

Abstract
Pyruvate phosphate dikinase (PPDK) is an essential enzyme of both the C4 photosynthetic pathway and cellular energy metabolism of some bacteria and unicellular protists. In C4 plants, it catalyzes the ATP‐ and Pi‐dependent formation of phosphoenolpyruvate (PEP) while in bacteria and protozoa the ATP‐forming direction is used. PPDK is composed out of three distinct domains and exhibits one of the largest single domain movements known today during its catalytic cycle. However, little information about potential intermediate steps of this movement was available. A recent study resolved a discrete intermediate step of PPDK's swiveling movement, shedding light on the details of this intriguing mechanism. Here we present an additional structural intermediate that possibly represents another crucial step in the catalytic cycle of PPDK, providing means to get a more detailed understanding of PPDK's mode of function.
Keywords: pyruvate phosphate dikinase, C4 photosynthesis, swiveling domain mechanism, catalytic intermediate
Short abstract
Interactive Figure 1; Interactive Figure 2 | PDB Code(s): 5LU4
Introduction
Pyruvate phosphate dikinase (PPDK) is a highly versatile enzyme catalyzing the interconversion between phosphoenolpyruvate (PEP) and pyruvate in bacteria, plants, and unicellular parasitic protists such as Giardia lamblia, Trichomonas vaginalis, or Entamoeba histolytica. While serving as a glycolytic enzyme in protists enhancing the energy efficiency in these organisms at energy‐limiting conditions,1 PPDK works in the opposite direction in chloroplasts of C4 plants where it catalyzes the ATP‐driven regeneration of the primary carboxylation substrate of the C4 pathway PEP.2 The active biological assembly in bacteria consists of a homodimer, while plant PPDK can form homotetramers as functional complex. Each PPDK monomer consists of three distinct structural and functional domains [Fig. 1(A)]: An N‐terminal nucleotide binding domain (NBD), a central domain (CD) housing the catalytic histidine residue that shuttles the phosphoryl group from the high‐energy phosphate substrates and a C‐terminal PEP/pyruvate binding domain (PBD). The substrate binding sites of the NBD and PBD are spaced 45 Å apart. Hence, a so‐called swiveling‐domain mechanism was proposed based on crystal structures from Clostridium symbiosum (Herzberg et al., 1996), Zea mays 3 and Trypanosoma brucei 4 to explain the transport of the phosphoryl group from the NBD to the PBD with the CD undergoing a large rotational (∼110°) and translational (∼40 Å) movement. Recent findings based on structures from the C4 and C3 plants Flaveria trinervia and Flaveria pringlei suggest that the swiveling motion proceeds with at least one discrete substep and might employ an alternate‐binding change mechanism.5 Here we present a novel crystal structure (PDB code 5LU4) of the C4‐PPDK from F. trinervia that has been crystallized in the presence of the glycolytic substrate pyruvate and the nucleotide inhibitor ADP. The structure represents a yet unknown potential conformational intermediate of the CD and further elucidates the sequential path of the swiveling motion in the PPDK catalytic cycle.
Figure 1.
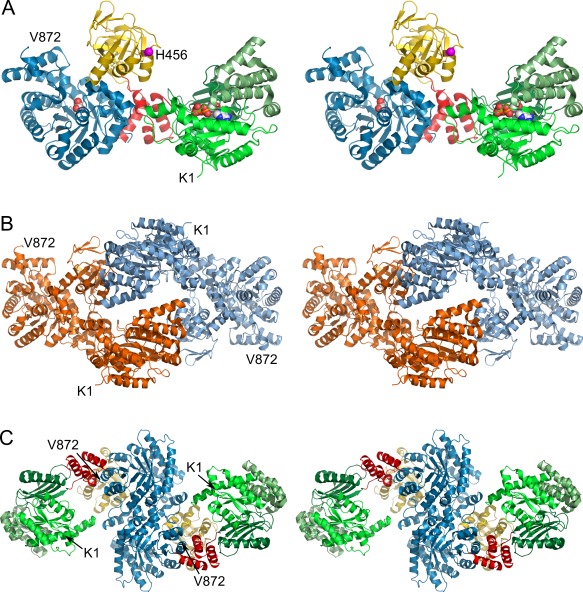
(A) Stereo cartoon representation of 5LU4 chain A illustrating the overall domain organization. The nucleotide binding domain (NBD, aa 1–340) and its three subdomains are colored in different greens. The PEP/pyruvate binding domain (PBD) is colored in blue (aa 534–872). The central domain (CD, yellow, aa 381–516) with the catalytic His456 (magenta, shown as sphere) is attached to both substrate binding domains via two short linker helices (red, aa 341–380 and 517–533). Pyruvate and ADP bound to the PBD and NBD respectively are depicted as spheres. (B) Dimeric assembly within the asymmetric unit (ASU). The dimer is formed by contacts between the NBDs and CDs of chains A and B, colored orange and blue respectively. (C) Biological assembly as identified by the program EPPIC 11 and reconstructed from crystal symmetry. The dimerization interface is formed by both PBDs as previously described 12. Individual domains are colored according to (A). An interactive view is available in the electronic version of the article
Results and Discussion
The pyruvate/ADP complex structure of FtPPDK (PDB code 5LU4) was solved using molecular replacement (MR) and was refined to a resolution of 2.90 Å with R/ values of 24.7%/28.6% (Table 1) with an estimated coordinate error of 0.52 Å. Coordinates of FtPPDK structure 5JVL chain D were used as a search model for MR. Crystals of 5LU4 belong to space group with the asymmetric unit (ASU) consisting of two FtPPDK monomers [Fig. 1(B)]. The inhibitory ADP molecule is bound to the NBD in the same manner as the ATP analogue 2′‐Br‐AppNHp in FtPPDK structures 5JVL and 5JVN [Fig. 2(C)] provoking a closed state of the NBD.5 Similar ADP‐bound conformations have also been observed for other proteins containing an ATP grasp fold such as the human citrate lyase (PDB code 3PFF) or the bacterial glycinamide ribonucleotide synthetase (PDB code 2XD4). In 5LU4, hydrogen bonds are formed between residues Lys25, Gln336, and the β‐phosphate of ADP as well as Thr108, Arg95, Lys25, and the α‐phosphate of ADP. The adenine ring is bound by hydrogen bonds formed by side chains of residues Ser93, Ser242, and the backbone of Val244. Glu324 is forming a hydrogen bond to the 2′‐OH group of the ribose moiety [Fig. 2(C)]. Compared to the previously resolved intermediate position of the CD in 5JVN and its NBD‐facing conformation in PPDK structures 2X0S (T. brucei) or 1KBL (C. symbiosum),6 the position of the CD in the newly resolved structure suggests domain swiveling along disparate axes to properly align the catalytic His456 for phosphoryl group transfer from the ATP substrate in the NBD to the pyruvate bound in the PBD. To this end, 5LU4 seems to reflect a potential consecutive conformational state following the CD intermediate resolved in 5JVN. Rotation of the CD towards the NBD along an axis defined by the linker helices connecting CD, NBD, and PBD in 5LU4 is similar to the previously resolved motion in the 5JVN intermediate. However, on top of this the CD is rotated by ∼104° around a second axis running almost perpendicular to the initial axis in the linker helices [Fig. 2(B)] in 5LU4 to further complete the catalytic cycle.
Table 1.
Data collection and refinement statistics
| 5LU4 | ||
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.9686 | |
| Space group |
|
|
| Cell dimensions | ||
| a, b, c (Å) | 74.16 126.52 219.00 | |
| α, β, γ (°) | 90, 90, 90 | |
| Resolution (Å) | 219.00–2.90 (3.00–2.90) | |
|
|
0.047 (0.499) | |
|
|
0.053 (0.564) | |
|
|
0.024 (0.242) | |
|
|
20.1 (3.19) | |
| Completeness (%) | 99.8 (99.7) | |
| Multiplicity | 4.9 (5.2) | |
| Wilson B (Å2) | 83.0 | |
| Model and refinement | ||
| Resolution (Å) | 109.79–2.90 (2.98–2.90) | |
| Reflections (unique/test) | 46418/2229 | |
| / (%) | 24.7/28.6 | |
| No. of atoms | ||
| Protein | 11954 | |
| Ligand/ion | 64 | |
| Water | 2 | |
| B‐factors (Å2) | ||
| Protein | 99.25 | |
| Ligands | 64.61 | |
| Water | 66.81 | |
| RMSD | ||
| Bonds lengths (Å) | 0.017 | |
| Bond angles (°) | 1.52 | |
| Ramachandran analysis | ||
| Favored regions (%) | 97.34 | |
| Allowed regions (%) | 2.43 | |
| Outliers (%) | 0.24 |
Highest resolution shell is shown in parentheses.
Figure 2.
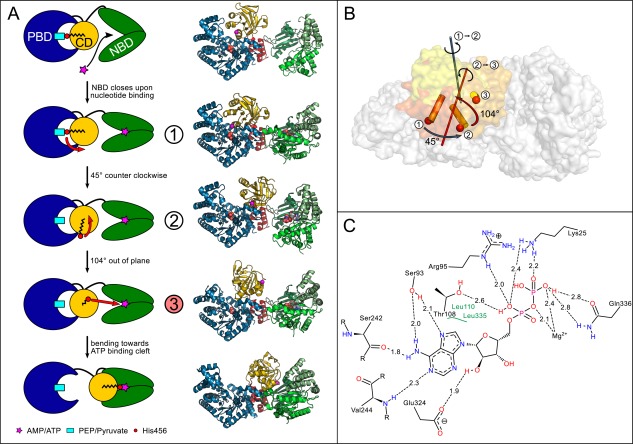
(A) Schematic model of the CD movement in the catalytic cycle taking into account currently known conformational intermediates. Helix 20 containing the catalytic His456 (red circle) at its N‐terminal end is drawn as black zig‐zag structure. The different CD conformations are numbered according to (B) with the newly solved intermediate structure highlighted in red. Corresponding crystal structures are shown on the right from top to bottom: 5JVJ chain A, 5JVL chain D, 5JVN, 5LU4 chain A 6, 1KBL 7. (B) Illustration of PPDK‐CD structural intermediates. CDs of published structures 5JVL chain C (1, dark‐orange), 5JVN (2, orange), and of newly resolved structure 5LU4 (3, yellow) as well as the corresponding rotational axes for transformation between the different intermediate states are shown. (C) Schematic representation of the nucleotide binding site in 5LU4 chain A containing tightly bound ADP. The distances between the bound nucleotide and interacting amino acids of the binding pocket are given in Å. An interactive view is available in the electronic version of the article
As for PPDK structures 5JVL and 5JVJ the ASU of 5LU4 contains two monomers. However, in contrast to the monomer arrangement in the ASU of previously published PPDK structures, where the functional homodimer is formed via their PBDs, monomers in the ASU of the swiveling domain intermediate 5LU4 interact directly via their NBDs or their CDs contacting the NBD in the other monomer, respectively [Fig. 1(B)]. Naturally, the biological assembly previously described for bacterial, plant, and protist PPDKs consisting of a PBD‐mediated dimer can be constructed from the crystallographic symmetry for the 5LU4 assembly, too [Fig. 1(C)]. Yet, the spatial arrangement of closely interacting NBDs in the ASU of 5LU4 preventing the CD from adopting its terminal NBD‐facing conformation was certainly a crucial factor to resolve the new potential swiveling intermediate. In addition to steric restrictions from the NBDs, the 5LU4 intermediate conformation is stabilized by a small number of polar interactions between the CD of monomer B and the NBD of monomer A in the ASU. The side chain of Glu290/A interacts with the backbone of residues Met453/B and Thr454/B. Furthermore, Asn250/A forms a hydrogen bond with Glu431/B. However, taking into account the overall dimensions and the high intrinsic flexibility of the protein as reflected by high B‐factors and the large domain movements in the catalytic cycle in CD and NBD, it is unlikely that these few interactions on their own have driven the CD in the intermediate conformation resolved in 5LU4. Therefore, the main reason for isolating this potential conformational intermediate of the CD swiveling motion in the crystal is probably related to the steric hindrance of the terminal CD transition path towards the NBD in the 5LU4 dimeric assembly. However, such a steric isolation of the swiveling intermediate does not preclude that the observed structural snapshot in fact represents a physiologically relevant though short‐lived conformational intermediate.
Similar restriction of the CD movement by symmetry‐related molecules has been observed in the PBD‐facing structure 2R82 of mutant C. symbiosum PPDK.7 Nonetheless, 2R82 clearly constitutes a plausible and well‐approved conformational state in the PPDK's swiveling domain mechanism. The potential physiological relevance of our trapped 5LU4 intermediate is further supported by the free energy landscape profile computed for nonphosphorylated FtPPDK.5 The transition path connecting the two extreme conformations of the CD in the swiveling motion, which are located themselves in local minima of the free energy landscape, follows a low energy valley. The previously resolved CD intermediate 5JVN is located near a shallow energy minimum along this path, while both, the extreme conformation of the CD resolved in PPDK of T. brucei (PDB code 2X0S)4 and the trapped 5LU4 intermediate of F. trinervia PPDK, are located on the edge of the proposed transition path at similar free energies (∼12 kcal mol−1) making both of them energetically feasible intermediates.
The combination of currently known crystal structures, including the conformational intermediate described here, allows to sketch a plausible path of the conformational shifts in the CD taking place in the proposed PPDK swiveling mechanism [Fig. 2(A)]: First, the CD is positioned with its catalytic His456 mediating phosphoryl group transfer between NBD and PDB in close proximity to the pyruvate binding site. At this initial state, the NBD is empty and in an open conformation (PDB code 5JVJ chain A). Subsequent nucleotide binding then triggers movement and closure of the NBD [PDB code 5JVL; Fig. 2(A), state 1]. In consequence, the CD is rotated towards the NBD by ∼45° around an axis located at the center of the CD running parallel to the linker helices that connect CD to NBD and PBD. The related swiveling motion places the CD in an intermediate conformation between both substrate binding domains as observed in 5JVN [Fig. 2(A), state 2]. To further align the catalytic His456 with the bound nucleotide substrate in the nucleotide binding cleft of the NBD, the CD then is rotated by ∼104° along an axis that is oriented perpendicular to the previous swiveling movement [Fig. 2(A), state 3]. Eventually the CD is tilted towards the nucleotide binding site, resulting in the NBD‐facing conformation, known from the 2X0S and 1KBL crystal structures of T. brucei and C. symbiosum.
Similar discrete substeps of rotary domain movements deduced form the ensemble of different PPDK conformational states have been described for other proteins exhibiting large rotational domain movements such as the F1‐ATPase,8 the bacterial flagellar motor,9 or the E. coli 5′‐nucleotidase.10 In these systems, the discovery of discrete substeps of a rotational movement — initially thought to be of rather continuous nature — has eventually led to deeper understanding of the underlying catalytic mechanism and a strong correlation between structure and molecular function. In the past, the CD swiveling motion of PPDK was well recognized as one of the largest single domain movements observed in enzyme catalysis, but has always been described as a smooth transition between two extreme conformational states.7, 12 Here we demonstrate, that this event involves two substeps at least. Noteworthy, a similar two‐stepped swiveling mechanism has been proposed for Enzyme I (EI) of the bacterial phosphoenolpyruvate:sugar phosphotransferase system (PTS). The PBD and CD of EI are structurally and functionally similar to their counterparts in PPDK and likewise catalyze a phosphoryl group transfer between distant substrate binding sites. Despite these similarities, the interdomain linker in EI adopts a different conformation as in PPDK. Remarkably, structural data on EI suggest a swiveling mechanism, where the CD is detached from the PBD by swiveling around an α‐helical linker as a first step, followed by the alignment of the catalytic histidine with its substrate histidine carrier proteine (HPr), implemented by a motion around a second linker segment.13 This mechanism is highly similar to the two‐stepped swiveling mechanism outlined in our study for PPDK as both proceed via a second step that is likely involved in the correct alignment of the catalytic histidine residue and the phosphoryl‐accepting substrate.
In summary, recent advances in our knowledge on discrete conformational intermediates of the CD swiveling motion found in crystal structures of PPDK from the C4 plant F. trinervia, now enable us to get a more detailed view on the conformational transitions of the protein in the catalytic cycle breaking with the paradigm that the proposed swiveling motion of the CD takes place as smooth transition between only two extreme conformations. But, on the contrary, the newly resolved intermediate conformations and related substeps provide a more detailed mechanistic understanding of a key enzyme of cellular energy metabolism in bacteria, protists, and plants.
Material and Methods
Expression and purification of recombinant FtPPDK
Codon‐optimized coding regions of PPDK from Flaveria trinervia stripped of the chloroplast transport sequence were cloned into the multiple cloning site of a pET‐16b vector (Novagen) containing a histidine10 tag and coding for a Tobacco Etch Virus (TEV) protease cleavage site. E. coli BL21 (DE3) cells (Agilent Technologies) transformed with this plasmid were grown in 2YT medium (5 g L−1 NaCl, 10 g L−1 yeast extract, 16 g L−1 peptone) at 30°C to an OD600 of 0.8. Protein expression was induced by the addition of 0.1 mM isopropyl‐β‐D‐thiogalactopyranoside (IPTG). Cells were harvested 18 h after induction by centrifugation. Harvested cells were suspended in lysis buffer (50 mM Tris/HCl pH 7.5, 300 mM NaCl, 10 mM imidazole, 10 mM MgSO4, 10% (w/v) glycerol, 5 mM DTT, 0.002% phenylmethanesulfonylfluoride) and disrupted using a cell disruptor (Constant Systems). PPDK was purified from the lysate using a nickel affinity chromatography column (GE Healthcare). Purification buffer (50 mM Tris/HCl pH 7.5, 300 mM NaCl, 10 mM MgSO4, 10% (w/v) glycerol, 5 mM DTT) and elution buffer (50 mM Tris/HCl pH 7.5, 300 mM NaCl, 500 mM imidazole, 10 mM MgSO4, 10% (w/v) glycerol, 5 mM DTT) were applied for purification. The loaded PPDK was washed with 50, 150, and 200 mM imidazol, before elution with 500 mM imidazole. Protein containing fractions were pooled and concentrated by ultrafiltration (30 kDa cutoff, Millipore). The buffer was exchanged to purification buffer using a PD‐10 column (GE Healthcare) before cleavage of the histidine tag by TEV protease was initiated at room temperature over night. Cleaved PPDK was separated from the affinity tag by reverse IMAC, the flow‐through was pooled, concentrated by ultrafiltration and the buffer changed for crystallization buffer (10 mM Tris/HCl pH 7.5, 5 mM MgSO4).
Crystallization
Initial crystals of the FtPPDK‐pyruvate‐ADP complex were obtained using the sitting drop vapor diffusion method. FtPPDK in crystallization buffer at a concentration of 10 mg mL−1 was incubated at room temperature for 20 min with 5 mM MgSO4, 5 mM pyruvate, and 5 mM ADP. The protein solution was mixed with precipitant at a 1:1 ratio resulting in a final volume of 200 nL and was equilibrated against 50 µL reservoir solution at 21°C. For crystal optimization the drop size was increased to 2 µL. The optimized precipitant contained 0.3 M MgCl2, 0.1 M MES pH 6.5, and 10% (w/v) PEG 4000. Crystals grew within two days to a size of 300 × 20 × 10 µm3. For cryoprotection ethylene glycol was added to the drop (final concentration 15%) before the crystals were flash‐frozen in liquid nitrogen.
X‐ray data collection and processing
X‐ray diffraction data were obtained at EMBL/DESY (Hamburg, Germany) beamline P13 using a wavelength of 0.9686 Å. The data set spans a total range of 270° with an oscillation range of 0.1° per image. The data set was processed with XDS,14 initial phases were determined by MR with Phaser15 using the coordinates of 5JVL chain D as starting model. The resulting structure was rebuilt using Bucaneer16 from the CCP417 suite, followed by several rounds of manual and iterative rebuilding using Coot18 and refinement with phenix.refine.19 Ligands were not modeled until near‐final refinement stages to reduce model bias. The atomic displacement parameters were refined individually and were partly described as groups of translation, libration, and screw‐motion (TLS).20 Feature‐enhanced maps (FEM) were used to enhance sensitivity for weak side chains.21 The structure was validated using tools provided by Coot and PHENIX, particularly MolProbity.22 Figures were generated using PyMOL23 and PoseView.24
Protein data bank accession code
Structure factors and coordinates of the pyruvate/ADP complex of FtPPDK have been deposited in the Protein Data Bank in Europe (PDBe) with the accession code 5LU4..
Acknowledgments
We thank the staff at EMBL/DESY (Hamburg) for their kind support, especially Isabel Bento for her support at beamline P13 during data collection. No conflict of interest declared. This work has been supported by Heinrich Heine University Düsseldorf (scholarships within the ‐Graduate Cluster for A.M).
References
- 1. Mertens E (1993) ATP versus pyrophosphate: glycolysis revisited in parasitic protists. Parasitol Today 9:122–126. doi: 10.1016/0169-4758(93)90169-g. [DOI] [PubMed] [Google Scholar]
- 2. Chastain CJ, Chollet R (2003) Regulation of pyruvate, orthophosphate dikinase by ADP‐/pi‐dependent reversible phosphorylation in C3 and C4 plants. Plant Physiol Biochem 41:523–532. doi: 10.1016/s0981-9428(03)00065-2. [DOI] [Google Scholar]
- 3. Nakanishi T, Nakatsu T, Matsuoka M, Sakata K, Kato H (2005) Crystal structures of pyruvate phosphate dikinase from maize revealed an alternative conformation in the swiveling‐domain motion. Biochemistry 44:1136–1144. doi: 10.1021/bi0484522. [DOI] [PubMed] [Google Scholar]
- 4. Cosenza LW, Bringaud F, Baltz T, Vellieux FM The 3.0 Å resolution crystal structure of glycosomal pyruvate phosphate dikinase from Trypanosoma brucei . J Mol Biol 318:1417–1432. doi: 10.1016/s0022-2836(02)00113-4. [DOI] [PubMed] [Google Scholar]
- 5. Minges A, Ciupka D, Winkler C, Höppner A, Gohlke H, Groth G (2017) Structural intermediates and directionality of the swiveling motion of pyruvate phosphate dikinase. Sci Rep 7:45389. doi: 10.1038/srep45389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herzberg O, Chen CCH, Liu S, Tempczyk A, Howard A, Wei M, Ye D, Dunaway‐Mariano D (2002) Pyruvate site of pyruvate phosphate dikinase: crystal structure of the enzyme‐phosphonopyruvate complex, and mutant analysis. Biochemistry 41:780–787. doi: 10.1021/bi011799+. [DOI] [PubMed] [Google Scholar]
- 7. Lim K, Read RJ, Chen CCH, Tempczyk A, Wei M, Ye D, Wu C, Dunaway‐Mariano D, Herzberg O (2007) Swiveling domain mechanism in pyruvate phosphate dikinase. Biochemistry 46:14845–14853. doi: 10.1021/bi701848w. [DOI] [PubMed] [Google Scholar]
- 8. Yasuda R, Noji H, Yoshida M, Kinosita K, Itoh H (2001) Resolution of distinct rotational sub‐steps by submillisecond kinetic analysis of F1‐ATPase. Nature 410:898–904. doi: 10.1038/35073513. [DOI] [PubMed] [Google Scholar]
- 9. Sowa Y, Rowe AD, Leake MC, Yakushi T, Homma M, Ishijima A, Berry RM (2005) Direct observation of steps in rotation of the bacterial flagellar motor. Nature 437:916–919. doi: 10.1038/nature04003. [DOI] [PubMed] [Google Scholar]
- 10. Schultz‐Heienbrok R, Maier T, Sträter N (2004) Trapping a 96° domain rotation in two distinct conformations by engineered disulfide bridges. Protein Sci 13:1811–1822. doi: 10.1110/ps.04629604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duarte JM, Srebniak A, Schärer MA, Capitani G (2012) Protein interface classification by evolutionary analysis. BMC Bioinformatics 13:334. doi: 10.1186/1471-2105-13-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herzberg O, Chen CC, Kapadia G, McGuire M, Carroll LJ, Noh SJ, Dunaway‐Mariano D (1996) Swiveling‐domain mechanism for enzymatic phosphotransfer between remote reaction sites. Proc Natl Acad Sci USA 93:2652–2657. doi: 10.1073/pnas.93.7.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Teplyakov A, Lim K, Zhu P, Kapadia G, Chen CCH, Schwartz J, Howard A, Reddy PT, Peterkofsky A, Herzberg O (2006) Structure of phosphorylated enzyme I, the phosphoenolpyruvate: sugar phosphotransferase system sugar translocation signal protein. Proc Natl Acad Sci USA 44:16218–16223. doi: 10.1073/pnas.0607587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kabsch W (2010) XDS. Acta Crystallogr D 66:125–132. doi: 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cowtan K (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D 62:1002–1011. doi: 10.1107/s0907444906022116. [DOI] [PubMed] [Google Scholar]
- 17. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50:760–763. doi: 10.1107/s0907444994003112. [DOI] [PubMed] [Google Scholar]
- 18. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D 60:2126–2132. doi: 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive python‐based system for macromolecular structure solution. Acta Crystallogr D 66:213–221. doi: 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Howlin B, Butler SA, Moss DS, Harris GW, Driessen HPC (1993) TLSANL: TLS parameter‐analysis program for segmented anisotropic refinement of macromolecular structures. J Appl Crystallogr 26:622–624. doi: 10.1107/s0021889893002729. [DOI] [Google Scholar]
- 21. Afonine P (2014) FEM: Feature enhanced map. Acta Crystallogr A 70:C318–C318. doi: 10.1107/s2053273314096818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2012) MolProbity: all‐atom structure validation for macromolecular crystallography. In: International Tables for Crystallography, pp. 694–701, International Union of Crystallography (IUCr), doi: 10.1107/97809553602060000884. [DOI] [PMC free article] [PubMed]
- 23.Schrödinger LLC, The PyMOL molecular graphics system, version 1.8.
- 24. Stierand K, Rarey M (2010) Drawing the PDB: Protein‐ligand complexes in two dimensions. ACS Med Chem Lett 1:540–545. doi: 10.1021/ml100164p. [DOI] [PMC free article] [PubMed] [Google Scholar]