

Abstract
The rate‐limiting step in prion diseases is the initial transition of a prion protein from its native form into a mis‐folded state in which the protein not only forms cell‐toxic aggregates but also becomes infectious. Recent experiments implicate polyadenosine RNA as a possible agent for generating the initial seed. In order to understand the mechanism of RNA‐mediated mis‐folding and aggregation of prions, we dock polyadenosine RNA to mouse and human prion models. Changes in stability and secondary structure of the prions upon binding to polyadenosine RNA are evaluated by comparing molecular dynamics simulations of these complexes with that of the unbound prions.
Keywords: prions, amyloid aggregation, protein misfolding, RNA, prion diseases
Introduction
Prions are proteins commonly found in many species including bacteria and fungi. In mammals and birds they are involved in the growth and maintenance of neuronal synapses1, 2, 3; and mis‐folded prions (usually addressed in this form as scrapie form or PrPSC, while prions in their functional form are called PrPC) assemble into cell‐toxic aggregates that are the cause for a number of neurodegenerative diseases in humans (Creutzfeldt‐Jacob, Kuru) and animals (Scrapie, Bovine Spongiform Encephalopathy).3, 4 While the structure of the infectious PrPSC state is not known, circular dichroism measurements indicate a lower helicity (43% in PrPC compared to 30% in PrPSC) and a larger β‐sheet content (3% in PrPC compared to 43% in PrPSC) than seen in the native PrPC structures that are resolved and deposited in the Protein Data Bank. In the often studied mouse prion, the loss of helicity results from the N‐terminal helix5, 6, 7, 8, 9, 10, 11, 12 (residues 143–161) converted into a β‐sheet, while the central helix B (172–196) and the C‐terminal helix C (200–229) stay intact. The now generally accepted protein‐only hypothesis of Stanley Prusiner states that mis‐folded prions are infectious, that is, a prion protein in its disease‐causing PrPSC form can convert an unfolded prion, or one that is in its native PrPC state, into its own form.13 This hypothesis implies that prion diseases progress by a nucleation mechanism where initial mis‐folded prions seed the spread of the cell‐toxic aggregates. There is evidence14, 15, 16 that such “infectiousness” of aggregates plays also a role in Alzheimer's and other amyloid diseases. Hence, the rate‐limiting factor in all these diseases is the formation of the initial seed that nucleates the growth of the toxic aggregates. An understanding of the initial stages of aggregation is especially important as the list of diseases associated with aggregation is rapidly growing.17 For instance, recent work18, 19 has pointed out a correlation between the pregnancy‐specific disorder preeclampsia and the appearance of protein aggregates and elevated levels of prion protein expression.20 This example therefore further underscores the importance of understanding the initial stages of prion aggregation for a wide array of disorders.
There is experimental evidence that polyanions, such as polyadenosine RNA (poly‐A‐RNA), can catalyze conversion of the native PrPC form into PrPSC seeds through interacting with the N‐terminal of the prion at either a segment made from residues 21–31 or at the segment made from residues 111–121.1, 21 For instance, the deletion of polybasic domain 21–31 creates prion proteins that do not undergo conversion to the infectious state in the presence of poly‐A‐RNA.22 However, it is not clear how interactions between the RNA and these two polybasic segments lead to unfolding of helix A and its re‐folding into a β‐sheet.
In the present article, we use molecular dynamics simulations to probe this mechanism and to identify early steps in the conversion of PrPC into PrPSC upon binding to poly‐A‐RNA. The mouse prion, for which the wealth of experimental data is available, is compared with the medically more relevant human prion protein allowing us to assess the effect of sequence on the mechanism of conversion. As the N‐terminal domain (residues 1–121) has not been resolved in the PrPC structures of mouse and human prion protein [deposited in the Protein Data Bank (PDB) under identifiers 2L39 (Mouse Sequence) and 2LSB (Human Sequence)], it is not possible to study directly in silico how docking of RNA to the PrPC structure modulates the stability of helix A and its conversion to a β‐sheet. For this reason, we have augmented the experimental structures with structure predictions of the N‐terminal domain. Comparing proposals from the well‐established MODELLER23, 24 and ITASSER25 – 27 programs, and validating them with the approach used by Hosen et al.,28 we generate initial protein structures for the docking of the prions in their PrPC form with poly‐A‐RNA fragments. The stability of such complexes, predicted by the docking software Autodock,29 is then evaluated in 300 ns long all‐atom molecular dynamics simulations with an explicit solvent. Through monitoring the protein's secondary structure, contacts formed with the RNA fragment and the protein (specifically helix A and polybasic domains of the N‐terminal region), and root mean square fluctuation of residues in both the bound and unbound state of prion proteins, we characterize how the interaction with poly‐A‐RNA triggers the transition from the cellular prion protein to its infectious scrapie form.
Results and Discussion
Visual inspection
Eight docked structures (one for each of the four binding sites as predicted by either ITASSER or MODELLER—see the method section) of the mouse prion—RNA complex, and six of the human prion—RNA complex, were followed each in three independent trajectories over 300 ns to evaluate the stability of the complexes. As a control, we also simulated each of the unbound mouse and human prion protein structures in three trajectories that have the same length and rely on the same protocols as used for the RNA–protein complexes. We start our analysis of these 42 trajectories with a visual inspection of the final configurations. Both docked and unbound structures are shown are shown for the human prion (where we found three binding sites) in Figure 1. The corresponding structures for the mouse prion, where we found four binding sites, are shown in Figure 2.
Figure 1.
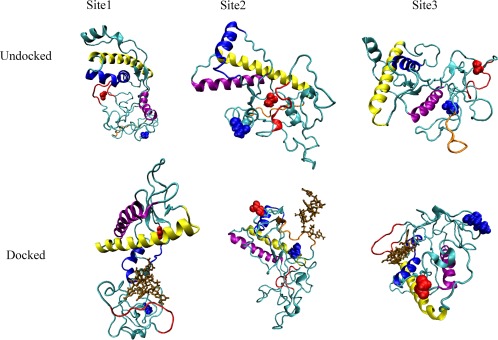
Binding motifs observed for the human prion protein. The upper row displays the bound systems and the lower row the unbound ones. Helix A is drawn in blue, helix B in yellow, the polybasic domain of residues 21–31 in red, and polybasic domain of residues 121–131 in orange (binding site 2 only). Blue spheres denote the N‐terminus while red spheres denote the C‐terminal region. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 2.
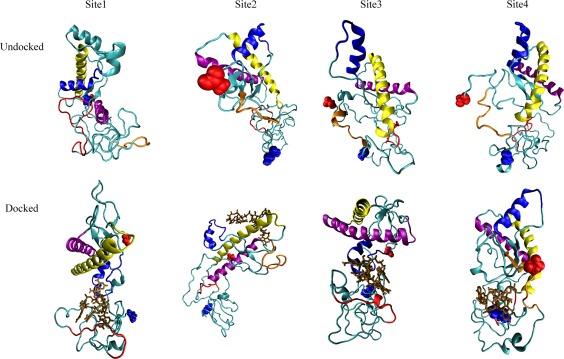
Binding motifs observed for the mouse prion protein. The upper row displays the bound systems and the lower row the unbound ones. Helix A is drawn in blue, helix B in yellow, the polybasic domain of residues 21–31 in red, and polybasic domain of residues 121–131 in orange (binding site 2 and 4 only). Blue spheres denote the N‐terminus while red spheres denote the C‐terminal region. [Color figure can be viewed at wileyonlinelibrary.com]
In the six trajectories that followed the time evolution of the RNA docked to the mouse prion protein at site 1 (the polybasic segment of residues 21–31) a pincer‐like structure between helix A (shown in blue) and the polybasic domain (shown in red) is quickly formed and encapsulates the RNA fragment (shown in brown). This pincer motif, characterized by distinct contacts between poly‐A‐RNA fragment and protein that we listed in Table 1, is also seen in the corresponding six trajectories for the human prion protein bound with the poly‐A‐RNA fragment at this segment. Note that this motif which does not differ between structures generated by MODELLER or ITASSER is not observed in the control simulations of the unbound proteins. In all trajectories of the prion–RNA complex where this motif appears, helix A dissolves over the course of the simulations, but does not in the control simulations of the unbound proteins. Since this loss of helicity in the bound protein occurs in the same region where it is seen in the conversion to the infectious state, we conjecture that the polybasic‐helical pincer motif initiates the conversion to the infectious state. The resulting direct interaction with helix A does not affect the secondary structure of the other helixes B and C, again in line with experimental results,7 while we observe in the neighborhood of helix A transient β structures that hint at the potential conversion to the PrPSC state, as shown in Figure 3. However, these structures are not consistently observed across all trajectories and when found oscillate between coil and strand conformations, indicating that our 300 ns trajectories are not sufficiently long to sample the conversion to the infectious state.
Table 1.
Contacts Between RNA and Prion That Define the Helix Pincer Motif
| 0–100 ns | 200–300 ns |
|---|---|
| Contacts with Helix A | |
| 144 ASP | 144 ASP |
| 145 TRP | 145 TRP |
| 147 ASP | 147 ASP |
| 148 ARG | 148 ARG |
| 139 HIS | |
| 140 PHE | |
| 146 GLU | |
| 149 TYR | |
| 150 TYR | |
| Contacts in the neighborhood of polybasic domain of residues 21–31 | |
| 25 ARG | 25 ARG |
| 27 LYS | 27 LYS |
| 34 GLY | 34 GLY |
| 35 GLY | 35 GLY |
| 33 THR | 33 THR |
| 41 GLN | 41 GLN |
Figure 3.
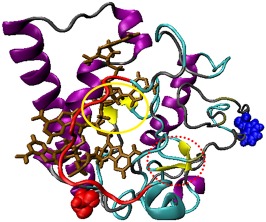
Secondary structure of the prion protein for system of RNA docked to site 1. Residues with β sheet secondary structure are indicated by yellow; α helices are indicated by purple; RNA is indicated by brown; coils are indicated by gray, and the polybasic domain 21–31 is indicated by red. Transient β‐strands around docking site are circled in a red dotted line while sheets that are from the PDB are circled in a yellow solid line. Blue spheres denote the N‐terminus while red spheres denote the C‐terminal region. [Color figure can be viewed at wileyonlinelibrary.com]
In the preliminary short trajectories, a strong binding of the poly‐A‐RNA molecule to the prion proteins was also observed for binding site 2, defined as the region around the polybasic domain of residues 111–121. In the 300 ns long trajectories of complexes formed with this binding site we do not observe the formation of the pincer‐motif seen in the simulation of complexes with binding site 1. This is likely because helix A (residues 140–161) is so close to the binding site that a pincer arrangement would lead to steric clashes. Consequently, secondary structure changes not more in the simulation of the poly‐A‐RNA–prion complex than in the trajectories of the unbound protein. Again we find little difference between human and mouse proteins, and between models generated with either ITASSER or MODELLER.
The poly‐A‐RNA fragment can also interact with mouse and human prion proteins at binding site 3, made of residues 135–145, which partially overlaps with helix A (residues 140–161). As with the complexes involving binding site 1, we observe in multiple trajectories the formation of the helical‐polybasic pincer motif, and the dissolution of helix A happens even faster when the RNA binds to site 3 than when it binds to site 1. This is because at site 3, the RNA binds directly to the helix A, and only later forms the helical‐polybasic pincer, which then traps the RNA in a region close to the helix A. This assumption is further supported by data from two trajectories for poly‐A‐RNA bound to the mouse ITASSER structure at site 3. While in two of the three trajectories the RNA fragment separated from the protein (at ∼75 ns and ∼95 ns, respectively), the complex persisted in one trajectory where, unlike in the other two trajectories, the pincer between the polybasic domain and helix A is formed and helix A dissolves.
Binding site 4 involves the poly‐A‐RNA fragment interacting with residues 1–5 of the N terminal domain and occurs only in mouse prion, despite that mouse and prion protein have the same sequence in this region. However, in the three structures where the RNA binds to the protein at site 4, the N‐terminal residues 1–5 are within 5 Å to the polybasic region 111–121 with which they form a pocket around the RNA fragment. As the unbound systems, prions with RNA bound at binding site 4 do not change secondary structure. Because of the close proximity to the polybasic domain of residues 111–121, one may consider binding site 4, which is only observed for a single structure predicted by MODELLER, as a variant of binding site 2. As most of the structures, predicted by MODELLER, have the first five N‐terminal residues within 5 Å to the polybasic domain 111–121, binding site 4 may be in artifact of this protein structure prediction software.
Our visual inspection of the 42 trajectories suggests that RNA can initiate conversion into the infectious scrapie form when binding to the prion protein at sites 1 and 3. Hence, we focus in our further analysis on these two binding sites. As we find little differences between ITASSER and MODELLER generated structures, we will no longer distinguish between them, but combine them in our analysis. Hence, in the following our statistics relies for human prion proteins bound to poly‐A‐RNA at site 1 on 6 trajectories, and 6 for the mouse prion bound to poly‐A‐RNA at site 1. For the human prion bound with RNA at site 3 we have also 6 trajectories and 6 trajectories for the corresponding mouse prion–RNA complexes.
RMSF analysis
One way to quantify the effects of docking RNA to specific regions of the prion protein is by comparing the root mean squared fluctuation (RMSF) of residues in the various RNA–prion complexes. These are shown in Figure 4 for all residues of the human and the mouse prion. The figure displays for each residue the ratio of RMSF measure for the bound protein divided by the corresponding value measured for the unbound prion. Hence, a value larger than one implies that a given residue is more flexible in a prion bound to RNA than in the unbound protein. In agreement with the previously discussed visual inspection of the various trajectories, we find for both human and mouse prion docked to either site 1 or 3 a characteristic spike in region of residues 150–180, which includes many residues of helix A (140–161), that is not seen for the other binding sites (data not shown). Binding to these two pockets leads for the human prion protein also to increased flexibility in the region of residues 35–90. This higher flexibility results from a loss of contacts between polar residues in this segment and helix A after binding of the poly‐A‐RNA fragment to the protein. The frequency of such contacts falls from 32.1% (7.2%) in undocked structures to 12.1% (2.9%) in the complexes. A similar decrease of frequency for such contacts is also observed for the mouse protein when the RNA is binding to site 1 and site 3, declining from 31.1% (4.4%) in undocked to 10.1 (3.3%) in docked structures, and consequently an increase in flexibility is observed for the segment of residues 35–90.
Figure 4.
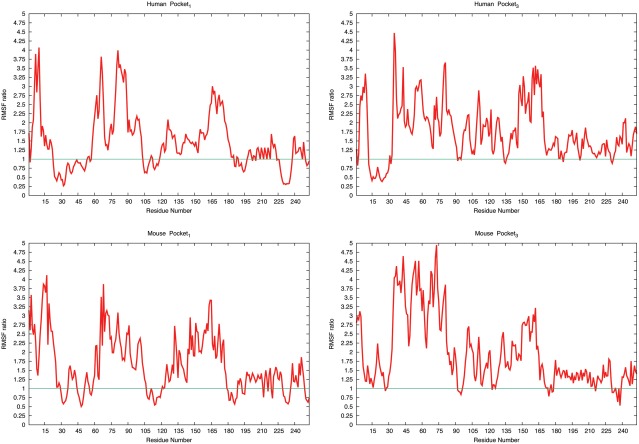
RMSF values measured for residues in docked structures divided by that measured in undocked structures. Ratios for the human prion protein are shown in the upper row, and ratios for the mouse protein in the lower row. The left column shows the ratios for structures docked to binding site 1, and the right column that for binding site 3. The ratio is calculated by dividing the average RMSF value for a given residues in the docked protein–RNA structure to the average RMSF of the undocked protein. The green line indicates a ratio of one. [Color figure can be viewed at wileyonlinelibrary.com]
The distribution of the residue root‐mean‐square‐fluctuations corroborates our previous observation that formation of the pincer‐like structure leads to dissolution of helix A. This dissolution is also seen in Table 2 where we show how the probability to find 1–4‐backbone hydrogen bonds, characteristic for α‐helices, changes along the trajectories. The helix starts dissolving by losing hydrogen bonds involving residues 144–148, which are also the ones that first form contacts with the RNA. As the simulations evolve, further helical backbone hydrogen bonds break, especially the bond between Tyr150 and Glu146, and TYR149 and ANS153; all residues that now form contacts with the poly‐A‐RNA fragment.
Table 2.
The Probability of a Given Helical Contact Averaged Over the Trajectories Where the Polybasic‐Helix Pincer Motif is Seen
| Backbone hydrogen bond | Control undocked | RNA docked 0–100 ns | RNA docked 200–300 ns |
|---|---|---|---|
| VAL161‐PRO157 | 50.0% (3.7) | 45.1% (4.6) | 10.1% (2.5) |
| VAL160‐HIS155 | 75.4% (1.2) | 55.4 (3.1) | 15.6% (2.5) |
| GLN159‐MET154 | 88.6% (3.6) | 61.5% (3.7) | 20.4% (4.1) |
| ASN158‐ASN153 | 81.7% (3.5) | 65% (4.4) | 19.5% (3.5) |
| PRO157‐ASN153 | 45.5% (4.1) | 43.4 (3.8) | 20.4 (4.0) |
| ARG156‐GLU152 | 78.7% (2.3) | 65.5% (7.7) | 20.9% (3.7) |
| HIS155‐ARG151 | 97.9% (4.2) | 81.0% (8.3) | 51.0% (3.4) |
| MET154‐TYR150 | 74.3% (3.1) | 65.6% (7.4) | 30.5% (4.4) |
| ASN153‐TYR149 | 86.1% (3.8) | 50.1% (4.2) | 0% (0) |
| GLU152‐ARG148 | 97.1% (2.1) | 71.70% (9.2) | 21.70% (4.2) |
| ARG151‐ASP147 | 94.7% (3.0) | 40.6% (5.3) | 20.6% (3.5) |
| TYR150‐GLU146 | 98.8% (1.3) | 60.1% (4.9) | 10% (2.1) |
| TYR149‐TRP145 | 91.5% (4.5) | 41.2% (4.1) | 15.0% (4.8) |
| ARG148‐ASP144 | 75.6% (5.1) | 42.5% (6.2) | 22.5% (4.9) |
| 147ASP‐143ARG | 96.9% (1.9) | 62.1% (4.8) | 25.6% (3.2) |
The net‐effect of the loss of backbone hydrogen bonds in helix A and the newly formed contacts of residues in this segment with the poly‐A‐RNA is an overall higher number of hydrogen bonds; that is, the binding of the RNA is despite the loss of helix‐stabilizing hydrogen bonds energetically favorable. This can be seen from Table 3 where this relative increase is shown for both human and mouse prion protein, at both binding sites. While in the control simulations the number of hydrogen bonds does not change it increases for the bound forms, with a larger growth for binding to site 1. In order to emphasize this point we show in Table 3 also the differences in the number of hydrogen bonds. Average over the first 100 ns there is only a weak signal, in that there is only an increase of about six hydrogen bonds; however, this number increases to about ten or more hydrogen bonds over the last 100 ns of the simulations. Hence, despite the dissolution of helix A additional backbone hydrogen bonds appear, possibly indicating the start of formation of another ordered structure.
Table 3.
Number of Main Chain–Main Chain Hydrogen Bonds Averaged Over Three Trajectories for Each Pocket of the Human Sequence
| 0–100 ns | 200–300 ns | |||||
|---|---|---|---|---|---|---|
| Ctrl | Docked | Ctrl | Docked | |||
| Name | Average number MC–MC H bonds | Average number MC–MC H bonds | Δ Bonds 0–100 ns | Average number MC–MC H bonds | Average number MC–MC H bonds | Δ Bonds 100–300 ns |
| Pocket 1 Human | 78.3 (2.9) | 84.2 (4.8) | 5.9 (3.5) | 78.9 (2.3) | 91.4 (3.0) | 12.5 (2.7) |
| Pocket 3 Human | 71.9 (3.4) | 78.2 (5.4) | 6.3 (4.2) | 84.6 (2.9) | 94.9 (3.0) | 10.3 (2.9) |
| Pocket 1 Mouse | 72.5 (4.0) | 77.6 (4.8) | 5.1 (4.1) | 71.4 (4.3) | 84.1 (3.7) | 12.7 (3.9) |
| Pocket 3 Mouse | 75.1 (4.5) | 79.1 (3.3) | 4.0 (3.5) | 74.7 (4.5) | 75.5 (3.9) | 10.8 (4.2) |
Δ Bonds is calculated as the difference between the docked and undocked average main chain–main chain hydrogen bonds.
The decrease in helicity resulting from the loss of stabilizing backbone hydrogen bonds in helix A is also seen in Table 4. While there is no change in the control, the average helicity decreases from about 43% to 32% for both proteins and binding sites. However, binding of the RNA fragment with the protein leads not only to a loss of helical structures but also to a gain in β arrangements. This effect is small but its significance becomes clear once one considers in addition the life times of such sheet‐like elements. For this reason, we list in Table 4 also the β‐strand occupancy, defined as the average amount of time a β‐strand is observed along the trajectory. While there is only an increase of ∼4.5% in total β‐strand propensity, the average life time of the transient β‐strands grows by approximately 25%, which may indicate the beginning conversion to the β‐sheet rich PrPSC structure.
Table 4.
Average Secondary Structure Content and β‐Strand Occupancy of the C Terminal Domain (residues 121 to 253) of All Trajectories of Structures With Polybasic‐Helix Pincer, i.e., Binding Sites 1 and 3
| Human | Mouse | |||
|---|---|---|---|---|
| Time | Control | Docked | Control | Docked |
| Helicity | ||||
| 0–100 ns | 43.1% (1.8) | 38.9% (2.7) | 39.2% (0.9) | 36.6% (2.2) |
| 200–300 ns | 41.9% (1.6) | 32.0% (2.3) | 42.7% (1.3) | 31.5% (2.4 |
| β Strands | ||||
| 0–100 ns | 4.3% (1.3) | 6.9% (1.6) | 4.2% (1.2) | 6.5% (1.7) |
| 200–300 ns | 4.5% (1.2) | 8.8% (1.8) | 4.4% (1.5) | 9.1% (1.4) |
| β Strand occupancy | ||||
| 0–100 ns | 26.7% (1.4) | 38.4% (4.6) | 26.2% (1.3) | 37.8% (4.0) |
| 200–300 ns | 27.9% (1.3) | 57.9% (4.2) | 28.2% (1.2) | 57.3% (4.5) |
The averages are calculated for the first and for the last 100 ns in order to show the evolution of the structures. Occupancy is defined as the percentage of time a given residue exists in a beta strand confirmation.
In order to emphasize the shift in hydrogen bonding and the corresponding structural rearrangements we show in Figure 5(A) contact map of the human prion (upper triangle) and mouse prion (lower triangle). For this figure, we measured the frequency of backbone–backbone hydrogen bonds in both docked and undocked structures, and the color coding marks the difference between these two frequencies. A reddish coloring indicates that a given hydrogen bond is more frequently seen in the bound structure than in the unbound one, and a greenish color that this hydrogen bond is more common in the unbound structure than in the bound one. We find the strongest signal for contacts involving residues 140–161, which are part of a helix in the native structure. In this region, there is a decline of helical contacts (see the greenish coloring of points parallel to the diagonal that mark 1–4 hydrogen bonds) and a corresponding increase in contacts that suggest β arrangements (the reddish colored data points orthogonal to the diagonal).
Figure 5.
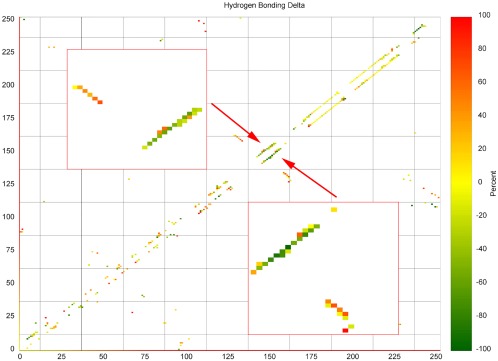
Difference between the frequency of backbone H‐bond contacts found in the bound prion protein and the frequency of such contacts found in the unbound protein. Diagonal elements correspond to helical contacts and linear clusters orthogonal to the diagonal correspond to β strand contacts. The upper triangle shows the contact map for the human prion protein and lower triangle the map for the mouse prion protein. The helix A region is shown enlarged in the inset. [Color figure can be viewed at wileyonlinelibrary.com]
Conclusion
We have simulated the effect of polyadenosine RNA bound to human and mouse prions on the stability and secondary structure of these proteins. Potential binding sites were predicted with Autodock, where unresolved parts of the experimental prion structures (in their PrPC form) are assumed to take structures predicted by standard software packages ITASSER and MODELLER. The poly‐A‐RNA fragment–prion complexes, generated in this way, were followed in long molecular dynamics simulations. In cases where the poly‐A‐RNA fragments binds with the N‐terminal polybasic segment 21–31, or directly with the N‐terminal helix A, we observe that binding of the RNA leads to formation of a pincer‐like structure between helix A and the polybasic domain that encapsulates the RNA. Because of steric clashes, the pincer cannot be formed when the RNA binds to the polybasic region 121–131, the other predicted binding site. Formation of the pincer seems necessary to recruit and trap the RNA, and it precedes dissolution of helix A, which starts with the N‐terminus of the helix and the helix subsequently unraveling toward its C‐terminal end. As the molecular dynamic trajectories proceed the helical contacts are replaced by short β‐strand arrangements that eventually will lead to the characteristic high β‐sheet content of disease‐causing mis‐folded PrPSC prion structure. This picture differs little between mouse and human prion protein, that is, is independent of the sequence differences between the two proteins. Hence, our results suggest a mechanism by that RNA binding to the prion protein at the segment 21–31 can trigger the conversion of the cellular prion protein structure PrPC to its infectious scrapie PrPSC form which afterwards becomes the seed for the formation of toxic amyloids. The shift in structure upon interaction with RNA from helix to coil could indicate the presence of intermediate states in the process of prion conversion. Recent evidence suggests that these intermediates can affect aggregation and conversion rates due to barriers between the intermediates and mis‐folded proteins.30 Furthermore, increasing the flexibility of an aggregate intermediate structure has been shown to reduce the time for conversion to a mis‐folded state for amyloid β.31 Amyloid β aggregates have also been shown to have α helical intermediate structures, that dissolve and convert to β‐sheets in the aggregation process.32 These studies therefore suggest a relationship between the initial stages of amyloid aggregation and prion conversion. While it is not clear how common or important this mechanism is in prion diseases, our results put an interesting twist on the “protein‐only” hypothesis.
Materials and Methods
Model generation
In mammals, prion proteins are anchored to the cellular membrane via glycosylphosphatidylinositol, added to the protein at the C‐terminus after cleavage of the last 24 residues.2, 3 Simulations of the protein anchored to the membrane are computationally expensive, and in order to explore efficiently the dynamics of RNA–prion interactions, we chose an unanchored prion model for our study. This requires to use in our simulations the non‐cleaved protein (of length 253 residues for human prions or 254 residues for mouse prions) to prevent the presence of an erroneously charged C terminus. In order to study the conversion of the PrPC form into the disease‐causing PrPSC state by molecular dynamic simulations, we need a suitable model of the mouse or human prion protein in its native PrPC structure. These structures have been only partially resolved by X‐ray diffraction, with the N‐terminal first 121 residues missing in the structures deposited in the Protein Data Bank (PDB) under PDB‐ID 2LSB (human) and 2L39 (mouse). As binding of the RNA to the protein is supposed to involve these N‐terminal residues, we propose to extend the PDB models by adding the N‐terminal first 121 residues, taking as their structure one predicted by the well‐established MODELLER or ITASSER programs.23, 24, 25, 26, 27 While both software packages rely on aligning a protein sequence to existing structures, they differ in the prediction protocols and protein models. Hence, by comparing two independent sets of predictions we hedge our study against potential biases. This structural prediction method is similar to that used by Hosen et al. to model Multidrug Resistance Protein‐6 (MRP6) and its interactions with various small molecules.28 MRP6 is a protein of sequence length 1,503 amino acids with intrinsically disordered regions that make its interactions difficult to predict and model. By using similar methods of model generation and refinement as Hosen et al., we expect to generate reasonable structures for modeling prion–RNA interaction.
For both packages we select the two structures that have the highest ranking, and allow them to relax in a short, 5 ns long molecular dynamic run at T = 310 K and 1 bar pressure. From each trajectory, we then collect ten evenly spaced configurations for quality assessment. Following the approach by Hosen et al.28 we evaluate the model quality by averaging the scores of web‐server, RAMPAGE,33 ERRAT,34 and ProQ,35 and comparing them to a cutoff value. We then find the RNA complexed structures using the docking software Autodock 25, bind to these configurations a five‐nucleotide snippet of poly‐A‐RNA, allowing free rotation around all single bonds in the poly‐A‐RNA. Autodock 25 was selected as the docking software as it has been used extensively to model docking to both large proteins and RNA.28, 36 We chose a fragment size of five nucleotides because this was in recent experiments the minimal size where photo‐degradation does not change the rate of conversion.37, 38
The predictions by either MODELLER or ITASSER lead to a total of two times ten docked structures for each software package (MODELLER/ITASSER) and protein (human/mouse), that are used as start point for short molecular dynamics simulations of 10 ns length at T = 310 K. In 29 (25) of the 40 runs for the human (mouse) prion protein, the RNA got detached from the protein, and we discarded the corresponding docked structures. Analyzing the remaining 11 docked structures for the human prion, we identified three stable binding sites for the human protein. Site 1 is found five times and corresponds to the binding between the RNA and the prion protein at the polybasic domain of residues 21–31. Site 2 involves the interaction with polybasic domain of residues 111–121 and is found four times, and site 3 (found three times) involves the residues 135–145 located in the N‐terminal helix A. For the mouse prion we find four binding sites in the 15 docked structures that did not dissolve in the molecular dynamics runs. These include the three binding sites seen for the human prion protein and a fourth one, involving residues 1–5 of the positively charged N‐terminal. Site 1 is found five times, site 2 three times, site 3 four times, and site 4 three‐times. Out of the docked structures that share the same binding site we then choose for each protein (human/mouse) and prediction algorithm (ITASSER/MODELLER) this structure for further analysis where the root‐mean‐square‐deviation over the 10 ns run was smallest, that is, the structure that appeared to be most stable. Note that in the case of the human prion we also repeated the above protocol for the post‐translated protein where the C‐terminal 23 residues are cleaved. This test let to the same binding sites. Since in the complexes with full‐sized protein the residues 230–253 do not form contacts with either the poly‐A‐RNA fragment or helix A, and the structure of this segment changes by ≈10 Å in the simulations described below, we conclude that the post‐translational cleavage does not affect the conversion between PrPC and PrPSC form of human and mouse prion.
Model confirmation
The quality of the predicted structures was assessed by following the approach of Hosen et al.28 and comparing the scores of three separate webservers: RAMPAGE,33 ERRAT,34 and ProQ.35 The cutoff used for the RAMPAGE score required the relaxed trajectory to have an aggregate of more than 85% of residues in favored regions and less than 0.8% (2 residues or less) in disfavored regions. Validation by ERRAT required the trajectories to have an average quality factor greater than 90%, as this suggests an acceptable model.33 ProQ predicts two scores, LGscore and MaxSub with cutoffs of 2.5 and 0.1, respectively.39 Averages for all analyzed structures are presented in Table 5.
Table 5.
Quality Scores of Trajectories Used for Docking Calculated From Three Separate Methods of Validation
| Sequence | Rampage favored | Rampage allowed | RAMPAGE disallowed | ERRAT | ProQ LgScore | ProQ MaxSub |
|---|---|---|---|---|---|---|
| Human Modeller | 88.6% (3.5) | 11.0% (2.1) | 0.6% (0.2) | 90.32% (2.1) | 0.265 (0.05) | 3.4 (0.4) |
| Human ITASSER | 90.7% (2.8) | 8.8% (2.8) | 0.4% (0.2) | 92.02% (3.2) | 0.350 (0.10) | 3.8 (0.6) |
| Mouse Modeller | 87.2% (4.0) | 12.4% (4.2) | 0.5% (0.3) | 90.03% (2.2) | 0.27 (0.20) | 3.5 (0.5) |
| Mouse ITASSER | 89.3% (3.0) | 10.3% (2.6) | 0.4% (0.1) | 91.91%(2.4) | 0.330 (0.08) | 3.7 (0.4) |
Docking confirmation
Binding site 3 involved the interaction with helix A without initial interaction to the N‐terminal region. This made it ideal to confirm that observed behavior was not due to bias in our model. Docking to the identified template structures, 2LSB and 2L40 for human and mouse respectively, we confirmed that binding to site 3 is the predominate site of interaction in the absence of N‐terminal polybasic domains. Simulation of these protein–RNA complexes leads to the same characteristic dissolution of α helical contacts (Supporting Information Fig. S1). Note that this unfolding of helix A is less pronounced in simulations without the N‐terminal domain.
Simulation protocol
The stability of the so‐derived 2 × 3 (2 × 4) docked structures predicted for the human (mouse) prion protein by either MODELLER or ITASSER was studied in longer molecular dynamics simulations, and compared with the outcome of control simulations of the two proteins (in their predicted configurations) without being docked to the RNA. These simulations rely on the GROMACS software package version 4.6.540 and utilize the CHARMM36 force field with associated nucleic acids parameters41, 42, 43, 44 and TIP3P water molecules,45, 46 a common choice for simulation of amyloid‐β systems which aggregate via pathways similar to that of prions.47, 48, 49 A cubic box with a side length of 12 Å is placed around the center of mass of the docked and undocked peptide systems. As this solvent box has periodic boundaries, electrostatic interactions are calculated using the PME algorithm.50, 51 The various RNA–prion complexes are first minimized by steepest descent, before being equilibrated in a succession of a 2 ns molecular dynamics run in an NVT ensemble and a 2 ns run in an NPT ensemble.
The six human prion and eight mouse prion protein structures, generated with the protocol described above, are the start point for long molecular dynamics simulations of 300 ns length in a NPT ensemble at 310 K and 1 bar pressure that allow us to probe the stability of the various systems. The equations of motions are integrated with a 2 fs time step, where hydrogen atoms are constrained by the LINCS algorithm52 and water using the Settle algorithm. The temperature is held constant at a physiological temperature of 310 K by a Parrinello–Donadio–Bussi thermostat53, 54 (τ = 0.1 fs), and pressure is similarly held constant at 1 bar by the Parrinello–Rahman algorithm (τ = 1 fs).55 As the simulation was not being performed on GPUs, a group cutoff scheme was used. A Verlet scheme would not lead to noticeable performance increases. Due to the system size, neighbor searching was handled on a grid with a cutoff of 1.5 nm. The long‐range electrostatic interactions were calculated with the Particle Mesh Ewald method, using cubic interpolation and the grid dimension set to 0.15 nm.
Generating different velocity‐distributions we follow each system in three trajectories in order to get a simple estimate for the statistical fluctuations. Data are saved every 4 ps for analysis with the tools available in GROMACS. Primarily, we measure the following quantities: root‐means‐square deviations of the Cα atoms (RMSD), secondary structure contents, contact distances, and hydrogen bonding footprint. Configurations are visualized using PYMOL.56
Supporting information
Supporting Information
Acknowledgment
The simulations in this work were done using XSEDE resources funded under project MCB160005 and on the BOOMER cluster of the University of Oklahoma.
References
- 1. Deleault NR, Harris BT, Rees JR, Supattapone S (2007) Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 104:9741–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Westergard L, Christensen HM, Harris DA (2007) The cellular prion protein (Prp(C)): its physiological function and role in disease. Biochim Biophys Acta 1772:629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, Aguzzi A (2010) Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 13:310–318. [DOI] [PubMed] [Google Scholar]
- 4. Caiati MD, Safiulina VF, Fattorini G, Sivakumaran S, Legname G, Cherubini E (2013) Prpc controls via protein kinase at the direction of synaptic plasticity in the immature hippocampus. J Neurosci 33:2973–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB (1993) Conversion of alpha‐helices into beta‐sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 90:10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Norstrom EM, Mastrianni JA (2006) The charge structure of helix 1 in the prion protein regulates conversion to pathogenic Prpsc. J Virol 80:8521–8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh J, Kumar H, Sabareesan AT, Udgaonkar JB (2014) Rational stabilization of helix 2 of the prion protein prevents its misfolding and oligomerization. J Am Chem Soc 136:16704–26707. [DOI] [PubMed] [Google Scholar]
- 8. Prusiner SB (1997) Prion diseases and the Bse crisis. Science 278:245–251. [DOI] [PubMed] [Google Scholar]
- 9. Singh J, Udgaonkar JB (2013) Dissection of conformational conversion events during prion amyloid fibril formation using hydrogen exchange and mass spectrometry. J Mol Biol 425:3510–3521. [DOI] [PubMed] [Google Scholar]
- 10. Schlepckow K, Schwalbe H (2013) Molecular mechanism of prion protein oligomerization at atomic resolution. Angew Chem Int Ed Engl 52:10002–10005. [DOI] [PubMed] [Google Scholar]
- 11. Tycko R, Savtchenko R, Ostapchenko VG, Makarava N, Baskakov IV (2010) The alpha‐helical C‐terminal domain of full‐length recombinant Prp converts to an in‐register parallel beta‐sheet structure in Prp fibrils: evidence from solid state nuclear magnetic resonance. Biochemistry 49:9488–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salamat K, Moudjou M, Chapuis J, Herzog L, Jaumain E, Beringue V, Rezaei H, Pastore A, Laude H, Dron M (2012) Integrity of helix 2‐gelix 3 domain of the Prp protein is not mandatory for prion replication. J Biol Chem 287:18953–18964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV (2016) New molecular insight into mechanism of evolution of mammalian synthetic prions. Am J Pathol 186:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berhanu WM, Alred EJ, Bernhardt NA, Hansmann UHE (2015) All‐atom simulation of amyloid aggregates. Phys Procdia 68C:61. [Google Scholar]
- 16. Berhanu WM, Alred EJ, Hansmann UH (2015) Stability of Osaka mutant and wild‐type fibril models. J Phys Chem B 119:13063–13070. [DOI] [PubMed] [Google Scholar]
- 17. Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366. [DOI] [PubMed] [Google Scholar]
- 18. Buhimschi IA, Nayeri UA, Zhao G, Shook LL, Pensalfini A, Funai EF, Bernstein IM, Glabe CG, Buhimschi CS (2014) Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci Transl Med 6:245ra92. [DOI] [PubMed] [Google Scholar]
- 19. Kouza M, Banerji A, Kolinski A, Buhimschi IA, Kloczkowski A (2017) Oligomerization of Fvflm peptides and their ability to inhibit beta amyloid peptides aggregation: consideration as a possible model. Phys Chem Chem Phys 19:2990–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang HS, Park SH, Park YW, Kwon HS, Sohn IS (2010) Expression of cellular prion protein in the placentas of women with normal and preeclamptic pregnancies. Acta Obstet Gynecol Scand 89:1155–1161. [DOI] [PubMed] [Google Scholar]
- 21. Miller MB, Geoghegan JC, Supattapone S (2011) Dissociation of infectivity from seeding ability in prions with alternate docking mechanism. PLoS Pathog 7:e1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zurawel AA, Walsh DJ, Fortier SM, Chidawanyika T, Sengupta S, Zilm K, Supattapone S (2014) Prion nucleation site unmasked by transient interaction with phospholipid cofactor. Biochemistry 53:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Webb B, Sali A (2014) Comparative protein structure modeling using Modeller. Curr Protoc Bioinform 47:1–32. [DOI] [PubMed] [Google Scholar]
- 24. Marti‐Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291–325. [DOI] [PubMed] [Google Scholar]
- 25. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I‐Tasser suite: protein structure and function prediction. Nat Methods 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roy A, Kucukural A, Zhang Y (2010) I‐Tasser: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y (2008) I‐Tasser server for protein 3d structure prediction. BMC Bioinform 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hosen MJ, Zubaer A, Thapa S, Khadka B, De Paepe A, Vanakker OM (2014) Molecular docking simulations provide insights in the substrate binding sites and possible substrates of the Abcc6 transporter. PLoS One 9:e102779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and Autodocktools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buchanan LE, Dunkelberger EB, Tran HQ, Cheng PN, Chiu CC, Cao P, Raleigh DP, de Pablo JJ, Nowick JS, Zanni MT (2013) Mechanism of Iapp amyloid fibril formation involves an intermediate with a transient beta‐sheet. Proc Natl Acad Sci USA 110:19285–19290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kouza M, Co NT, Nguyen PH, Kolinski A, Li MS (2015) Preformed template fluctuations promote fibril formation: insights from lattice and all‐atom models. J Chem Phys 142:145104. [DOI] [PubMed] [Google Scholar]
- 32. Rojas AV, Liwo A, Scheraga HA (2011) A study of the alpha‐helical intermediate preceding the aggregation of the amino‐terminal fragment of the beta amyloid peptide (Abeta(1‐28)). J Phys Chem B 115:12978–12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2:1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lovell SC, Davis IW, Arendall WB, III , de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 50:437–450. [DOI] [PubMed] [Google Scholar]
- 35. Wallner B, Elofsson A (2003) Can correct protein models be identified? Protein Sci 12:1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peltonen K, Colis L, Liu H, Trivedi R, Moubarek MS, Moore HM, Bai B, Rudek MA, Bieberich CJ, Laiho M (2014) A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 25:77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Piro JR, Supattapone S (2011) Photodegradation illuminates the role of polyanions in prion infectivity. Prion 5:49–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piro JR, Harris BT, Supattapone S (2011) In situ photodegradation of incorporated polyanion does not alter prion infectivity. PLoS Pathog 7:e1002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maiti R, Van Domselaar GH, Zhang H, Wishart DS (2004) Superpose: a simple server for sophisticated structural superposition. Nucleic Acids Res 32:W590–W594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) Gromacs 4.5: a high‐throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph‐McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz‐Kuczera J, Yin D, Karplus M (1998) All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 42. MacKerell AD, Jr , Feig M, Brooks CL, III (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas‐phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25:1400–1415. [DOI] [PubMed] [Google Scholar]
- 43. Denning EJ, Priyakumar UD, Nilsson L, Mackerell AD, Jr (2011) Impact of 2′‐hydroxyl sampling on the conformational properties of RNA: update of the Charmm all‐atom additive force field for RNA. J Comput Chem 32:1929–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, Mackerell AD, Jr (2012) Optimization of the additive Charmm all‐atom protein force field targeting improved sampling of the backbone Phi, Psi and side‐chain Chi(1) and Chi(2) dihedral angles. J Chem Theory Comput 8:3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mahoney MW, Jorgensen WL (2000) A five‐site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J Chem Phys 112:8910–8922. [Google Scholar]
- 46. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. [Google Scholar]
- 47. Kutzner C, Grubmuller H, de Groot BL, Zachariae U (2011) Computational electrophysiology: the molecular dynamics of ion channel permeation and selectivity in atomistic detail. Biophys J 101:809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zachariae U, Schneider R, Briones R, Gattin Z, Demers JP, Giller K, Maier E, Zweckstetter M, Griesinger C, Becker S, Benz R, de Groot BL, Lange A (2012) Beta‐barrel mobility underlies closure of the voltage‐dependent anion channel. Structure 20:1540–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jang H, Connelly L, Arce FT, Ramachandran S, Kagan BL, Lal R, Nussinov R (2013) Mechanisms for the insertion of toxic, fibril‐like beta‐amyloid oligomers into the membrane. J Chem Theory Comput 9:822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Darden T, York D, Pedersen L (1993) Particle Mesh Ewald: an N⋅Log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. [Google Scholar]
- 51. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593. [Google Scholar]
- 52. Hess B (2008) P‐Lincs: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput 4:116–122. [DOI] [PubMed] [Google Scholar]
- 53. Miyamoto S, Kollman PA (1992) Settle—an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem 13:952–962. [Google Scholar]
- 54. Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:7. [DOI] [PubMed] [Google Scholar]
- 55. Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. [Google Scholar]
- 56. DeLano WL (2002) The PyMOL User's Manual, DeLano Scientific, San Carlos. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information