

Abstract
The extracellular adherence protein (Eap) plays a crucial role in pathogenesis and survival of Staphylococcus aureus by inhibiting the classical and lectin pathways of complement. We have previously shown that Eap binds with nanomolar affinity to complement C4b and disrupts the initial interaction between C4b and C2, thereby inhibiting formation of the classical and lectin pathway C3 pro‐convertase. Although an underlying mechanism has been identified, the structural basis for Eap binding to C4b is poorly understood. Here, we show that Eap domains 3 and 4 each contain a low‐affinity, but saturable binding site for C4b. Taking advantage of the high lysine content of Eap, we used a zero‐length crosslinking approach to map the Eap binding site to both the α′‐ and γ‐chains of C4b. We also probed the C4b/Eap interface through a chemical footprinting approach involving lysine modification, proteolytic digestion, and mass spectrometry. This identified seven lysines in Eap that undergo changes in solvent exposure upon C4b binding. We found that simultaneous mutation of these lysines to either alanine or glutamate diminished C4b binding and complement inhibition by Eap. Together, our results provide insight into Eap recognition of C4b, and suggest that the repeating domains that comprise Eap are capable of multiple ligand‐binding modes.
Keywords: complement, protein‐protein interactions, Staphylococcus aureus, extracellular adherence protein
Introduction
The complement system fulfills an essential role in initiating the inflammatory response, and in clearance of foreign, diseased, or damaged cells (reviewed in Ref. 1). Though it is composed of nearly 30 proteins, the complement system can be divided conceptually into two distinct stages that consist of (i) initiation, amplification, and opsonization followed by (ii) assembly of the terminal complement complex and effector function. The initial stage of complement activity begins with ligand‐binding by a series of pattern recognition receptors and leads to assembly of multi‐subunit, surface‐associated serine proteases known as C3 convertases. Depending upon the specific biochemical stimuli which initiate complement activation, the surface‐bound C3 convertases that result may exist in two different isoforms. The so‐called Classical (CP) and Lectin (LP) Pathway C3 convertase is composed of the activated fragment of complement C4 (i.e., C4b) bound to the activated form of complement C2 (i.e., C2a). Proteolysis of the central complement component C3 by C4b2a leads to generation of C3b; this surface‐linked product triggers assembly of the alternative pathway (AP) C3 convertase, which is composed of C3b bound to the activated form of fB (i.e., Bb). The self‐amplifying effect of the AP C3 convertase fosters extensive opsonization of target surfaces with C3b, leads to activation of complement component C5, and drives downstream events that contribute to the effector functions of complement. Chief among these are assembly of the terminal complement complex (i.e., C5b‐9) and recruitment of inflammatory cells such as neutrophils to the site of complement activation.
Many successful pathogenic and/or parasitic species are capable of disrupting or escaping the immune response of their hosts. Though these so‐called immune evasion strategies are highly diversified, their specific modes of action and overall functional outcome are ultimately dictated by the route of infection and tissue tropism of the invading organism in question. As a prototypic endovascular pathogen, the Gram‐positive bacterium Staphylococcus aureus secretes multiple inhibitory proteins that act on the central innate immune events of opsonization, recruitment of neutrophils, and phagocytosis.2, 3, 4 Because the complement system plays a vital role in each of these processes, there appears to have been heavy selective pressure on S. aureus to evolve efficient means for interfering with complement activity. Indeed, studies from throughout the last decade have identified and characterized over a dozen individual complement evasion proteins from S. aureus. On balance, this work suggests that the complement evasion arsenal of S. aureus is likely the most diverse of any pathogen examined.
While the number of complement inhibitors expressed by S. aureus is noteworthy, the unique mechanisms of action employed by these proteins have also garnered attention. In this regard, S. aureus heavily targets formation, function, and stability of the surface‐bound, multi‐subunit C3 and C5 convertases which drive amplification of the complement response and opsonization of the bacterial cell.5, 6, 7, 8 We recently characterized the S. aureus Extracellular Adherence Protein (Eap) as a novel inhibitor of both the classical (CP) and lectin (LP) complement pathways.8 Eap binds to complement component C4b and thereby prevents binding of the pro‐protease C2 to C4b. Since formation of the CP/LP C3 pro‐convertase complex (i.e., C4b2) is required to assemble the fully‐active C3 convertase (i.e., C4b2a) that the CP and LP share, Eap is capable of simultaneously disrupting the two most potent routes for initiating complement activity. In doing so, Eap not only prevents opsonization of the S. aureus cell by C3b, it also blocks both phagocytosis and killing of S. aureus by neutrophils.
Eap is secreted by nearly all strains of S. aureus, though it is found in isoforms that vary in the number of ∼100 residue EAP repeats contained within the polypeptide.9, 10 Domain deletion studies on the four‐domain isoform of Eap from S. aureus strain Mu50 have demonstrated that the C4b‐binding and the CP/LP inhibitory activity of Eap lie within its two C terminal‐most repeats.8 Whereas full‐length Eap binds C4b with low‐nanomolar affinity (K D=185 nM), a truncated form of the protein consisting of domains 3 and 4 (Eap34) binds only ∼2.8‐fold more weakly (K D=525 nM). Importantly, inclusion of 1 μM Eap34 in either a CP or LP‐specific ELISA assay inhibits formation of the terminal complement complex similarly to full‐length Eap. By contrast, Eap domains 1 and 2 (Eap12) bind C4b weakly (K D ∼10 μM) and inhibit CP and LP activity to only a minor extent.8 While these results strongly suggest that there is specificity for C4b binding within the individual domains of the Eap protein, the structural basis for this specificity remains undefined.
The genome of S. aureus strain Mu50 contains three unique coding sequences (eap, eaph1, and eaph2) for proteins that adopt the EAP fold. Eap itself is composed of a tandem array of four non‐identical EAP domains, while both EapH1 and EapH2 consist of a single domain, giving rise to a total of six unique EAP domains expressed by S. aureus Mu50.10, 11 Although EAP domains share a significant level of structural identity as a group10 they are much more divergent at the sequence level when compared to one another (26–83% pairwise identity among the six). Despite this diversity, we recently discovered that recombinant forms of each individual EAP domain in isolation can act as a potent and selective inhibitor of neutrophil serine proteases (NSPs), as typified by neutrophil elastase (NE).11 Our subsequent crystal structure of NE bound to EapH1 revealed that the inhibitory site in EapH1 is composed of a pentagon‐shaped surface in the EapH1 tertiary structure,11 and which is formed from two separate stretches of amino acids that are separated by more than 20 residues in the EapH1 sequence. The regions of EapH1 that comprise its NE‐binding site are not highly conserved among other EAP domains [Fig. 1(A)]. This not only raised questions as to how the remaining EAP domains interact with NSPs, it also implied that the NE/EapH1 structure on its own would provide little insight into C4b binding by Eap. Furthermore, these observations led to the hypothesis that EAP domains as a whole may be capable of interacting with their ligands through multiple distinct binding modes.
Figure 1.
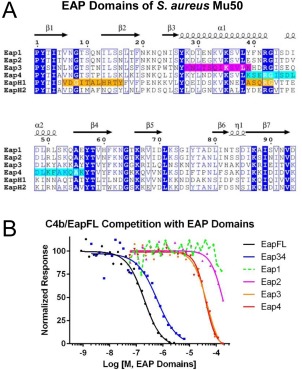
Domains Eap3 and Eap4 of the extracellular adherence protein from S. aureus Mu50 each contain a C4b‐binding site. (A) Sequence alignment of the four EAP repeats from S. aureus Mu50, along with the single‐domain EAP proteins, EapH1 and EapH2. Invariant positions are depicted in inverse blue typeface, while positions conserved in at least 4 of 6 sequences are drawn inside blue boxes. The secondary structure elements for a canonical EAP domain are shown above the sequence, and are derived from the 1.35 Å resolution structure of Eap2 (PDB Accession Code 1YN310). EapH1 positions that comprise the NE‐binding site in the NE/EapH1 co‐crystal structure (PDB Accession Code 4NZL11) are shaded orange. Regions in Eap3 and Eap4 spanning the lysines which exhibit a >50% change in solvent accessibility in the presence of C4b are shaded magenta and cyan, respectively. (B) The ability of untagged full‐length Eap (EapFL), Eap34, and the individual Eap repeats to compete the AlphaScreen signal generated by myc‐EapFL and C4b‐biotin was assessed over a logarithmic dilution series. Data presented here are taken from a representative trial of at least three independent experiments. Legend is inset.
To investigate these issues in greater detail, we developed a combinatorial strategy of zero‐length crosslinking, foot‐printing by mass spectrometry, site directed mutagenesis, and functional assays to obtain structural insights into C4b/Eap binding. In this manuscript, we present the outcome of this series of experiments which demonstrate the importance of key lysine residues in Eap domains 3 and 4 in mediating an interaction with the C4b α′‐ and γ‐chains. We also present a model for the C4b/Eap34 complex that is consistent with the experimental restraints identified during this work, as well as a recently published solution analysis of the C4b2 complex.12 Together, these data provide a physical basis for understanding the previously described competition between Eap and C2 for binding to C4b.8 This study not only expands our understanding of Eap's impact on formation of the CP/LP C3 pro‐convertase, it broadens our appreciation of the structure/function relationships of EAP domains as well.
Results
The Eap3 and Eap4 domains each contain a complement component C4b binding site and inhibit the classical and lectin pathways
We have previously shown that both full‐length Eap and Eap34 display dose‐dependent competition of a luminescence signal generated via site‐specifically biotinylated C4b binding to c‐myc epitope‐tagged Eap.8 In that same study, we observed that an equimolar mixture of the four individual domains from S. aureus Mu50 Eap failed to compete in a similar fashion even at concentrations up to 10 μM (i.e., 2.5 μM each domain). Because linking two weakly binding individual ligands into a single larger molecule can provide an increase in apparent affinity, we decided to quantitatively assess the C4b‐binding affinities of the individual domains of Eap in isolation. Using the competition‐based binding assay described above [Fig. 1(B)], we found that both Eap3 and Eap4 bound C4b with a K D value of ∼40 μM. While this value is nearly two orders of magnitude weaker than that of Eap34 (K D=525 nM),8 both Eap3 and Eap4 were nevertheless able to compete with full length Eap for C4b binding, in a dose‐dependent and saturable manner. Furthermore, we observed substantially weaker/no competition when either Eap1 or Eap2 were used in this assay, which indicated that the competitive effect was specific for Eap3 and Eap4.
Although Eap is a potent inhibitor of C3 activation, the mechanistic basis for this activity lies its ability to bind C4b and disrupt formation of the C4b2 pro‐convertase.8 But because the C4b2 pro‐convertase is shared by both the CP and LP, Eap manifests a similar inhibitory effect regardless of whether CP or LP activity is examined in various functional assays.8 Thus, to simplify our structure/function studies here, we chose to investigate complement activation via the LP as our primary analytical platform. Since our previous study examined the activity of various Eap truncations at a fixed concentration of both inhibitor (i.e., 1 μM) and serum [i.e., 1% (v/v) pooled human serum], we decided to test whether domain‐deleted forms of Eap also exhibited dose‐dependent inhibition of the LP using a fixed concentration of serum. Consistent with our previous results, Eap34 displayed IC50 values closest to full‐length Eap in ELISA‐based assays designed to measure the deposition of either C3b (227 vs 71.1 nM, respectively) or C5b‐9 (163 vs 74.9 nM, respectively) under conditions specific for activating the LP [Fig. 2(A) and Table S1, Supporting Information]. Likewise, we found that the individual domains of Mu50 Eap inhibited the activity of the LP [Fig. 2(B) and Table S1, Supporting Information], albeit at much higher concentrations than required for either the full‐length protein or Eap34. Nevertheless, the activity of the individual domains in this assay was consistent with their affinities for C4b [Fig. 1(B)], as both Eap3 and Eap4 blocked LP activity with IC50 values in the range of 5–15 μM, while neither Eap1 nor Eap2 completely inhibited complement activity in either assay. Together, these studies on individual EAP domains demonstrated that both Eap3 and Eap4 contain a specific C4b‐binding site that is capable of mediating inhibition of the LP, and therefore of the CP as well.
Figure 2.
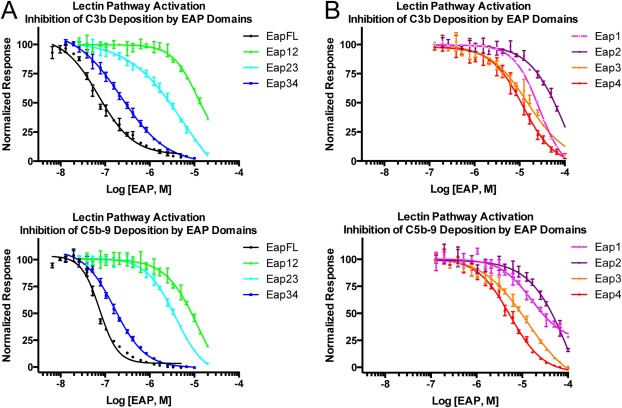
Domains Eap3 and Eap4 are sufficient to inhibit the lectin pathway of complement. Lectin pathway complement activity was assessed over a logarithmic dilution series of various Eap proteins. 1% (v/v) pooled human serum was used as a source of complement components, and each assay point was repeated in triplicate prior to fitting each series to a dose‐response curve. Legends are inset. (A) Lectin pathway activity in the presence full‐length Eap, Eap12, Eap23, or Eap34 was assessed by ELISA specific for either C3b (top panel) or C5b‐9 (bottom panel). (B) Lectin pathway activity in the presence of Eap1, Eap2, Eap3, or Eap4 was assessed by ELISA specific for either C3b (top panel) or C5b‐9 (bottom panel).
Eap34 binds to the α′‐ and γ‐chains of C4b
While we previously established that both full‐length Eap and Eap34 inhibit C2 binding to C4b,8 the structural basis for this activity remains unknown because the Eap binding site on C4b is undefined. C4b is composed of three distinct polypeptides that assemble into a disulfide‐linked heterotrimer. The identities of these chains are α′ (75.5 kDa, pI = 4.99), β (71.6 kDa, pI = 8.69), and γ (33.1 kDa, pI = 6.37), and they are readily resolved by SDS‐PAGE when samples are prepared under reducing conditions. Separately, the amino acid sequence of S. aureus Mu50 Eap is noteworthy for its high lysine content (∼15%, or 68 of 446 residues in the predicted full‐length protein). Since these lysines are distributed roughly equally throughout the Eap protein [Fig. 1(A)], we predicted that lysine‐specific chemistries might provide a relatively high level of positional accuracy for mapping the Eap binding site on C4b.
To test this idea, we utilized a “zero‐length” crosslinking strategy that is highly selective for amines and carboxylates within contact distance of one another.13 We first used 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide (EDC) and N‐hydroxysulfosuccinimide (sulfo‐NHS) to convert the carboxylate groups of C4b into amine‐reactive esters. Then we added an equimolar concentration of Eap34, and allowed crosslinking to occur over the course of 180 min. Separation of the reaction products by 10% tris‐tricine SDS‐PAGE under reducing conditions revealed a time‐dependent change in the electrophoretic mobility of the γ‐chain in the presence of Eap34 when compared to C4b control [Fig. 3(A, C)]. This change in mobility can also be seen when compared to a control experiment wherein purified C4b was subjected to a similar reaction sequence in the absence of Eap34 [Fig. S1(A), Supporting Information]. Notably, the staining intensity of the γ‐chain decreased throughout the course of the reaction, while a new species of mobility distinct from either the γ‐chain or Eap34 appeared. Although not directly correlated to the addition of mass of Eap34 to the γ‐chain, the mobility of this putative Eap34/γ‐chain adduct could be explained by a non‐uniform charge/mass ratio for this species when compared to other polypeptides in the gel. As an independent approach to understand the change in electrophoretic mobility of the γ‐chain, we excised the putative adduct band from the 180 min time point, proteolyzed the polypeptides in‐gel, and characterized the digestion products by MALDI‐TOF mass spectrometry. Peptide fingerprint analysis revealed the presence of both Eap34 and the C4b γ‐chain at coverage levels of 94.8 and 54.3%, respectively [Fig. S1(B), Supporting Information]. Together, these results established the identity of this species as an Eap34/γ‐chain adduct, and strongly suggested that Eap34 interacts with the C4b γ‐chain.
Figure 3.
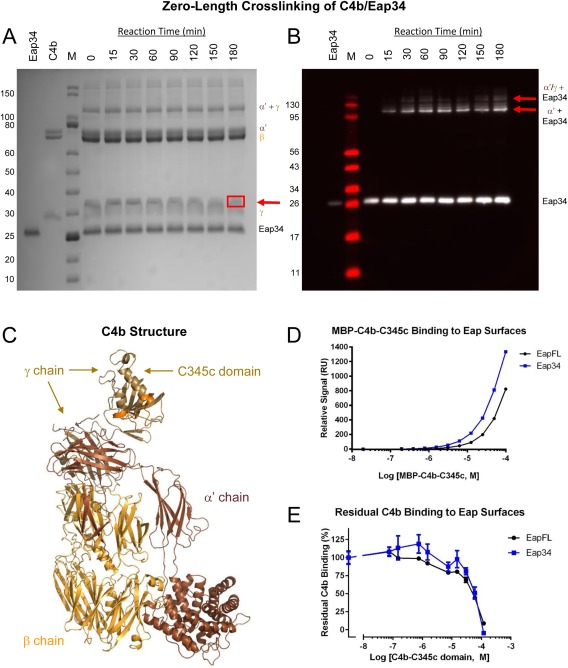
The Eap34 binding site involves both the α′‐ and γ‐chains of C4b. (A) Time‐dependent, zero‐length crosslinking between free amines of Eap34 and activated carboxylates of C4b. C4b was activated by treatment with EDC and NHS prior to incubation with Eap34. Aliquots were withdrawn over the course of 180 min, and the contents were denatured, reduced, and separated by SDS‐PAGE. While no time‐dependent changes were visible for either the α′ or β‐chains of C4b, loss of staining intensity of the γ‐chain correlated with the appearance of a band with altered mobility from that of the γ‐chain or Eap34 (red arrow). This band contained both Eap34 and γ‐chain, as judged by peptide fingerprinting analysis [Fig. S1(B), Supporting Information]. (B) An identical reaction series to that shown in panel A was prepared, separated by SDS‐PAGE under reducing conditions, and transferred to membranes for analysis by immunoblotting. A polyclonal anti‐Eap antisera raised in sheep readily detected recombinant Eap34, and additional adduct bands of higher apparent molecular weight corresponding to the α′‐chain + Eap34 and the α′/γ‐chains + Eap34 (red arrows). (C) A representation of the C4b structure (PDB Accession Code 4XAM23) is provided for clarity in interpreting SDS‐PAGE and blotting images. Each distinct polypeptide is colored differently, with the α′‐chain in brown, β‐chain in yellow‐orange, and γ‐chain in beige. The MIDAS‐acceptor site of the C345c domain, which mediates metal‐dependent binding of the vWF domain of C2 to C4b, is shaded orange. (D) Analysis of the dose‐response sensorgram data shown in Figure S2, Supporting Information, using a steady‐state binding model. (E) Competition binding between recombinant C4b‐C345c and native C4b, using the same biosensor surfaces shown in panel D. A fixed concentration of C4b was incubated with increasing concentrations of C4b‐C345c, and the residual binding of C4b was determined following co‐injection in triplicate. Notably, the midpoint of both curves in this study correspond well to the affinities observed in the MBP‐C4b‐C345c direct binding studies of panel D and Figure S2, Supporting Information.
To further characterize the products of the crosslinking reaction, we repeated the experiment and processed the samples for Western blotting. We probed the reaction series membrane with a sheep polyclonal anti‐Eap antibody followed by detection using an HRP‐conjugated secondary antibody. Although this procedure did not reveal anti‐Eap immunoreactivity in the region of the membrane corresponding to the Eap34/γ‐chain adduct, this could arise from masking or reaction of residues essential for antibody recognition in the crosslinked species [Fig. 3(B, C)]. Nevertheless, Western blotting revealed clear anti‐Eap immunoreactivity at a molecular weight corresponding to Eap34 coupled to the C4b α′‐chain [Fig. 3(B), bottom red arrow, 3(C)]. A species corresponding to Eap34 coupled to the crosslinked α′ + γ‐chains was also detected, albeit at a far lower abundance [Fig. 3(B), top red arrow, 3(C)]. Thus, analysis of the crosslinking reaction by Western blotting strongly suggested that Eap34 recognizes the C4b α′‐chain in addition to the γ‐chain, as described above.
The C345c domain lies at the C terminus of the C4b γ‐chain and constitutes nearly one‐half of the γ‐chain molecular weight [Fig. 3(C)]. To determine if this domain contributes to Eap34 binding, we expressed C4b‐C345c as a fusion with E. coli maltose‐binding protein (i.e., MBP‐C4b‐C345c). We took advantage of the enhanced solubility that this fusion protein provides relative to C4b‐C345c alone to achieve sufficient concentrations for direct binding studies. MBP‐C4b‐C345c exhibited clear, dose‐dependent binding to biosensor surfaces composed of either full‐length Eap or Eap34 [Fig. 3(D) and Fig. S2, Supporting Information]. Although we could not reach sufficient concentrations of MBP‐C4b‐C345c to completely saturate either surface, both sensorgram series fit to nearly identical K D values of 63 and 69 μM when analyzed by a steady‐state model [Fig. 3(C)]. In an alternative approach, we also tested whether excess C4b‐C345c protein inhibited binding of saturating levels of C4b to either full‐length Eap or Eap34 biosensor surfaces. Increasing levels of C4b‐C345c in the presence of a fixed concentration of C4b led to a gradual loss of C4b binding by both surfaces [Fig. 3(E)]. Importantly, the midpoint of both competition series matched well with the K D values obtained for MBP‐C4b‐C345c binding to full‐length Eap and Eap34 [Fig. 3(D, E)]. In summary, our results suggest that the C4b γ‐chain and more specifically, the C4b‐C345c domain, as the binding site of a single domain for Eap34. This interpretation is consistent with the affinities of single EAP domains for C4b [Fig. 1(B)] and the relative affinity of the C4b‐C345c domain for Eap34 [Fig. 3(D, E) and Fig. S2, Supporting Information]. Additional contributions are also made through a single domain of Eap34 binding to the C4b α′‐chain [Fig. 3(B, C)]. Such a multipartite interaction with distinct regions of C4b explains the enhanced affinity of Eap34 relative to the individual domains [Fig. 1(B)].8
Specific lysine residues mediate Eap34 binding to C4b and inhibition of the classical and lectin pathways
Our discovery that Eap3 and Eap4 bind C4b and inhibit the LP [Figs. 1(B) and 2(B), and Table S1, Supporting Information] indicated that both of these domains contain a specific C4b binding site. Furthermore, the detection of zero‐length crosslinks between Eap34 lysine sidechains and C4b α′‐ and γ‐chain carboxylates strongly suggested that specific lysines in Eap34 were critical to forming the C4b/Eap34 complex (Fig. 3 and Fig. S1, Supporting Information). To identify the lysine residues of Eap34 that mediated interaction with C4b, we employed a chemical footprinting/protection strategy where mass spectrometry of chymotryptic peptides was used to assess ligand‐dependent changes in sidechain acetylation following exposure to N‐acetyl‐hydroxysuccinimide (NHS‐Ac).14 Chymotrypsin digestion of Eap34, followed by characterization of the products by MALDI‐TOF MS resulted in identification of approximately 60 peptides that covered ∼80% of the Eap34 sequence, including 33 of the 37 lysine residues [Fig. S3(A), Supporting Information]; the four C‐terminal‐most lysine residues were not observed. A repeat of this experiment following exposure to NHS‐Ac revealed that all of the observed lysines were acetylated, and therefore surface/solvent‐exposed [Fig. S3(A), Supporting Information].
We carried out an analogous NHS‐Ac labeling experiment on a sample of C4b/Eap34. We used concentrations of each monomer (10 μM C4b and 7 μM Eap34) in sufficient excess of the K D for the C4b/Eap34 interaction to ensure nearly 90% occupancy of the complex, but low enough in concentration to avoid generation of non‐specific, higher‐order aggregates. Analysis of four replicates of Eap34 chymotryptic peptides produced in this manner revealed clear trends in protection differences across the Eap34 sequence [Figs. 4(A) and Fig. S3(B), Supporting Information]). Whereas twelve lysines (i.e., K331 and K335 in Eap3 and all lysines in Eap4 from K446 to the C terminus) were acetylated in at least 75% of total observations, seven lysines (i.e., K297, K304, and K308 in Eap3, and K413, K424, K427, and K430 in Eap4) were acetylated in fewer than 33% of total observations. Of these, K304, K427, and K430 were acetylated less than 10%. Significantly, we found residues exhibiting high levels of protection from acetylation in both the Eap3 and Eap4 domains. Thus, these chemical footprinting results were consistent with the C4b‐binding data presented above [Fig. 1(B)].
Figure 4.
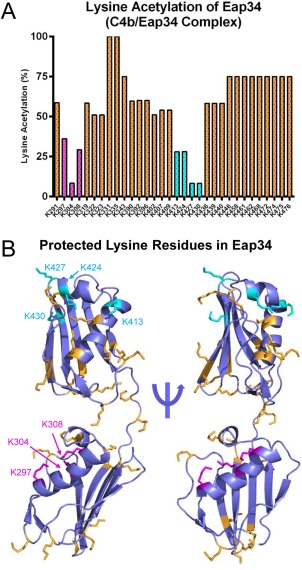
Specific lysine residues in Eap34 experience changes in solvent accessibility upon C4b binding. The solvent accessibility of Eap34 lysine residues was examined in the absence or presence of C4b through a chemical footprinting approach. A chymotrypsin digestion coverage map was first obtained for Eap34 as described in the Materials and Methods section, and changes in m/z of various peptides were characterized in an identical sample that had been treated with NHS‐Ac. Alterations in these acetylation modification patterns were studied for a pre‐formed C4b/Eap34 complex. The results of four independent experiments were merged to arrive at a total number of observations for each individual lysine residue. (A) Relative protection status of each observable Eap34 lysine in samples of C4b/Eap34. Positions K297, K304, and K308 of Eap3 (magenta), and K413, K424, K427, and K430 in Eap4 (cyan) were acetylated in less than 33% of all observations, implying they were less solvent‐accessible than other Eap34 lysines (orange) when bound to C4b. (B) Structural context of the lysine residues in Eap34. All lysine sidechains are rendered in ball‐and‐stick convention. Residues that were readily acetylated in the experiment described above are colored orange, while those identified as protected from acetylation when bound to C4b are colored as magenta (Eap3) and cyan (Eap4), respectively.
Although there are no crystal structures available for Eap3 or Eap4, there do exist high‐resolution structures for Eap2, EapH1, and EapH2, which show a high‐level of identity to one another (∼90% of the residues in each domain align with an r.m.s.d < 1Å).10 Using these structures as a template, we previously constructed a model for full‐length Eap based on small angle X‐ray scattering data and energy minimization.15 Inspection of this model revealed that the lysines which contribute to two C4b contact sites lie in different regions of their respective domains [Fig. 4(B)]. While the protected residues of Eap3 are associated with its large α‐helix, the key lysines of Eap4 primarily lie near the smaller α‐helix and the turn that connects it with the β‐strand that follows. Importantly, the locations of these residues within the Eap3 and Eap4 domains differ from those of EapH1 that contact NE in a co‐crystal structure [Fig. 1(A), orange highlight].11 This observation suggests that EAP domains are capable of diverse modes of binding to their respective ligands.
We used site‐directed mutagenesis as a strategy for independently assessing the role of these Eap34 lysine residues in C4b binding and complement inhibition. We overexpressed and purified two distinct mutants; the first mutant had all seven of the protected lysine residues identified above changed to alanine (i.e., Eap34‐KΔA), while the second mutant had the same residues changed to glutamate (i.e., Eap34‐KΔE). Circular Dichroism spectropolarimetry on both mutants revealed no significant structural alterations [Fig. 5(A)]. However, while both mutants maintained activity in a competition binding assay, each was diminished in its affinity for C4b [Fig. 5(B)]. Specifically, the apparent K D values of 1.53 μM for Eap34‐KΔA and 3.75 μM for Eap34‐KΔE were decreased by 2.6‐ and 6.4‐fold, respectively, when compared to wild‐type Eap34. To investigate the functional consequences of these mutations, we examined the ability of each protein to inhibit generation of C3b in an ELISA‐based assay specific for the LP [Fig. 5(C)]. Both the Eap34‐KΔA (IC50=984 nM) and Eap34‐KΔE (IC50=2.24 μM) mutants were less effective inhibitors of the LP when compared to wild‐type (IC50=227 nM). Significantly, the magnitude of these changes in inhibitory activity (4.3‐ and 9.9‐fold loss) was consistent with that observed in the C4b‐binding studies above. In sum, these results established that specific lysine residues in Eap34 play important roles in mediating binding to C4b and inhibition of complement activity.
Figure 5.
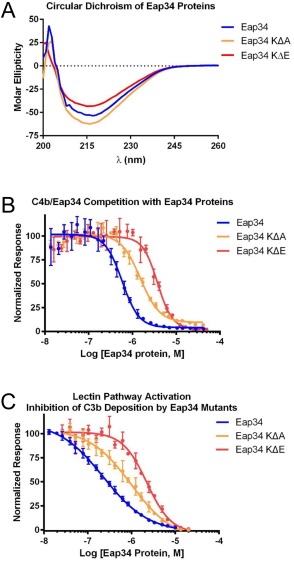
Eap34 lysine residues identified by chemical footprinting contribute to C4b binding. The contribution of Eap34 lysines to C4b binding was explored by site‐directed mutagenesis, wherein the seven protected lysines were mutated in concert to either alanine (Eap34‐KΔA) or glutamate (Eap34‐KΔE). (A) CD Spectropolarimetry was used to assess the secondary structure content of Eap34, as well as the Eap34‐KΔA and Eap34‐KΔE mutants. (B) The ability of untagged Eap34 and both mutants to compete the AlphaScreen signal generated by myc‐Eap34 and C4b‐biotin was assessed over a logarithmic dilution series. All assay points were conducted in triplicate prior to fitting to a dose‐response curve. (C) Lectin pathway activity in the presence of Eap34 and both mutants was assessed by ELISA specific for C3b. All assay points were conducted in triplicate prior to fitting to a dose‐response curve. Legends are inset.
Eap binds C4b at a different site than the Group B Streptococcus complement inhibitory protein
We previously determined that C4b/Eap binding impedes assembly of the CP/LP C3 pro‐convertase.8 More recently, Pietrocola et al. identified a novel complement inhibitory protein (i.e., CIP) from Group B Streptococcus that also binds C4b and blocks assembly of the CP/LP C3 pro‐convertase.16 Since Eap and CIP appear to share a similar mechanism of action, and since they both act directly on C4b, we hypothesized that Eap and CIP might also share the same C4b binding site. To test this possibility, we investigated whether CIP could compete the luminescence signal generated by biotinylated‐C4b binding to either c‐myc epitope‐tagged full‐length Eap or Eap34 (Fig. 6). Even though the reported K D for C4b/CIP binding is ∼95 nM,16 we observed essentially no competition in either assay at CIP concentrations as high as 10 μM, and only partial competition at the highest levels of CIP attainable. Such weak competition between Eap and CIP strongly suggests that these proteins recognize different binding sites on C4b even though they manifest inhibition of complement activity through an apparently similar mechanism.
Figure 6.
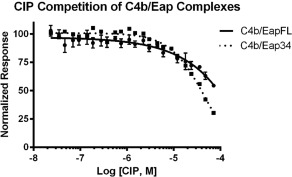
CIP from group B Streptococcus does not compete with S. aureus Eap for C4b binding. The ability of untagged CIP to compete the AlphaScreen signal generated by C4b‐biotin and either myc‐tagged full‐length Eap or Eap34 was assessed over a logarithmic dilution series. All assay points were conducted in triplicate prior to fitting to a dose‐response curve. The weak and incomplete competitive effect displayed by CIP only at the highest concentrations tested suggests that the CIP binding site on C4b is distinct from that from Eap.
Discussion
Since its discovery in the early 1990s, Eap has stood out from other staphylococcal immune evasion proteins by virtue of its reported interactions with multiple host‐derived ligands.17, 18, 19, 20, 21, 22 This impressive list of binding partners includes large glycoproteins found in the extracellular matrix, such as fibronectin, fibrinogen, vitronectin, and collagen, as well as cell surface receptors, including ICAM‐1. More recently, we discovered that Eap binds to complement component C4b8 as well as to the neutrophil granule proteases elastase (NE), cathepsin‐G (CG), and proteinase‐3 (PR3).11 Significantly, these latter interactions have been characterized through direct bio‐physical/chemical approaches, such as surface plasmon resonance and isothermal titration calorimetry (ITC). These results have permitted rigorous quantitative validation of both the affinity and specificity of Eap's interactions with C4b and NSPs.8, 11 Such information is essential for understanding the functional consequences of Eap/ligand interactions. It can also help guide structural studies of Eap, the independently folding EAP domains which comprise it, and the various complexes they form.
A co‐crystal structure of the Eap homolog, EapH1, bound to the NSP, NE, has been solved.11 To date, this represents the only structure of an EAP domain‐containing protein bound to its target. While the NE/EapH1 co‐crystal structure identified a flexible loop region in EapH1 which contributes to the majority of the NE contact site, poor levels of conservation in this region across various EAP domain sequences suggests that alternative binding modes may be found in other EAP/NSP complexes. Nevertheless, the relatively high‐affinity of the NE/EapH1 complex (K D=20 nM, as judged by ITC11) along with its co‐crystal structure, provided strong evidence that certain EAP domains can form robust interactions with their respective targets. The case of Eap binding to C4b appears to be somewhat more complex. Not only does C4b bind full‐length Eap nearly 10‐fold weaker than NE/EapH1,8, 11 the individual domains that comprise its minimally active fragment (i.e., Eap3 and Eap4) bind C4b with K D values some 200‐fold weaker still [Fig. 1(B)]. Although the interactions of Eap3 and Eap4 with C4b are saturable, specific, and are sufficient to manifest complement inhibitory activities characteristic of the intact Eap molecule [Figs. 1(B) and 2 and Table S1, Supporting Information], joining the two domains together has a profound effect on their overall affinity and inhibitory activity [Figs. 1(B) and 2 and Table S1, Supporting Information].8 This observation suggests that combinations of adjacent EAP domains may generate composite binding surfaces and synergistically enhance properties that are relatively weak in isolation. Indeed, this sort of structure/function relationship was proposed following solution structural analysis of full‐length Eap.15 Collectively, these results suggest that EAP domains can manifest multiple high‐affinity protein‐protein interaction modes, both as individuals and in combination with one another.
Our understanding of how multiple EAP domains working in tandem can generate a high‐affinity ligand would be greatly enhanced by a crystal structure of Eap34 bound to C4b. Although we successfully crystallized a closely related complex (i.e., C4c/Eap34), these crystals diffracted X‐rays poorly and ultimately proved unsuitable for in‐depth studies. Subsequent attempts to characterize this complex in the solution state by SAXS also failed, primarily as a result of its modest affinity and a tendency of the sample to aggregate non‐specifically at higher concentrations. To circumvent these limitations, we used an acetylation footprinting strategy to identify positions of contact between Eap34 and C4b (Fig. 4 and Fig. S3, Supporting Information). While a limitation of this approach is that it only reports on free amino groups, the unusually high percentage of lysines found not only in Eap34, but all EAP domains, makes it particularly suitable for studying these molecules. Indeed, we successfully identified seven lysines in Eap34 that experienced substantial changes in acetylation protection in the presence of C4b (Fig. 4 and Fig. S3, Supporting Information). Concurrent mutagenesis of these lysine positions to alanine diminished the C4b‐binding affinity and complement inhibitory properties of the protein (Fig. 5). This loss of affinity and inhibitory activity was augmented by changing the same positions to glutamate. On the basis of these results, we conclude that the specific lysine residues identified by acetylation footprinting collectively contribute to C4b binding and complement inhibition by Eap34. Moreover, these data also suggest that the positive charge of the lysine sidechain is important at these positions. To further define whether specific chemical features of lysine or positive charge alone was more important, we attempted to prepare a mutant where these seven positions had been concurrently changed to arginine. Unfortunately, this mutant could not be produced in a soluble form.
Considerations of specific residues aside, our acetylation protection data have also identified additional interaction sites on EAP domains [Fig. 4(B)]. In particular, our results show that the C4b‐binding site of Eap34 involves the large α‐helix in Eap3 and a region including the smaller α‐helix in Eap4. While it is noteworthy that these two sites are removed from the prominent loop region of EapH1 that contacts NE in the NE/EapH1 co‐crystal structure [Fig. 1(A)],11 it is important to recognize that mutation of these positions diminished, but did not completely abrogate the C4b binding or complement inhibitory activity of Eap34 (Fig. 5). We believe that this argues for the involvement of additional Eap34 positions in contacting C4b. In this regard, residues nearest the lysines discussed above would be among obvious candidates. Future work that makes use of complimentary approaches (e.g., scanning mutagenesis) will likely be needed to identify these additional residues and to define their contributions to C4b binding.
The studies described here also help explain how Eap inhibits formation of the CP/LP C3 pro‐convertase, since our zero‐length crosslinking studies and supporting biochemical data can now be placed in a three‐dimensional context [Fig. 3(A‐C) and Fig. S1, Supporting Information]. Shortly after our initial characterization of the C4b/Eap interaction and its functional consequences,8 Mortensen et al. reported the first crystal structure of C4b.23 More recently, these same authors described an extensive investigation of the CP/LP C3 pro‐convertase in solution.12 Together, their work has provided critical insights on the formation of the CP/LP C3 pro‐convertase, including description of the domain orientations of C4b bound to both C2 and C2a. Their data, along with our results showing that Eap34 competes with C2 for C4b binding8 and that Eap34 binds the C4b α′ and γ‐chains (Fig. 3, and Figs. S1 and S2, Supporting Information), has allowed us to construct an experimentally constrained structural model for the C4b/Eap34 complex (Fig. 7 and Fig. S4, Supporting Information).
Figure 7.
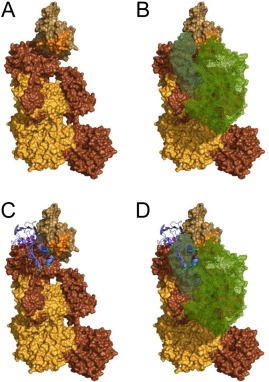
An experimentally constrained structural model for the C4b/Eap34 complex. An experimentally constrained model for the C4b/Eap34 complex was generated by the ClusPro server as described in the Materials and Methods section. (A) Surface rendering of C4b where each distinct polypeptide is shaded as follows: α′‐chain in brown, β‐chain in yellow‐orange, and γ‐chain in beige. The MIDAS‐acceptor site of the C345c domain, which mediates metal‐dependent binding of the vWF domain of C2 to C4b, is shaded orange. (B) Space filling model of the CP/LP C3 pro‐convertase, C4b2. C4b is colored as described in panel A, while the two regions of C2 (wire mesh) are colored with C2b in forest green and C2a in light green. (C) C4b/Eap34 structural model where C4b is colored as in panels A and B, while Eap34 is shown as a purple ribbon. The locations of key lysine residues in Eap34 are highlighted in magenta (Eap3) and cyan (Eap4), consistent with Figure 4(B). (D) Superposition of the structures shown in panels B and C. Note the similar locations of the C2b and Eap34 binding sites on C4b, and the potential for extensive steric clash if both proteins were present. This model is therefore in good agreement with the previously described ability of Eap34 to potently compete with C2 for C4b binding.8
Although it is difficult to account for the flexibility of the residues which link the individual domains in the Eap34 protein and any changes they might experience upon binding to C4b, our structural model nevertheless satisfies several restrictions established via lysine footprinting data (Fig. 4). In the case of Eap3, the protected lysines K297, K304, and K308 all lie within close proximity to sidechains derived from positions E763, E764, and D765 near the C4b α′‐chain N‐terminus, all of which could participate in the zero‐length crosslinking chemistry that we employed here. While the basis for Eap4 crosslinking to C4b is not as obvious, our model suggests that the protected lysines K413 and K424 may approach contact distances with the C‐terminus of the C4b γ‐chain. Since the terminal carboxylate of a polypeptide can also be modified by EDC and converted into amine‐reactive esters of NHS, the extreme C‐terminus of C4b could also represent a site of crosslinking between Eap34 and C4b.
We have previously shown that full‐length Eap inhibits the initial interaction between the C2b region of C2 and C4b.8 Considering the structural homology between the AP C3 pro‐convertase C3bB and the CP/LP C3 pro‐convertase C4b2, along with the fact that the smaller fragments of fB (i.e., Ba) and C2 (i.e., C2b) are responsible for the initial association of these pro‐proteases with C3b and C4b, respectively,12, 24 our model suggests that the competitive effects of full‐length Eap on C2b binding to C4b can be explained by contacts between Eap34 and both the C4b α′‐ and γ‐chains (Figs. 3 and 7, and Fig. S4, Supporting Information). Specifically, the pocket on C4b that forms between its γ‐chain C345c domain and the neighboring macroglobulin‐like domain of the C4b α′‐chain is too small to accommodate C2b when Eap34 is bound (Fig. 7). It is also worth noting that the character of C4b in this region is thought to be influenced by an unusual stretch of 10 residues at the C‐terminus of the α′‐chain, which consists of 3 sulfated tyrosines along with 7 aspartates and glutamates. Although this sequence is not visible in the C4b crystal structure, SAXS‐constrained modeling of this sequence in C4b is consistent with its presence at or near the proposed Eap34 binding site (Fig. 7 and Fig. S4, Supporting Information).23 Thus, the proposed Eap34 binding site is ideal not only for disturbing interactions between the C4b and its ligand, C2, it may also facilitate recruitment of the highly positively charged Eap molecule (pI = 9.96) to this particular binding site on C4b in the first place.
Many powerful complement evasion mechanisms act at the level of C3 convertases.25 Formation of active C3 convertases is predicated upon assembly of their corresponding pro‐convertases, however, which has rendered these latter structures very effective points for execution of evasion strategies.25, 26 Indeed, both S. aureus Eap and Group B Streptococcus CIP share the ability to block initial interaction between C4b and C2, and thereby inhibit formation of the CP/LP C3 pro‐convertase.16, 25 Even though Eap and CIP manifest similar activities, CIP surprisingly failed to compete with either Eap or Eap34 for C4b binding (Fig. 6). This observation suggests the structural basis for CIP function is substantially different than that of Eap. In this regard, the results we present here demonstrate that the Eap binding site lies on the C4b α′‐ and γ‐chains (Figs. 3 and 7 and Figs. S1, S2, and S4, Supporting Information), and are broadly consistent with orthosteric inhibition of pro‐convertase formation. By contrast, comparatively little is known regarding the structural basis for C4b/CIP binding. Although structural information for CIP is limited, Pietrocola et al. suggested that CIP might share structure/function similarity to the S. aureus AP inhibitors, Efb‐C and Ehp.16, 26 While neither Efb‐C nor Ehp bind C4 or its activated fragment C4b, both of these proteins bind a native complement protein (i.e., C3), its major activation product (i.e., C3b), and inhibit pro‐convertase formation through an allosteric mechanism.7, 26 By analogy, these authors' suggestion also predicts that (i) CIP binds C4b in an altogether different site than Eap, and that (ii) occupation of this binding site alters C4b conformational states in a way that disturbs normal C2 binding. While additional structural information is clearly needed to draw accurate conclusions on these matters, the lack of competition between CIP and Eap for C4b binding which we report here suggests that an allosteric mechanism is likely for CIP.
Materials and Methods
Native and recombinant proteins
Human serum proteins C4, C4b, C1s, C4b‐binding protein (C4BP), and Factor I were obtained in purified form from Complement Technologies (Tyler, TX). Site‐specific biotinylation of C4b was carried out using a previously described method.8 All recombinant S. aureus proteins, CIP from Group B Streptococcus, as well as C4‐C345c and the MBP fusion MBP‐C4‐C345c were overexpressed and purified according to previously described methods.27 Site‐directed mutagenesis was used to construct lysine to alanine (KΔA) and lysine to glutamate (KΔE) forms of Eap34 using gBlocks® Gene Fragments (Integrated DNA Technologies) with SalI and NotI restriction sites added at the 5′ and 3′ end, respectively. Following PCR, the mutagenic product was subcloned into the prokaryotic expression vector, pT7HMT, and sequenced to confirm its integrity.27 Protein expression and purification was carried out as described for the wild‐type Eap34 protein.8
AlphaScreen binding assays
An AlphaScreen competition‐based binding assay was performed using a previously published protocol.8, 28 In short, C4b/EapFL, C4b/Eap34, and C4b/CIP competition based assays were done using a total volume of 25 μL, in a buffer composed of HBS [20 mM HEPES (pH 7.4), 140 mM NaCl, 0.1% (w/v) BSA, 0.01% (v/v) Triton‐X 100]. Each component was added to the final concentrations: 50 nM myc‐EapFL (50 nM myc‐Eap34), 5 nM C4b biotin, 20 μg/μL anti‐c‐myc AlphaScreen acceptor beads, and 20 μg/μL AlphaScreen donor beads. A twofold dilution series was prepared for each unlabeled competitor protein and allowed to equilibrate with myc‐Eap/myc‐Eap34/myc‐CIP and C4b biotin for 1 h. Following this, the acceptor beads were added, incubated for 1 h, and then the donor beads were added and incubated for an additional 0.5 h. Reactions were then transferred to half‐Area 96‐well plates and measured using an EnSpire multimode plate reader (Perkin Elmer Life Sciences). Data analysis and curve fitting were carried out as previously described.28
Activity of the lectin pathway of complement on an artificial surface
Functional activity of the lectin pathway (LP) was determined using a previously described method.8, 29 In short, 96‐well polystyrene high bind microplates (Corning Life Sciences) were coated overnight to specifically activate the LP (20 μg/mL Saccharomyces cerevisiae mannan [Sigma‐Aldrich]). Plates were blocked with 1% (w/v) BSA, in PBS (pH 7.4) with 0.05% (v/v) Tween 20 for 1 h at 37°C. In order to obtain IC50 values for each inhibitor, a two‐fold dilution series was done by diluting the protein 1:1 in LP buffer (50 mM HEPES (pH 7.5), 140 mM NaCl, 0.1% (w/v) Gelatin, 0.1% (w/v) BSA, 2 mM CaCl2, 0.5 mM MgCl2, 1% (v/v) Pooled Complement Human Serum [Innovative Research Inc.]) before direct application to the coated, blocked ELISA plate and incubated for 1 h at 37°C. Deposited C3b and C5b‐9 were detected with 0.333 μg/mL C3d Antibody (003–05): sc‐58928 and 0.2 μg/mL C5b‐9 (aE11): sc‐58935 (Santa Cruz Biotechnology, Inc.), respectively, diluted in PBS (pH 7.4), 0.1% (w/v) BSA, 0.05% (v/v) Tween‐20 and incubated at RT for 0.5 h. Finally, the Goat anti‐Mouse IgG, IgM (H + L) Cross Absorbed Secondary Antibody, HRP conjugate (Thermo Scientific) was diluted to 1.6 μg/mL in equivalent dilution buffer and added to each well for 0.5 h at RT. HRP‐labeled Abs were detected with 50 μL of 1‐Step Ultra TMB‐ELISA (Thermo Scientific), the reaction was stopped by the addition of an equal volume of 2 M H2SO4, and the absorbance at 450 nm was measured using a VERSAMAX microplate reader. Data were fit to a four‐parameter (variable slope) dose‐response—inhibition curve software (GraphPad, La Jolla, CA).
EDC/sulfo‐NHS zero‐length crosslinking of Eap34 and C4b
A zero‐length crosslinking procedure was adapted from a previously described method.13 In short, purified complement C4b (Complement Tech) and recombinant Eap34 were buffer exchanged into 0.05 M MES (pH 6.0), 0.5 M NaCl (reaction buffer), and 0.1 M sodium phosphate (pH 7.5), respectively. C4b (5 μM) was activated by the addition of 2 mM 1‐Ethyl‐3‐(3‐dimethylaminopropyl)‐carbodiimide (EDC) and 5 mM N‐hydroxysulfosuccinimide (sulfo‐NHS) at RT for 15 min. The reaction was quenched by quickly desalting the protein in reaction buffer using a 30 kDa centrifugal filter. Eap34 was added in 5% excess of a 1:1 molar ratio in a 1:10 volume (i.e., 5 μL Eap34 to make 50 μL total volume) and incubated for 180 min at RT. After quenching and addition of Eap34, 7.5 μL aliquots were removed at time points t = 0, 15, 30, 60, 90, 120, 150, and 180 min, added to an equal volume reducing Laemmli sample buffer, and 10 μL of each reaction were separated by 10% SDS‐PAGE. The t = 180 min time point, cross‐linked gel band corresponding to the red box in Figure 3(A) was excised, extracted, digested with 30 ng/μL sequencing grade Trypsin (Promega) and subjected to MALDI‐TOF (Bruker Daltonics Ultraflex III) mass spectrometry analysis. A similar SDS‐PAGE gel was run with 3 μL reaction volume and subjected to Western blot analysis using a specific polyclonal sheep anti‐Eap antibody (gift from Prof. J.‐I. Flock, Karolinska Institute, Sweden), followed by detection with an HRP‐conjugated anti‐sheep secondary antibody. Eap34 gel bands were visualized on a FluorChem M system (Protein Simple) following reaction with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) according to manufacturer's suggestions.
Surface plasmon resonance experiments
C4b‐C345c direct binding and competition studies were done using a Biacore 3000 or T‐200 instrument (GE Healthcare) at 25°C. Experiments were run at a flow rate of 20 μL min−1 in HBS‐T (20 mM HEPES (pH 7.4), 140 mM NaCl, 0.005% (v/v) Tween‐20) supplemented with 5 mM NiCl2. Although nickel is not a physiologically relevant cation, it has been shown to stabilize complement proteins and C3 convertases for in vitro analysis.12, 30, 31 C4b‐C345c direct binding and C4b residual binding experiments were carried out by immobilization of full‐length Eap (4000–7000 RU) and Eap34 (4400–5900 RU) by amine coupling to a CM‐5/CMD‐200M sensor chip (GE Healthcare/Xantec). Dose‐response experiments were performed using an MBP fusion of C4b‐C345c in order to approach saturating concentrations. MBP‐C4‐C345c was injected for 2 min with 1 min dissociation, followed by a 1 min regeneration injection of 2 M NaCl. The average response for the 5–15 s preceding the injection stop was plotted versus concentration of MBP‐C4b‐C345c and fit using a nonlinear regression, log (agonist) vs. response—variable slope curve in GraphPad Prism6.
Using the sensor chip configuration described above, an assay was done to assess competition between C4b‐C345c and C4b for binding to Eap. In short, a flow rate of 10 μL min−1 in the same buffer described above was used for all injections. C4b was injected in triplicate at a concentration of 500 nM to establish a basal level of C4b binding, and this concentration was held constant for the competition experiments. C4b and subsequent samples of C4b plus C4b‐C345c (varying concentrations from 75 nM to 120 μM) were injected for 2 min with 2 min dissociation, and regenerated by two 1 min injections of 2 M NaCl. To calculate residual C4b binding, an injection of C345c alone was subtracted from the [C4b + C345c] injection, normalized to the basal C4b response, and plotted against the concentration of C345c.
Lysine‐acetylation footprinting MALDI‐TOF mass spectrometry of Eap34 and C4b
The general approach was adapted from Chen et al.14 Specifically, a 10 μL solution of Eap34 (7 μM) and complement C4b (10 μM) was incubated for 20 min at RT in HBS (pH 7.4). These concentrations allowed the reaction to be 10‐fold above the K D as well as have excess C4b to eliminate any free‐Eap34. 5 μL of a 1:10 dilution of 2 M N‐acetyl‐hydroxysuccinimide (synthesized as previously described14) in acetonitrile (ACN) was added to the incubated proteins and allowed to react for 20 min at RT. The reaction was stopped by the addition of 10 μL of 1 M NH4HCO3 (pH 8.0) for 20 min at RT. Eap34 and C4b were separated by SDS‐PAGE performed under non‐reducing conditions. The gel was zinc‐imidazole stained (0.3 M ZnCl2, 0.2 M imidazole) and the Eap34 bands were excised and subjected to in‐gel digestion overnight at 37°C by 10 ng/μL sequencing grade Chymotrypsin (Promega). Digested peptides were extracted by subsequent additions of 200 μL 0.1% (v/v) TFA and 100 μL ACN, 0.1% (v/v) TFA and lyophilized. Lyophilized peptides were re‐suspended in 50 μL of 0.1% (v/v) TFA and purified by C4‐ZipTip (EMD Millipore). Eluted peptides were diluted 1:10, mixed 1:1 in matrix buffer (50 mg/mL DHB in 1:1 ACN:0.1% (v/v) TFA), and subjected to MALDI‐TOF (Bruker Daltonics Ultraflex III) mass spectrometry analysis.
Mass spectra were analyzed by mMass Version 5.5.0 (http://www.mmass.org). Spectra were calibrated to known high intensity peptides. Peaks were matched with a tolerance of less than 0.4 Da, with a mass range of 500–3500 Da, and were subjected to envelope fitting to determine the measured versus the computer‐generated model. A mass shift of 42.0106 Da was observed for each acetylated lysine residue. Analysis of the final, calibrated peptide coverage map was done using a scoring system based on percentage of acetylation for each individual lysine residue. As an example, a peptide containing 4 lysines with a peak shift equating to 3 acetylations (126.0318 Da), was scored by the relationship (# of acetyl groups/# of lysine residues in peptide) × 100 = % acetylation per lysine residue or (3/4) × 100 = 75% acetylation/lysine.
Circular dichroism (CD)
Far‐UV CD spectropolarimetry was used to assess the secondary structure content of Eap34 mutants in comparison to the wild‐type protein. Samples were dissolved in 20 mM HEPES (pH 7.4), 140 mM NaCl at a concentration of 1 mg/mL (∼40 μM). A buffer control was also collected. Spectra were collected across a 190–260 nm range, at 50 nm min−1, using 0.5 nm pitch, 1 s response, and a 1 nm bandwidth. All data was collected on a Jasco J‐815 instrument using a cylindrical small volume quartz cuvette (1 mm path length) (Starna Cells, Inc., Atascadero, CA).
Statistical analyses
Analysis was performed using GraphPad Prism 6.0 and mMass Version 5.5.0.
Structural modeling
The ClusPro server32, 33 was used to construct a structural model for the C4b/Eap34 complex. The structure of human C4b (PDB accession code 4XAM23) and a SAXS‐derived model of Eap3415 were used as inputs. Modeling was restrained with experimentally derived parameters that restricted docked complexes to include the α′ and γ‐chains of C4b, and designated Eap34 residues K297, K304, K308, K413, K424, K427, and K430 as participating in intermolecular contacts. The resulting model was compared with that of a C4b2 complex generated by superimposing the individual structures of C4b, C2a (PDB accession code 2I6S34) and C2b (PDB accession code 3ERB35) onto the structure of the AP C3 pro‐convertase, C3bB (PDB accession code 2XWJ24). This procedure takes advantage of the global architectural similarity between the C3 pro‐convertases of the AP and CP/LP.12 All images of protein structures were rendered using PyMol.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors thank Drs. Brandon Garcia, Sofia Mortensen, Gregers Andersen, Daphne Stapels, Suzan Rooijakkers, and Michal Hammel for helpful discussions regarding this project. The authors also acknowledge Dr. Jan‐Ingmar Flock for generously providing anti‐Eap antiserum. Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article. Author contributions: J.L.W. designed and performed experiments, analyzed data, and wrote the manuscript; K.X.R. assisted with experimental design, performance, and data analysis; B.B.K. assisted with experimental design, performance, and data analysis; J.K.W. provided essential reagents for experiments; B.V.G. designed this study, analyzed data, and wrote the manuscript.
STATEMENT: The extracellular adherence protein (Eap) from Staphylococcus aureus binds with nanomolar affinity to complement component C4b, blocks formation of the classical and lectin pathway C3 pro‐convertase, and thereby inhibits complement activity. Here, we employ a series of solution biochemical methods to more completely describe the Eap binding site on C4b, and vice‐versa. This work enhances our understanding of pathogen‐derived complement regulators, and further defines the structure/function relationships that underlie the broad ligand‐binding capacity of Eap.
References
- 1. Ricklin D, Hajishengallis G, Yang K, Lambris JD (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11:785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Laarman A, Milder F, van Strijp J, Rooijakkers S (2010) Complement inhibition by gram‐positive pathogens: molecular mechanisms and therapeutic implications. J Mol Med 88:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thammavongsa V, Kim HK, Missiakas D, Schneewind O (2015) Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13:529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lambris JD, Ricklin D, Geisbrecht BV (2008) Complement evasion by human pathogens. Nat Rev Microbiol 6:132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, van Strijp JA (2005) Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol 6:920–927. [DOI] [PubMed] [Google Scholar]
- 6. Jongerius I, Kohl J, Pandey MK, Ruyken M, van Kessel KP, van Strijp JA, Rooijakkers SH (2007) Staphylococcal complement evasion by various convertase‐blocking molecules. J Exp Med 204:2461–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen H, Ricklin D, Hammel M, Garcia BL, McWhorter WJ, Sfyroera G, Wu YQ, Tzekou A, Li S, Geisbrecht BV, Woods VL, Jr , Lambris JD (2010) Allosteric inhibition of complement function by a staphylococcal immune evasion protein. Proc Natl Acad Sci USA 107:17621–17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woehl JL, Stapels DA, Garcia BL, Ramyar KX, Keightley A, Ruyken M, Syriga M, Sfyroera G, Weber AB, Zolkiewski M, Ricklin D, Lambris JD, Rooijakkers SH, Geisbrecht BV (2014) The extracellular adherence protein from Staphylococcus aureus inhibits the classical and lectin pathways of complement by blocking formation of the C3 proconvertase. J Immunol 193:6161–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hussain M, Becker K, von Eiff C, Peters G, Herrmann M (2001) Analogs of Eap protein are conserved and prevalent in clinical Staphylococcus aureus isolates. Clin Diagn Lab Immunol 8:1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geisbrecht BV, Hamaoka BY, Perman B, Zemla A, Leahy DJ (2005) The crystal structures of EAP domains from Staphylococcus aureus reveal an unexpected homology to bacterial superantigens. J Biol Chem 280:17243–17250. [DOI] [PubMed] [Google Scholar]
- 11. Stapels DA, Ramyar KX, Bischoff M, von Kockritz‐Blickwede M, Milder FJ, Ruyken M, Eisenbeis J, McWhorter WJ, Herrmann M, van Kessel KP, Geisbrecht BV, Rooijakkers SH (2014) Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc Natl Acad Sci USA 111:13187–13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mortensen S, Jensen JK, Andersen GR (2016) Solution structures of complement C2 and its C4 complexes propose pathway specific mechanisms for control and activation of the complement proconvertases. J Biol Chem 291:16494–16507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grabarek Z, Gergely J (1990) Zero‐length crosslinking procedure with the use of active esters. Anal Biochem 185:131–135. [DOI] [PubMed] [Google Scholar]
- 14. Chen H, Schuster MC, Sfyroera G, Geisbrecht BV, Lambris JD (2008) Solution insights into the structure of the Efb/C3 complement inhibitory complex as revealed by lysine acetylation and mass spectrometry. J Am Soc Mass Spectrom 19:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hammel M, Nemecek D, Keightley JA, Thomas GJ, Jr , Geisbrecht BV (2007) The Staphylococcus aureus extracellular adherence protein (Eap) adopts an elongated but structured conformation in solution. Protein Sci 16:2605–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pietrocola G, Rindi S, Rosini R, Buccato S, Speziale P, Margarit I (2016) The group B Streptococcus‐secreted protein CIP interacts with C4, preventing C3b deposition via the lectin and classical complement pathways. J Immunol 196:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGavin MH, Krajewska‐Pietrasik D, Ryden C, Hook M (1993) Identification of a Staphylococcus aureus extracellular matrix‐binding protein with broad specificity. Infect Immun 61:2479–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boden MK, Flock JI (1992) Evidence for three different fibrinogen‐binding proteins with unique properties from Staphylococcus aureus strain Newman. Microb Pathog 12:289–298. [DOI] [PubMed] [Google Scholar]
- 19. Palma M, Haggar A, Flock JI (1999) Adherence of Staphylococcus aureus is enhanced by an endogenous secreted protein with broad binding activity. J Bacteriol 181:2840–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chavakis T, Hussain M, Kanse SM, Peters G, Bretzel RG, Flock JI, Herrmann M, Preissner KT (2002) Staphylococcus aureus extracellular adherence protein serves as anti‐inflammatory factor by inhibiting the recruitment of host leukocytes. Nat Med 8:687–693. [DOI] [PubMed] [Google Scholar]
- 21. Flock M, Flock JI (2001) Rebinding of extracellular adherence protein Eap to Staphylococcus aureus can occur through a surface‐bound neutral phosphatase. J Bacteriol 183:3999–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kreikemeyer B, McDevitt D, Podbielski A (2002) The role of the map protein in Staphylococcus aureus matrix protein and eukaryotic cell adherence. Int J Med Microbiol 292:283–295. [DOI] [PubMed] [Google Scholar]
- 23. Mortensen S, Kidmose RT, Petersen SV, Szilagyi A, Prohaszka Z, Andersen GR (2015) Structural basis for the function of complement component C4 within the classical and lectin pathways of complement. J Immunol 194:5488–5496. [DOI] [PubMed] [Google Scholar]
- 24. Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, Gros P (2010) Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 330:1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia BL, Zwarthoff SA, Rooijakkers SH, Geisbrecht BV (2016) Novel evasion mechanisms of the classical complement pathway. J Immunol 197:2051–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garcia BL, Ramyar KX, Ricklin D, Lambris JD, Geisbrecht BV (2012) Advances in understanding the structure, function, and mechanism of the SCIN and Efb families of Staphylococcal immune evasion proteins. Adv Exp Med Biol 946:113–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geisbrecht BV, Bouyain S, Pop M (2006) An optimized system for expression and purification of secreted bacterial proteins. Protein Expr Purif 46:23–32. [DOI] [PubMed] [Google Scholar]
- 28. Garcia BL, Summers BJ, Lin Z, Ramyar KX, Ricklin D, Kamath DV, Fu ZQ, Lambris JD, Geisbrecht BV (2012) Diversity in the C3b [corrected] contact residues and tertiary structures of the staphylococcal complement inhibitor (SCIN) protein family. J Biol Chem 287:628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roos A, Bouwman LH, Munoz J, Zuiverloon T, Faber‐Krol MC, Fallaux‐van den Houten FC, Klar‐Mohamad N, Hack CE, Tilanus MG, Daha MR (2003) Functional characterization of the lectin pathway of complement in human serum. Mol Immunol 39:655–668. [DOI] [PubMed] [Google Scholar]
- 30. Fishelson Z, Muller‐Eberhard HJ (1982) C3 convertase of human complement: enhanced formation and stability of the enzyme generated with nickel instead of magnesium. J Immunol 129:2603–2607. [PubMed] [Google Scholar]
- 31. Villiers MB, Thielens NM, Colomb MG (1985) Soluble C3 proconvertase and convertase of the classical pathway of human complement. Conditions of stabilization in vitro. Biochem J 226:429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20:45–50. [DOI] [PubMed] [Google Scholar]
- 33. Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: a fully automated algorithm for protein‐protein docking. Nucleic Acids Res 32:W96–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Milder FJ, Raaijmakers HC, Vandeputte MD, Schouten A, Huizinga EG, Romijn RA, Hemrika W, Roos A, Daha MR, Gros P (2006) Structure of complement component C2A: implications for convertase formation and substrate binding. Structure 14:1587–1597. [DOI] [PubMed] [Google Scholar]
- 35. Krishnan V, Xu Y, Macon K, Volanakis JE, Narayana SV (2009) The structure of C2b, a fragment of complement component C2 produced during C3 convertase formation. Acta Cryst D65:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information