 198) |
|---|---|---|
| All causality | ||
| Totala | 136 (68.3) | 127 (64.1) |
| Neoplasm progression | 32 (16.1) | 36 (18.2) |
| Somnolence | 24 (12.1) | 8 (4.0) |
| Nausea | 19 (9.5) | 16 (8.1) |
| Vomiting | 18 (9.0) | 13 (6.6) |
| Dizziness | 16 (8.0) | 9 (4.5) |
| Constipation | 10 (5.0) | 13 (6.6) |
| Treatment-relatedb | ||
| Totala | 64 (32.2) | 41 920.7) |
| Somnolence | 18 (9.0) | 6 (3.0) |
| Dizziness | 15 (7.5) | 6 (3.0) |
| Nausea | 10 (5.0) | 8 (4.0) |
Patients with adverse events in multiple system organ classes were counted only once towards the total.
Treatment-emergent adverse events judged by the investigator to be at least potentially related to study treatment.
In study 2, 60% of patents reported a TEAE during Part A, while 72% of Sativex-treated patients and 62% of placebo-treated patients reported at least one TEAE in Part B (Table 5).
Table 5.
Treatment-emergent adverse events in ≥5% of Sativex patients in study 2.
| Event, n (%) | Sativex | Placebo |
|---|---|---|
| Part A: Single-blind treatment | ||
| N | 404 | – |
| All causality | ||
| Totala | 241 (59.7) | – |
| Neoplasm progression | 48 (11.9) | – |
| Somnolence | 46 (11.4) | – |
| Dizziness | 27 (6.7) | – |
| Nausea | 25 (6.2) | – |
| Vomiting | 22 (5.4) | – |
| Decreased appetite | 20 (5.0) | – |
| Treatment-relatedb | ||
| Totala | 128 (31.7) | – |
| Somnolence | 42 (10.4) | – |
| Nausea | 21 (5.2) | – |
| Dizziness | 21 (5.2) | – |
| Part B: Double-blind randomized treatment | ||
| N | ||
| All causality | 103 | 103 |
| Totala | 74 (71.8) | 64 (62.1) |
| Neoplasm progression | 30 (29.1) | 15 (14.6) |
| Weight decreased | 7 (6.8) | 4 (3.9) |
| Anaemia | 6 (5.8) | 7 (6.8) |
| Asthenia | 6 (5.8) | 6 (5.8) |
| Decreased appetite | 6 (5.8) | 3 (2.9) |
| Somnolence | 6 (5.8) | 1 (1.0) |
| Treatment-relatedb | ||
| Totala | 16 (15.5) | 12 (11.7) |
| Somnolence | 6 (5.8) | 0 (0.0) |
Patients with adverse events in multiple system organ classes were counted only once towards the total.
Treatment-emergent adverse events judged by the investigator to be at least potentially related to study treatment.
In part B, the most common TEAE in the Sativex-treated group was somnolence, which occurred in 6 (5.8%) Sativex-treated patients; treatment-related somnolence was not observed in placebo-treated patients. All other TEAEs occurred at an incidence of <5% within either treatment group. Across both studies, neoplasm progression (all cases unrelated to study treatment) was the most common TEAE. The most common TEAEs (≥5%) in the Sativex group are listed in Tables 4 and 5.
In study 1, serious TEAEs occurred in 35/199 (17.6%) Sativex-treated patients versus 44/198 (22.2%) placebo-treated patients. The most common event in both treatment groups was neoplasm progression (Sativex, 11.6%; placebo, 15.7%; all considered treatment-unrelated). Two patients who received Sativex reported treatment-related serious TEAEs: one case of constipation in a patient on high doses of opioids (360 mg/day morphine equivalents); and one case of moderate disorientation and moderate somnolence that manifested on the fourth day of treatment in a patient on an average daily dose of Sativex of 2.5 sprays.
In study 2, serious TEAEs occurred in 80/404 (19.8%) Sativex-treated patients in part A and in 33/103 (32.0%) Sativex-treated and 16/103 (15.5%) placebo-treated patients in part B. As in study 1, treatment-unrelated neoplasm progression was the most common serious TEAE in part A (Sativex, 10.1%) and part B (Sativex, 27.2%; placebo, 10.7%). In both studies, no serious TEAE occurred with a frequency greater than 2.0%.
Generally, in both studies, most TEAEs were balanced in the Sativex and placebo groups, although somnolence, a typical side effect of CB therapy, was numerically higher in the Sativex groups in both studies. Specifically, in the Sativex- and placebo-treated groups, somnolence occurred in 18 (9%) versus 6 (3%) patients, respectively, in studies 1 and 6 (5.8%) versus 1 (1%) patients, respectively, in study 2.
In total, 43 (10.8%) patients in study 1 developed TEAEs resulting in death, including 19 (9.5%) in the Sativex group, and 24 (12.1%) in the placebo group. None were considered to be treatment-related. Neoplasm progression was the cause of death in 17 Sativex- and 23 placebo-treated patients. One other patient in the Sativex group with metastatic colorectal cancer died from metabolic acidosis. The remaining two deaths were due to cerebrovascular accident in a Sativex-treated patient with metastatic gastric cancer; and to bronchitis in a placebo-treated patient with metastatic uterine cancer and metastatic lung cancer.
In study 2, 42 (10.4%) patients died during part A. None of the deaths were considered to be treatment-related. Neoplasm progression was the cause of death in 35 patients. Other TEAEs resulting in death included tumour pain in one patient, intestinal perforation in a patient with stomach cancer, general physical health deterioration in a patient with metastatic lung cancer, acute respiratory failure and sepsis in a patient with metastatic cervical cancer, meningitis listeria in a patient with metastatic liver cancer and renal failure in two patients with metastatic rectal cancer. In part B, a total of 32 (15.5%) patients died during treatment, including 21 (10.2%) who died during the treatment period (12 in the Sativex group and 9 in the placebo group). In addition, 8 patients (7.8%) in the Sativex group died after treatment and before the follow-up visit, and 3 patients (2.9%) in the Sativex group died after the follow-up visit. None of the deaths were considered treatment-related. Neoplasm progression was responsible for 22/23 deaths in the Sativex group and 9/9 in the placebo group. One additional Sativex-treated patient with metastatic oesophageal cancer died of pneumonia. More than double the number of patients in the Sativex-treated group discontinued treatment due to AEs compared to patients in the placebo-treated group (14 (13.6%) Sativex-treated patients vs. 6 (5.8%) placebo-treated patients).
No treatment-emergent suicidal ideations or behaviour developed in study 1 in patients treated with Sativex, whereas 4 patients on placebo developed treatment-emergent suicidality. In Part A of study 2, treatment-emergent suicidal ideation was reported in two patients receiving Sativex. In part B, no treatment-emergent suicidal ideations or behaviour were observed in either treatment group.
Discussion
Building on earlier exploratory studies,14–16 the two randomized phase 3 trials described in this report assessed whether adjunctive Sativex would provide efficacious analgesia in advanced cancer patients with chronic pain unalleviated by optimized opioid therapy. Overall, 303 patients were randomized to Sativex and 302 to placebo during the parallel-group treatment phases in the two trials. Across studies, the treatment groups were well balanced in terms of age, gender, race, type of cancer, baseline pain level, duration of pain, and baseline opioid use. Contrary to hypothesis, the primary efficacy endpoint (percent improvement (study 1) and mean change (study 2) in average daily pain NRS scores) was not met in either study. Subsequent analyses on the per-protocol population, which excluded patients with protocol violations, also found no superiority of Sativex for the primary endpoints (data not shown). Since more study withdrawals occurred in the Sativex group than the placebo group (11.4% more in tudy 1 and 9.7% more in study 2), a larger fraction of patients in the Sativex group were assigned a primary endpoint value of 0 (where patients demonstrated an improvement prior to their withdrawal) or a negative percentage change from baseline (where patients demonstrated a worsening of their pain prior to withdrawal). Nonetheless, analyses using different imputation methods (missing not at random (MNAR) and last observation carried forward (LOCF)) to deal with missing data or no imputation methods failed to find statistically significant differences between treatment groups for the primary endpoints in either trial.
Several factors may have contributed to the negative outcomes observed here. In both trials, a significant number of participants withdrew from the study (32% and 20.6% in study 1 and 24.3% and 14.6% in study 2 in the therapy and placebo arms, respectively). Moreover, the relatively high mortality rates observed in both studies (10% and 12.6%, and 22.3% and 9%) further increased the number of lost patients. Missing data are a well-known factor that negatively impacts study outcomes.18 In addition, the nature of the primary endpoints may have affected outcomes. Self-reported NRS scores can associate with day-to-day variations in mood, a phenomenon expected to have a significant impact in a fragile population of advanced cancer patients. Thus, development and validation of more objective tools to measure pain may be necessary for full pain control assessment in advanced cancer patients. Finally, although the number of sprays of Sativex and placebo were measured, the effectiveness of patient dosing, which affects absorption and pharmacokinetic factors (e.g. oromucosal delivery versus gastric delivery due to swallowing) was unknown in the present study. CBs are well known to undergo extensive first-pass metabolism following oral administration.
In accordance with the adopted hierarchical testing procedure, no formal statistical tests of significance were conducted for the key secondary endpoints. However, in study 1, both Sativex and placebo were associated with similar improvements in average pain, worst pain and sleep disruption NRS scores, while in study 2, worst pain and sleep disruption scores increased more in the placebo group relative to the Sativex group. The tendency, but lack of significance, of a slight opioid-sparing effect of Sativex in the trials may have been attributable to the advanced stage of disease and the small incremental pain control given by Sativex administration. It is also important to note that, per protocol, all other medications prescribed for pain were to be continued during the study period at a stable dose. Additional studies are warranted to investigate ways to reduce opioid use in this patient population.
The greatest effect of Sativex in these secondary outcome analyses appeared to be on clinic-conducted questionnaires (SGIC, PGIC and PSQ) in study 1, an interesting observation given that similar effects were not observed on the direct NRS pain instruments in the same study. This perhaps indicates that the relatively simple, single-question NRS instruments were too blunt to capture the full ramifications of pain in patients with advanced cancer. Future studies might benefit from exploring whether general well-being and overall day-to-day functionality in advanced cancer patients are appropriate targets for therapeutic intervention.
Intriguingly, in post hoc exploratory analyses, the subset of patients who were from the United States and were <65 years of age showed a better percent improvement in average pain NRS score compared to patients of the same age enrolled from sites outside of the US (Figure 4). Analysis of data from study-1 showed that US patients <65 years old had slightly higher pain, lower opioid dose at the baseline, more exposure to cannabis in the past and fewer reported deaths (Table 6). It is possible that a lower use of opioids may have led to a reduced down-regulation of opioid receptors and enhanced synergy between the CB and opioid receptors, a well-known interaction described in the literature,19 leading to a more favourable outcome in these patients. So potentially, lower and more balanced use of opioids for pain control at baseline resulted in more favourable clinical outcome from adjunct therapy with Sativex, including better control of chronic and breakthrough pain scores and extended duration of analgesia. Additionally, earlier and better-planned pain treatment in US cancer patients may have allowed for better overall pain control. Lastly, it is possible that the US subgroup might have less advanced disease due to timely established diagnosis, or possible cultural differences in pain perception (although difficult to quantify) may have accounted for the difference in primary endpoint outcome. These issues are explored in depth in a parallel report on Sativex in advanced cancer patients with chronic pain. Additional clinical studies may be needed to further validate the results of this post hoc analysis.
Figure 4.
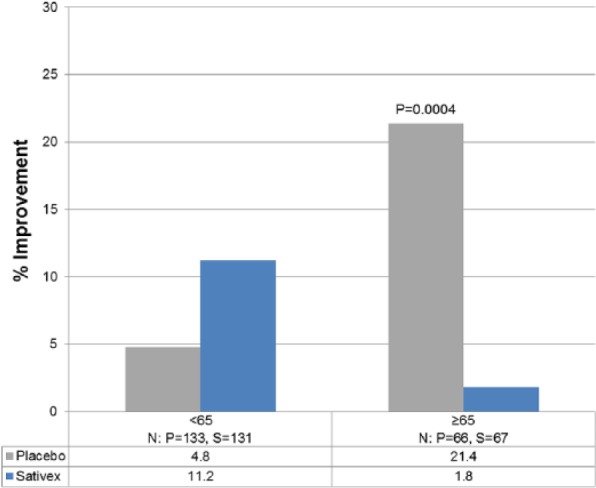
Median percent improvement from baseline in NRS average pain score by age.
Table 6.
Demographic and Baseline Characteristics of Subjects From the US and the Rest of the World.
| US (N = 120) |
ROW (N = 279) |
|||
|---|---|---|---|---|
| Sativex (n = 58) | Placebo (n = 62) | Sativex (n = 142) | Placebo (n = 137) | |
| Age ≥65, n (%) | 15 (26) | 14 (23) | 52 (37) | 52 (38) |
| Average pain (mean) | 6.2 | 6.4 | 5.5 | 5.5 |
| Total opioid* (mean/median) | 149/120 | 158/120 | 220/193 | 230/210 |
| Maintenance | 121/90 | 131/90 | 191/180 | 206/180 |
| Breakthrough | 29/5 | 27/3 | 29/16 | 25/10 |
| Prior use of cannabis, n (%) | 14 (24) | 11 (18) | 3 (2) | 1 (1) |
*morphine equivalence (mg).
The most common all-causality and treatment-related TEAEs associated with Sativex therapy were gastrointestinal disorders (nausea and vomiting) and nervous system disorders (somnolence and dizziness), as found in earlier studies on the agent. The vast majority of serious TEAEs, TEAEs leading to study discontinuation, and deaths were attributable to the patients’ underlying cancer, which was expected given the patient population. None of the deaths were treatment-related in either study. A disproportionate number of deaths occurred in patients randomized to Sativex compared with placebo (23 (22.3%) vs. 9 (8.7%)) during part B of study 2, indicating a significant correlation between treatment group and survival status (hazard ratio = 2.682; 95% CI: 1.239, 5.805; p = 0.012). The time to death by log-rank test also demonstrated a statistically significant correlation between treatment group and survival status (p = 0.009). While imbalances in withdrawal rates due to adverse events or consent withdrawal were most likely consequences of the specific administered medication, the observed imbalance in mortality rates in part B of study 2 remains unexplained. When the immediate post-follow-up study period was taken into account, the imbalance between the treatment group and the placebo group largely disappeared, to the extent that an independent safety monitoring board concluded as follows: ‘the committee finds no safety issues that would impede continued development of the study agent Sativex’. Due to the specificity of expression of the CB1 and CB2 receptors in central nervous system (CNS) areas associated with pleasure and reward and in immune cells, positing that the imbalance is a consequence of off-target effects on other organs seems unlikely. Moreover, as the imbalance of deaths in study 2 occurred exclusively post-treatment (11 (10.7%) in the Sativex arm vs. 0 (0.0%) in the placebo arm), a plausible causal relationship with Sativex treatment may be unlikely, consistent with safety results from across the Sativex developmental programme in multiple disease states.
Conclusion
In conclusion, this report described two phase 3 randomized placebo-controlled studies investigating the analgesic efficacy of Sativex oromucosal spray in cancer patients with chronic pain despite optimized opioid therapy. While Sativex did not demonstrate superiority to placebo in reducing self-reported average pain NRS scores, the findings were robust and bring valuable information to cancer pain research. Intriguingly, Sativex treatment was numerically better than placebo in US patients, especially in those less than 65 years for average pain NRS score, an issue that is examined in more detail in a parallel study. The safety profile of Sativex was consistent with earlier studies, and no new safety concerns or evidence of abuse or misuse were identified. In retrospect, results from patient-reported outcome questionnaires suggest that patient function may be an appropriate target to assess in future pain trials in this population.
Supplementary Material
Acknowledgments
The authors would like to thank Roman Wiklacz, MD (Centrum Onkologii, Warszawa, Poland); Jerzy Jarosz, MD (Fundacja Hspicjum Onkologiczne sw.Krzysztofa, Warszawa, Poland) and David Norris, PhD (Ecosse Medical Communications, Falmouth, MA) for editorial assistance provided during the preparation of this report.
Footnotes
Conflict of interest: The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding: Funding for the study was obtained from Otsuka Pharmaceutical Development & Commercialization, Inc., Rockville, MD, USA.
References
- 1. Portenoy RK, Lesage P. Management of cancer pain. Lancet 1999; 353(9165): 1695–1700. [DOI] [PubMed] [Google Scholar]
- 2. Van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, et al. Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Ann Oncol 2007; 18(9): 1437–1449. [DOI] [PubMed] [Google Scholar]
- 3. Breivik H, Cherny N, Collett B, et al. Cancer-related pain: a pan-European survey of prevalence, treatment, and patient attitudes. Ann Oncol 2009; 20(8): 1420–1433. [DOI] [PubMed] [Google Scholar]
- 4. Bruera E, Kim HN. Cancer pain. JAMA 2003; 290(18): 2476–2479. [DOI] [PubMed] [Google Scholar]
- 5. Smith TJ, Saiki CB. Cancer pain management. Mayo Clin Proc 2015; 90(10): 1428–1439. [DOI] [PubMed] [Google Scholar]
- 6. World Health Organization. Cancer pain relief: with a guide to opioid availability, http://apps.who.int/iris/handle/10665/37896 (1996, accessed October 2016).
- 7. World Health Organization. WHO’s cancer pain ladder for adults, http://www.who.int/cancer/palliative/painladder/en/ (accessed October 2016).
- 8. Cleeland CS, Gonin R, Hatfield AK, et al. Pain and its treatment in outpatients with metastatic cancer. N Engl J Med 1994; 330(9): 592–596. [DOI] [PubMed] [Google Scholar]
- 9. Jacox A, Carr DB, Payne R. New clinical-practice guidelines for the management of pain in patients with cancer. N Engl J Med 1994; 330(9): 651–655. [DOI] [PubMed] [Google Scholar]
- 10. Campbell FA, Tramer MR, Carroll D, et al. Are cannabinoids an effective and safe treatment option in the management of pain? A qualitative systematic review. BMJ 2001; 323(7303): 13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pertwee RG. Pharmacological actions of cannabinoids. In: Pertwee RG. (ed.) Cannabinoids. Berlin: Springer, 2005, pp. 1–51. [DOI] [PubMed] [Google Scholar]
- 12. Electronic Medicines Compendium. Sativex Oromucosal Spray, http://www.medicines.org.uk/emc/medicine/23262
- 13. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol 2011; 163(7): 1344–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnson JR, Burnell-Nugent M, Lossignol D, et al. Multicenter, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer-related pain. J Pain Symptom Manage 2010; 39(2): 167–179. [DOI] [PubMed] [Google Scholar]
- 15. Johnson JR, Lossignol D, Burnell-Nugent M, et al. An open-label extension study to investigate the long-term safety and tolerability of THC/CBD oromucosal spray and oromucosal THC spray in patients with terminal cancer-related pain refractory to strong opioid analgesics. J Pain Symptom Manage 2013; 46(2): 207–218. [DOI] [PubMed] [Google Scholar]
- 16. Portenoy RK, Ganae-Motan ED, Allende S, et al. Nabiximols for opioid-treated cancer patients with poorly-controlled chronic pain: a randomized, placebo-controlled, graded-dose trial. J Pain 2012; 13(5): 438–449. [DOI] [PubMed] [Google Scholar]
- 17. Dworkin RH, Turk DC, Farrar JT, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain 2005; 113(1–2): 9–19. [DOI] [PubMed] [Google Scholar]
- 18. Koch GG, Wiener LE. Commentary for the Missing Data Working Group’s perspective for regulatory clinical trials, estimands, and sensitivity analyses. Stat Med 2016; 35(17): 2887–2893. [DOI] [PubMed] [Google Scholar]
- 19. Abrams DI, Couey P, Shade SB, et al. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther 2011; 90(6): 844–851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.