

Abstract
Lipopolysaccharide (LPS) and the periplasmic protein, LptA, are two essential components of Gram‐negative bacteria. LPS, also known as endotoxin, is found asymmetrically distributed in the outer leaflet of the outer membrane of Gram‐negative bacteria such as Escherichia coli and plays a role in the organism's natural defense in adverse environmental conditions. LptA is a member of the lipopolysaccharide transport protein (Lpt) family, which also includes LptC, LptDE, and LptBFG2, that functions to transport LPS through the periplasm to the outer leaflet of the outer membrane after MsbA flips LPS across the inner membrane. It is hypothesized that LPS binds to LptA to cross the periplasm and that the acyl chains of LPS bind to the central pocket of LptA. The studies described here are the first to comprehensively characterize and quantitate the binding of LPS by LptA. Using site‐directed spin‐labeling electron paramagnetic resonance (EPR) spectroscopy, data were collected for 15 spin‐labeled residues in and around the proposed LPS binding pocket on LptA to observe the mobility changes caused by the presence of exogenous LPS and identify the binding location of LPS to LptA. The EPR data obtained suggest a 1:1 ratio for the LPS:LptA complex and allow the first calculation of dissociation constants for the LptA–LPS interaction. The results indicate that the entire protein is affected by LPS binding, the N‐terminus unfolds in the presence of LPS, and a mutant LptA protein unable to form oligomers has an altered affinity for LPS.
Keywords: LptA, LPS, lipopolysaccharide, LPS binding protein, EPR spectroscopy, dissociation constants, periplasmic protein
Introduction
Escherichia coli, and many other important pathogens classified as Gram‐negative bacteria, have an inner membrane (IM) and an asymmetric outer membrane (OM) that protect the cytoplasm and act as highly selective permeability barriers. The outer leaflet of the OM mainly comprises lipopolysaccharide [LPS or endotoxin; Fig. 1(B)], which is a large lipid essential for the survival of nearly all Gram‐negative bacteria and a major factor in the protection of bacteria from adverse environmental stresses such as toxins and the hostile environments found during infection or host colonization.
Figure 1.
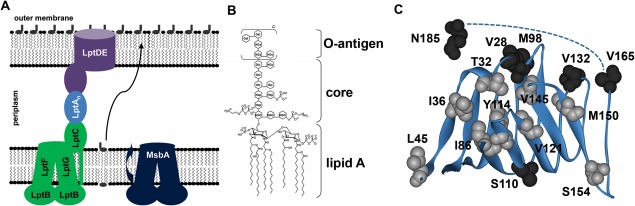
Structures of LptA and LPS and cartoon of the LPS transport proteins. (A) Cartoon showing the eight proteins involved in the transport of LPS (gray lipids) from the IM, across the periplasm, and to the outer leaflet of the OM in Gram‐negative bacteria. Multiple LptA proteins may comprise the bridge. (B) General structure of E. coli O111:B4 LPS. The lipid A base contains 4–7 acyl chains, and the O‐antigen composition varies by strain and repeats up to n = 40.1, 16 (C) LptA as observed in the 2R1A crystal structure13 with studied sites facing the interior (gray) and exterior (black) of the protein highlighted. The C‐terminal tail residues (166–185) are not resolved in the crystal structure and are represented by a dotted line.
The base of LPS (lipid A) is synthesized in the cytoplasm and inserted into the inner leaflet of the IM,1 where the core sugars are added before the ABC transporter MsbA flips the lipid to the outer leaflet of the IM2, 3, 4 and the O‐antigen is attached by WaaL to create LPS.3 The Lpt (LPS transport) system, composed of the IM ATPase cassette LptFGB2, the IM‐associated protein LptC, the periplasmic protein LptA (18.1 kDa), and the OM LptDE complex [Fig. 1(A)], is known to be involved in the transport of LPS across the periplasm and into the outer leaflet of the OM,5, 6, 7, 8, 9 yet it is not known how LPS is transported by these proteins.
It is possible that one or more LptA proteins bridges the periplasm to form a large, connected protein complex; this is supported by our data on the oligomerization of E. coli LptA10 and from the Kahne lab.11, 12 The acyl chains of LPS are hypothesized to bind to the interior of the LptA protein fold to protect the hydrophobic region of the lipid as it crosses the water‐soluble periplasm.8, 13 In vivo cross‐linking studies showing that interior‐facing sites T32, I36, S95, Y114, and L116 substituted with the unnatural amino acid pPBA could cross‐link with LPS in vivo,12 confirming the hypothesis that LPS binds to the interior pocket of LptA. Other sites that bind LPS and the LptA–LPS binding affinity have yet to be determined. Therefore, the location of the LPS binding site(s) and the LptA–LPS dissociation constants are studied here by monitoring the environment of spin labels attached to 15 specific positions on LptA using site‐directed spin‐labeling electron paramagnetic resonance (EPR) spectroscopy. A major strength of the EPR spectroscopy technique is its ability to detect and follow changes in local structure due to intramolecular and intermolecular conformational changes or dynamic interactions with other proteins or substrates based on spin label mobility changes or distance changes between two spin labels.14 The introduced spin label side chain is an excellent reporter of local packing, protein–ligand, and protein–protein interaction sites.
Results
LptA sites affected by LPS binding
To determine the location and effect of LPS binding on LptA, 15 single cysteine mutations were introduced, with nine sites facing the hypothesized LPS binding pocket and six sites facing the exterior surface of LptA [Fig. 1(C)]. Each mutant protein was expressed, purified, spin labeled at the unique cysteine to form the R1 side chain, and tested for changes in the mobility of R1 due to the presence of exogenous LPS.
According to light scattering data, LptA oligomerizes in a concentration‐dependent manner.10 LptA is an average of a trimer in solution at 2 µM, and a considerably higher order oligomerization state (25 mers) is predicted at a protein concentration of 100 µM.10 Continuous wave (CW) X‐band EPR spectra for 2 µM (black) LptA samples in the absence of LPS are shown in Figure 2. The combined use of a high‐Q resonator and custom‐designed AquaStar tubing15 allowed for the collection of EPR spectroscopy samples containing 2 µM purified LptA protein, a low concentration of protein that still yields high‐quality spectral data. In addition, the mobility of each R1 at 100 µM (green) LptA is also presented in Figure 2 to showcase the mobility differences between lower and higher order oligomerization at various locations on LptA, as the shape of the spectral line reflects the mobility of each R1.14 With the exception of N185R1, each site studied across LptA shows a motional change upon change in protein concentration. N185R1, the final amino acid in the LptA protein, is in an unstructured tail region according to the lack of electron density found in the crystal structures.16 The EPR data shown here clearly support that finding and indicate the mobility of this tail region is not affected by oligomerization. Sites located in the very center of the LptA protein show small changes in motion upon higher order oligomerization (light purple, Supporting Information, Fig. S1). The remaining sites show large decreases in mobility upon oligomerization (dark purple, Supporting Information, Fig. S1). The slow motion observed for the N‐terminal and C‐terminal edge sites, therefore, has some contribution from quaternary interactions between LptA protomers within the oligomeric structure. The addition of 30% Ficoll to limit the tumbling of the protein in solution did not affect the motion of R1 (data not shown); therefore, the motional changes observed by EPR spectroscopy are due to local side chain motions and are not due to a change in the overall tumbling rate of the protein. To minimize the effects of oligomerization in the EPR spectra and focus on the changes associated with LPS binding, and enable the largest excess of LPS to be included in the samples, 2 µM LptA was used in the LPS‐binding experiments.
Figure 2.

Overlays of X‐band CW EPR spectra of spin‐labeled apo LptA at the positions indicated at 2 µM (black) and 100 µM (green) protein concentration. Spectra are normalized to the same center line height and are 100 G wide.
Many of the spectra recorded indicate more than one motional component, while the spectra of the R1 side chain at positions I86, Y114, V121, V145, and M150 in 2 µM LptA show large populations of very slow motion in the apo state (Figs. 2 and 3). Side chain motions are indicative of the tertiary and quaternary environment of each position, and these sites show restricted movement of the R1 side chain likely due to tertiary interactions with neighboring side chains, consistent with their location within the interior pocket of LptA. In contrast, R1 at T32, S154, V165, and N185 moves rapidly, indicative of free movement of the side chain. This is especially evident at C‐terminal sites N185 and V165: N185R1 does not show a concentration dependence to its motion (Fig. 2), consistent with its unresolved location within the crystal structure (pdb:2R1A, pdb:2R19, 7) and suggestive of its location away from an oligomeric interface, while V165R1 shows a small degree of concentration‐dependent decrease in motion consistent with its location on an oligomeric interface. The spectra for I36R1, L45R1, S110R1, and V132R1 at 2 µM show intermediate motion. The amount of endogenous LPS present in the purified and spin‐labeled samples was determined to be <0.2 µM (Supporting Information, Fig. S2). Therefore, the spectra shown in black in Figure 3 do, in fact, represent the apo state of the protein.
Figure 3.
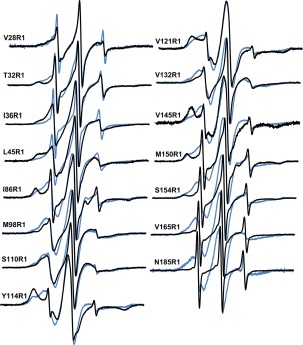
Overlays of X‐band CW EPR spectra of 2 µM LptA in the absence (black) and presence (blue) of 198 µM LPS. Spectra are normalized to the same center line height and are 100 G wide.
To determine the location and effect of LPS binding on LptA, 198 µM LPS was added to each 2 µM singly labeled protein sample. Motional changes were observed at each position studied. For sites V28, T32, I36, L45, I86, V121, V145, and M150, R1 became more mobile upon the addition of LPS (Fig. 3). Based on the crystal structure, these sites are all expected to face the putative LPS‐binding pocket (Supporting Information, Fig. S3); thus, the increased mobility indicates that tertiary interactions are relieved, suggesting that the protein opens upon LPS binding. In addition, it is notable that in the presence of LPS, R1 on N‐terminal sites V28, T32, I36, and L45 each show unrestricted motion indicative of a freely moving domain, suggesting that the N‐terminal helix and edge strand move away from the core of the protein or become unstructured upon LPS binding. Conversely, R1 became more immobilized at positions M98, S110, V132, S154, V165, and N185, with decreases in motion at the externally facing sites and in the C‐terminus of LptA (Fig. 3). No site was unaffected by the addition of LPS (Fig. 3 and Supporting Information, S3).
The spectra for each of these mutants was deconvoluted using spectral subtraction methods to quantify the percent of LptA proteins affected by addition of 198 µM LPS at each site studied. Interestingly, when the percentage of LptA protein affected by LPS is plotted versus residue number, the general trend is toward increased population of LptA affected by LPS binding from the N‐terminus of LptA to the C‐terminus (Supporting Information, Fig. S4).
Seven of the LptA mutations, located at different positions across the protein, were further analyzed at various LPS concentrations to generate LPS binding curves. Figure 4 shows the binding curves resulting from the collection of EPR spectra of spin‐labeled LptA in the presence of varying amounts of LPS and quantifying the percentage of LptA in each sample affected by the addition of LPS. The resulting data were best fit to a single‐site ligand binding function to generate dissociation constants for the LptA–LPS interaction. For sites in the center and C‐terminal regions of LptA, the dissociation constants for LPS binding were clustered in the 7–34 µM range, and the B max values ranged from 69–103% (Table 1). These values suggest a 0.7:1 to 1:1 ratio for the LPS:LptA complex. Surprisingly, the K d values for the N‐terminal edge sites studied are consistently weaker, with L45R1 data showing a K d value >200 µM due to a linear data set unable to be fit by a saturable binding curve and T32R1 and I36R1 data resulting in K d values of 119 µM and 137 µM, respectively.
Figure 4.
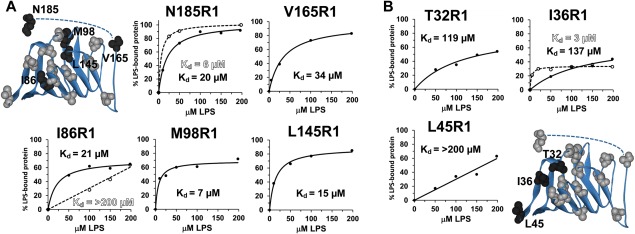
Plots of the data points generated from the deconvolution of the EPR spectra containing varying amounts of LPS and the resulting fits using a single‐site ligand binding model in SigmaPlot (Systat Software, San Jose, CA). Data are plotted for (A) Center and C‐terminal and (B) N‐terminal LptA sites in the WT (solid circles) and Q148A/K149A LptA (open circles) backgrounds. K d values are indicated and listed in Table 1 with standard errors and B max values; L45R1 data do not yield finite values. Protein structures highlight in black the locations of the LptA sites studied for each panel.
Table 1.
LptA–LPS Binding Data As Determined by EPR Spectroscopy
| LptA mutant | K d (µM) | B max (%) |
|---|---|---|
| T32R1 | 119 ± 42 | 86 ± 14 |
| I36R1 | 137 ± 52 | 72 ± 14 |
| I36R1/Q148A/K149A | 3 ± 1 | 34 ± 1 |
| L45R1 | >200 | ND |
| I86R1 | 21 ± 7 | 71 ± 4 |
| I86R1/Q148A/K149A | >200 | ND |
| M98R1 | 7 ± 2 | 69 ± 4 |
| V145R1 | 15 ± 2 | 89 ± 2 |
| V165R1 | 34 ± 5 | 97 ± 4 |
| N185R1 | 20 ± 3 | 103 ± 3 |
| N185R1/Q148A/K149A | 6 ± 0 | 103 ± 1 |
LptA oligomerization and LPS binding
To determine the effect of oligomerization on LPS binding to LptA, three additional LPS binding curves were generated for LptA proteins that are unable to form oligomers (Fig. 4, open circles). Previously, we generated the K148A/Q149A mutant of LptA, which was shown to disrupt LptA oligomerization by EPR spectroscopy.17 SEC‐LS data (Supporting Information, Fig. S5) also show that at 2 µM, the I36C/K148A/Q149A protein is expected to be a monomer. The CW EPR spectrum of I36R1/K148A/Q149A shows no concentration‐dependent decrease in motion.17 Remarkably, I36R1, which is located on the N‐terminal edge strand that unfolds upon binding to LPS, has a dramatically increased affinity for LPS when LptA is unable to oligomerize (Fig. 4). That is, the N‐terminal strand unfolds at a considerably lower LPS concentration as a monomer than as an oligomer. Conversely, I86R1, which faces directly into the interior of the hydrophobic pocket of LptA, has a higher affinity for LPS when LptA no longer is able to form oligomers. That is, the interior of the protein binds LPS at lower LPS concentrations when in an oligomeric form; more LPS molecules are required to induce a conformational change at I86R1 when LptA cannot oligomerize. The C‐terminal residue 185 shows a somewhat increased but similar affinity for LPS independent of the oligomerization state.
Discussion
The entire LptA protein appears to be affected by LPS binding, including the N‐terminal edge residues, sites facing the putative LPS binding pocket, external facing sites, and the very C‐terminal residue; these data suggest a major rearrangement within the protein and significant shifting of tertiary contacts within the protein fold. The mobility of R1 increases upon LPS binding for the sites facing the interior of the protein, which is the putative LPS binding pocket, supporting the hypothesis that the protein opens to bind LPS. Contacts between the acyl chains and R1 are expected to be less restricting than tertiary contacts with amino acid side chains; thus, our data are consistent with the hypothesis that not only does the protein open to relieve tertiary interactions but also the acyl chains of LPS may bind within the hydrophobic interior pocket of LptA and still allow intermediate motion of R1.
All five of the LptA sites shown to cross‐link with LPS in vivo, T32, I36, F95, Y114, and L116,12 were initially included in this study. The expression of LptA F95C and L116C mutant proteins was unsuccessful. CW EPR data for T32R1, I36R1, and Y114R1 support the in vivo findings that these sites are directly affected by LPS binding. Unexpectedly, the N‐terminal edge sites become unfolded in the presence of LPS. In addition, I36R1, which is located on the N‐terminal edge strand, binds LPS with greater affinity when LptA is a monomer. This may be due to the fact that this edge is more accessible in the monomeric form because it is not confined by the oligomeric interface and, therefore, is more readily able to recognize and respond to LPS. Even external facing sites (e.g., 98, 110, 132) show changes in motion upon LPS binding. This is due either to local rearrangements in packing due to the opening of the protein or to the O‐antigen wrapping around the protein, especially given that none of the exterior facing sites in the in vivo study12 was able to cross‐link with LPS. Interestingly, the motion of the very last residue, N185R1, which is located in a highly unstructured region of the protein, becomes restricted upon LPS binding. Our data suggest that the C‐terminal tail may lock down on LPS and, thus, adopt a more localized position than in the apo state. The addition of 30% Ficoll to the protein solution did not affect the motion of R1 (data not shown); thus, the changes observed by EPR spectroscopy are due to local side chain motions and are not due to a shift in the tumbling rate of LptA upon binding of the large LPS moiety.
The LPS‐induced motional changes are distinct from the concentration‐dependent motional changes observed by EPR spectroscopy. Clearly, increased tertiary contacts are created throughout the protein upon oligomerization, based on the large decreases in spectral motion for most sites studied when the concentration is changed from 2 to 100 µM. This increase in end‐to‐end oligomerization size has a great effect across all but the ends and the very center of each protomer. The motion of I36R1, L45R1, M150R1, S154R1, and V165R1, β‐strand edge sites, is expected to be affected by the increase in oligomerization number due to their location on the oligomeric interface. In contrast, the N‐terminal helix, given its position away from the oligomeric interface, and the highly flexible C‐tail are expected to be largely unaffected by oligomerization, as is observed by the EPR spectroscopy data. Remarkably, large motional changes are observed for sites I86R1 and V121R1, which are located near the center of the protein facing inside the pocket, and for sites V145R1 and V132R1, which are located on the tips of the loops between the β‐strand adjacent to the C‐terminal edge strand. These data suggest that there are rearrangements of the protein upon oligomerization beyond the protein–protein contact surfaces. Therefore, the motional changes observed are a result of tertiary rearrangements within the protomer. Only sites located in the very center of the protein are largely unaffected (V28R1, T32R1, M98R1, S110R1, and Y114R1).
The N‐terminal edge strand (I36R1) and the C‐terminal end site (N185R1) show increased affinities for LPS when the protein cannot form oligomers. In contrast, the internal pocket of LptA (I86R1) shows a dramatically lower affinity for LPS in the absence of oligomerization (Fig. 4). These LPS binding data for monomeric LptA indicate that the N‐terminal edge strand and the C‐terminal tail may be involved in the initial LPS recognition step and that oligomerization is required to efficiently move LPS through the protein.
On the basis of K d values obtained (Table 1) and the total percentage of R1‐labeled protein affected by LPS (Supporting Information, Fig. S4), the N‐terminal sites (28, 32, 36, and 45) and Y114R1 are less responsive to LPS binding than other sites studied on the protein; it is possible that the cysteine substitution or the spin label interferes with LPS binding. Given the 3 µM K d value for I36R1/Q148A/K149A and LPS, it is unlikely that the added label is the cause of the weak K d value for the I36R1–LPS interaction. Excluding the N‐terminal sites, data for the LptA‐LPS interaction using EPR spectroscopy results in K d values of 7–35 µM, which are somewhat stronger than the 28–71 µM apparent dissociation constants calculated for the lipo‐oligosaccharide (LPS without the O‐antigen)–LptC interaction using fluorescence techniques.18 It has been hypothesized that LptA and LptC have different affinities for LPS to enable the transfer of the lipid from LptC to LptA; it is unclear if the observed affinity differential is sufficient to drive LPS transfer from LptC to LptA. The B max values for all sites studied in the oligomer were near 100%, suggesting a 1:1 ratio for the LPS:LptA complex. This is consistent with the expected 25 Å width of the acyl chains of one LPS molecule fitting within the approximately 33 Å wide putative binding pocket of LptA. Interestingly, the N‐terminal strand unfolds upon recognition and binding of LPS, so this may result in LPS slipping into a binding pocket formed by the remaining folded strands of approximately 28 Å wide, nearly perfectly suited to fit the LPS acyl chains.
Materials and Methods
Protein expression and purification
Single cysteine mutations were introduced into the WT LptA protein using QuikChange mutagenesis (Stratagene, La Jolla, CA) and verified by sequencing (Retrogen, San Diego, CA) as described.10, 17 LptA was expressed in E. coli BL21(DE3)pLysS cells from a pET21b (Novagen, EMD Millipore, Billerica, MA) vector with a C‐terminal 6×His tag and purified using cobalt affinity chromatography (Clontech, Mountain View, CA), as described previously.10, 17 Purified protein was spin labeled at the unique cysteine using a 10‐fold excess of the sulfhydryl‐specific spin probe, 2,2,5,5‐tetramethylpyrroline‐3‐yl‐methanethiosulfonate spin label (Toronto Research Chemicals, New York, ON), to generate the R1 side chain and then dialyzed to remove excess label and imidazole. Protein was concentrated using Microcon YM‐10 centrifugal concentrators. Protein concentration was determined using the Thermo Scientific Pierce BCA Protein Assay Kit (Rockford, IL) using lysozyme (14 kDa) as the protein standard.
Detection of endogenous LPS
The Thermo Scientific Pierce LAL Chromogenic Endotoxin Quantitation Kit was utilized to quantitate the amount of LPS retained in purified LptA. The LptA WT, I36C, and V165C‐purified proteins were tested following the manual instructions at 4 and 20 pM concentrations with the addition of 1 pM LPS to WT LptA serving as a positive control (Supporting Information, Fig. S2). Briefly, a standard curve was created ranging between 0 and 1.0 EU/mL with 0.5 EU equaling 1 pM LPS. Reagents were prepared according to the manufacturer's instructions. The assay was performed in a 96‐well plate maintained at 37°C with a heat block. After the assay was completed, the absorbance was measured at 405 nm on a Thermo Fisher Scientific Varioskan Flash microplate reader, the standard curve was calculated, and LPS concentration of each sample was determined.
CW EPR spectroscopy
CW EPR spectroscopy was carried out at room temperature on a Bruker (Bruker Biospin, Billerica, MA) E500 using a Bruker ER4122 SHQE‐W1 cavity, a 42 s scan time with 100–400 averages, and a 1.5 G modulation amplitude under nonsaturating power. Spin‐labeled protein samples were contained in custom extruded AquaStar tubing (2 µM).15 LPS from Escherichia coli O111:B4 (List Biological Laboratories, Inc, Campbell, CA) was resuspended in 50 mM NaPO4, 300 mM NaCl, pH 7 buffer at a concentration of 200 µM, the highest concentration attainable given its solubility limit. The percentage of protein affected by LPS binding in each sample was determined using spectral subtraction methods. The corresponding apo spectrum was manually subtracted from each composite spectrum. The double integration values of the resulting spectra (i.e., the number of spins affected by the presence of LPS) were compared to the double integration value of each composite spectrum (i.e., the number of total spins in the sample) to obtain the percentage of proteins affected by LPS binding.
Supplementary Material
Illustrative figures and supporting data can be found in the Supplementary Materials file (LPS binding to LptA_Supplemental Material.pdf).
Supporting information
Supporting Information
Acknowledgments
The authors thank Jackie Merten, Adriana Newson, and Matthew Fischer for laboratory assistance; Jason Sidabras for helpful discussions; and Jimmy Feix for critically reading the manuscript.
References
- 1. Raetz CR (1990) Biochemistry of endotoxins. Annu Rev Biochem 59:129–170. [DOI] [PubMed] [Google Scholar]
- 2. Karow M, Georgopoulos C (1993) The essential Escherichia coli msbA gene, a multicopy suppressor of null mutations in the htrB gene, is related to the universally conserved family of ATP‐dependent translocators. Mol Microbiol 7:69–79. [DOI] [PubMed] [Google Scholar]
- 3. Raetz CR, Whitfield C (2002) Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CR (1998) Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J Biol Chem 273:12466–12475. [DOI] [PubMed] [Google Scholar]
- 5. Chng SS, Gronenberg LS, Kahne D (2010) Proteins required for lipopolysaccharide assembly in Escherichia coli form a transenvelope complex. Biochemistry 49:4565–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruiz N, Kahne D, Silhavy TJ (2009) Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat Rev Microbiol 7:677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sperandeo P, Lau FK, Carpentieri A, De Castro C, Molinaro A, Deho G, Silhavy TJ, Polissi A (2008) Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli . J Bacteriol 190:4460–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tran AX, Trent MS, Whitfield C (2008) The LptA protein of Escherichia coli is a periplasmic lipid A‐binding protein involved in the lipopolysaccharide export pathway. J Biol Chem 283:20342–20349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Villa R, Martorana AM, Okuda S, Gourlay LJ, Nardini M, Sperandeo P, Deho G, Bolognesi M, Kahne D, Polissi A (2013) The Escherichia coli Lpt transenvelope protein complex for lipopolysaccharide export is assembled via conserved structurally homologous domains. J Bacteriol 195:1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Merten JA, Schultz KM, Klug CS (2012) Concentration‐dependent oligomerization and oligomeric arrangement of LptA. Protein Sci 21:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freinkman E, Okuda S, Ruiz N, Kahne D (2012) Regulated assembly of the transenvelope protein complex required for lipopolysaccharide export. Biochemistry 51:4800–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okuda S, Freinkman E, Kahne D (2012) Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli . Science 338:1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suits MD, Sperandeo P, Deho G, Polissi A, Jia Z (2008) Novel structure of the conserved gram‐negative lipopolysaccharide transport protein A and mutagenesis analysis. J Mol Biol 380:476–488. [DOI] [PubMed] [Google Scholar]
- 14. Klug CS, Feix JB, Methods and applications of site‐directed spin labeling EPR spectroscopy In: Correia JJ, Detrich HW, Eds. (2008) Biophysical Tools for Biologists, Volume One: In Vitro Techniques. Oxford, UK: Academic Press, pp 617–658. [DOI] [PubMed] [Google Scholar]
- 15. Sidabras JW, Mett RR, Hyde JS (2017) Extruded dielectric sample tubes of complex cross section for EPR signal enhancement of aqueous samples. J Magn Reson 277:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Magalhaes PO, Lopes AM, Mazzola PG, Rangel‐Yagui C, Penna TC, Pessoa A Jr. (2007) Methods of endotoxin removal from biological preparations: a review. J Pharm Pharm Sci 10:388–404. [PubMed] [Google Scholar]
- 17. Schultz KM, Feix JB, Klug CS (2013) Disruption of LptA oligomerization and affinity of the LptA‐LptC interaction. Protein Sci 22:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sestito SE, Sperandeo P, Santambrogio C, Ciaramelli C, Calabrese V, Rovati GE, Zambelloni L, Grandori R, Polissi A, Peri F (2014) Functional characterization of E. coli LptC: interaction with LPS and a synthetic ligand. ChemBioChem 15:734–742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information