

Abstract
A subset of B‐cell lymphoma patients have dominant mutations in the histone H3 lysine 27 (H3K27) methyltransferase EZH2, which change it from a monomethylase to a trimethylase. These mutations occur in aromatic resides surrounding the active site and increase growth and alter transcription. We study the N‐terminal trimethylase NRMT1 and the N‐terminal monomethylase NRMT2. They are 50% identical, but differ in key aromatic residues in their active site. Given how these residues affect EZH2 activity, we tested whether they are responsible for the distinct catalytic activities of NRMT1/2. Additionally, NRMT1 acts as a tumor suppressor in breast cancer cells. Its loss promotes oncogenic phenotypes but sensitizes cells to DNA damage. Mutations of NRMT1 naturally occur in human cancers, and we tested a select group for altered activity. While directed mutation of the aromatic residues had minimal catalytic effect, NRMT1 mutants N209I (endometrial cancer) and P211S (lung cancer) displayed decreased trimethylase and increased monomethylase/dimethylase activity. Both mutations are located in the peptide‐binding channel and indicate a second structural region impacting enzyme specificity. The NRMT1 mutants demonstrated a slower rate of trimethylation and a requirement for higher substrate concentration. Expression of the mutants in wild type NRMT backgrounds showed no change in N‐terminal methylation levels or growth rates, demonstrating they are not acting as dominant negatives. Expression of the mutants in cells lacking endogenous NRMT1 resulted in minimal accumulation of N‐terminal trimethylation, indicating homozygosity could help drive oncogenesis or serve as a marker for sensitivity to DNA damaging chemotherapeutics or γ‐irradiation.
Keywords: NRMT1, NRMT2, N‐terminal methylation, mutation, methyltransferase, DNA damage, cancer, EZH2, aromatic cage
Introduction
Lysine methylation is an important post‐translational modification (PTM) for regulating protein function. This PTM plays crucial roles in chromatin organization, DNA repair, and transcriptional regulation.1, 2 The ε‐amino groups of lysine side chains can be monomethylated, dimethylated, and trimethylated, and each methylation state has a distinct functional readout, dependent on the residue methylated.1, 3, 4, 5, 6, 7 These functional readouts are generally accomplished by reader proteins, which contain PTM‐specific recognition domains.8, 9, 10 Readers binding to methyllysine commonly have chromatin organization modifier domains (chromodomains) but can also contain Tudor, MBT, PWWP, PHD finger domains or Ankyrin or WD repeats.11 These methyllysine binding domains are specific for distinct lysine residues and distinct methylation states (mono‐, di‐, or tri‐).12 Recognition of methylated lysines by methyllysine readers leads to recruitment of protein complexes, such as chromatin‐remodeling complexes, transcriptional machinery, or DNA repair holoenzymes.3, 4, 5, 6, 13
As methylation governs such diverse processes, altering methylation levels, or the degree of methylation, can be deleterious. Recent work demonstrated that a subset of B‐cell lymphoma patients have dominant mutations in the histone H3 lysine 27 (H3K27) methyltransferase EZH2, the catalytic component of the polycomb repressive complex 2 (PRC2).14, 15 These mutations occur in residues that create an aromatic cage in the active site and are conserved in the majority of methyltransferases.15 One of the most commonly mutated residues in EZH2 is tyrosine 641 (Y641).15 Mutation of this tyrosine to phenylalanine (Y641F) or asparagine (Y641N) shifted the H3K27 methylation pattern, promoting trimethylation over monomethylation.15 The dominant Y641 mutations changed the size of the EZH2 aromatic cage, and thus altered its catalytic specificity.15 As a result of the shift in methylation state, transcriptional profiles were altered and cellular proliferation rates and colony formation ability increased.15, 16
N‐terminal methylation of the free α‐amino group is another type of protein methylation, and it has been established as a regulator of protein–DNA interactions.17 Loss of N‐terminal methylation of regulator of chromatin condensation 1 (RCC1) results in its mislocalization from chromatin and multipolar spindle formation,17 while loss of N‐terminal methylation of the DNA repair protein DNA‐binding protein 2 (DDB2) impairs its recruitment to damaged DNA, and subsequently, nucleotide excision repair (NER).18 Our work focuses on the homologous N‐terminal methyltransferases NRMT1 (N‐terminal RCC1 methyltransferase 1) and NRMT2 (N‐terminal RCC1 methyltransferase 2), which following cleavage of the initiating methionine, methylate the α‐amine of the first N‐terminal residue of their targets.19, 20 They differ in catalytic specificity in that NRMT2 is a monomethylase, and NRMT1 is a trimethylase.19, 20 NRMT1 is a distributive trimethylase, as it binds its substrate and adds one methyl group at a time, dissociating from the substrate after the addition of each methyl group.19 NRMT2 primes substrates with the first methyl group, thereby increasing trimethylation rates of NRMT1.19 NRMT1 and NRMT2 are 50% identical and 75% similar and share an N‐terminal X‐P‐K consensus sequence.19, 21 Based on this consensus sequence, it is predicted that these methyltransferases target over 300 substrates in humans.21
Y641 of EZH2 aligns with similar tyrosines in the active sites of other methyltransferases, including G9a and SETD7, and confers trimethylase activity to both these methyltransferases when mutated to phenylalanine or alanine.22, 23 The corresponding aromatic residues in NRMT1 and NRMT2 are Y19 and F75, respectively.19 Given that mutation of tyrosine to phenylalanine has been shown to change catalytic specificity,15 we hypothesized these aromatic residues were responsible for the differing catalytic activities of NRMT1 and NRMT2. In addition to Y19 and F75, the active sites of NRMT1 and NRMT2 have differing aromatic residues at positions W20 and Y76, respectively.19 We also tested the effect of these residues on the catalytic specificities of NRMT1 and NRMT2. Lastly, both NRMT1 and NRMT2 mutations are found in human cancers (Cosmic Catalogue of Somatic Mutations in Cancer; http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). While mutations of Y19 or F75 have yet to be identified, we tested whether other mutations nearby structurally (NRMT1 Q144H—lung cancer) or in the adjacent peptide‐binding channel (NRMT1 N209I—endometrial cancer; NRMT1 P211S—lung cancer; and NRMT2 V224L—breast cancer) alter catalytic activity (Cosmic Catalogue of Somatic Mutations in Cancer).
We have shown that loss of NRMT1 results in oncogenic phenotypes such as increased proliferation, cell invasion and migration, and an increased sensitivity to DNA damaging agents.24, 25 These phenotypes suggest that methylation by NRMT2 alone is insufficient to functionally compensate for the loss of trimethylation,24, 25 and indicates a decrease in NRMT1 trimethylase activity will result in similar oncogenic phenotypes. Determining which cancer mutations alter the levels of N‐terminal methylation can help to determine their role in promoting tumor progression and also provide a marker for tumors more sensitive to DNA damaging agents. Studying mutations in the conserved aromatic residues of the active site will also tell us whether these residues can universally control catalytic specificity or if alternate structural motifs contribute to the catalytic specificity of NRMT1 and NRMT2.
Results
Aromatic cage mutants do not exhibit altered methylation patterns
Y19 is the NRMT1 tyrosine residue that most closely aligns with the position of Y641 in the methyltransferase active site of EZH2. In NRMT2, this residue is replaced by F75. As mutation of EZH2 Y641 to phenylalanine switches its catalytic activity, we hypothesized this amino acid substitution between NRMT1 and NRMT2 might be the cause of their differing catalytic activities. To test this hypothesis, recombinant proteins were made for NRMT1 Y19F and NRMT2 F75Y, and their ability to mono/dimethylate or trimethylate full‐length recombinant RCC1 over 1 h was assayed by Western blot. Unexpectedly, neither mutation significantly affected methyltransferase activity [Figs. 1(A,B) and 2(A,B)]. Similar to wild type (WT) NRMT1, NRMT1 Y19F exhibited only trimethylase activity [Fig. 1(A,B)]. Similar to WT NRMT2, NRMT2 F75Y exhibited only monomethylase activity [Fig. 2(A,B)].
Figure 1.
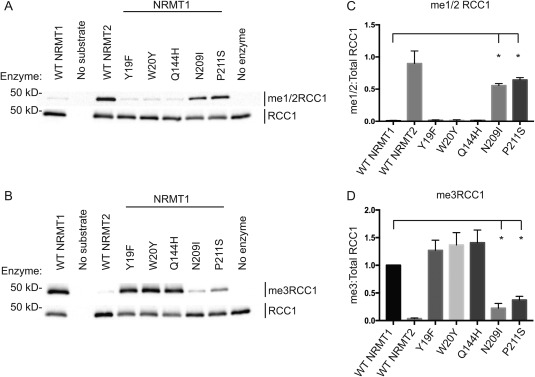
N‐terminal methylation patterns of wild type and mutant NRMT1. The directed mutation of aromatic residues Y19 and W20 in NRMT1 and the Q144H mutation found in human lung cancer showed no effects on (A) monomethylase/dimethylase (me1/2RCC1) or (B) trimethylase (me3RCC1) activity. However, the NRMT1 mutants N209I (endometrial cancer) and P211S (lung cancer) exhibit (A) increased monomethylation/dimethylation of RCC1 and (B) decreased trimethylation of RCC1 as compared to wild type (WT). Total RCC1 is shown as loading control. (C) Densitometry analysis of panel A. Ratio of me1/2RCC1:Total RCC1 band intensity. (D) Densitometry analysis of panel B. Ratio of me3RCC1:Total RCC1 band intensity, normalized to WT NRMT1. Each data point represents the mean ± SEM of three independent experiments. * denotes P < 0.05, determined by an unpaired two‐tailed Student's t‐test. Bands were quantified using ImageJ software (NIH).
Figure 2.
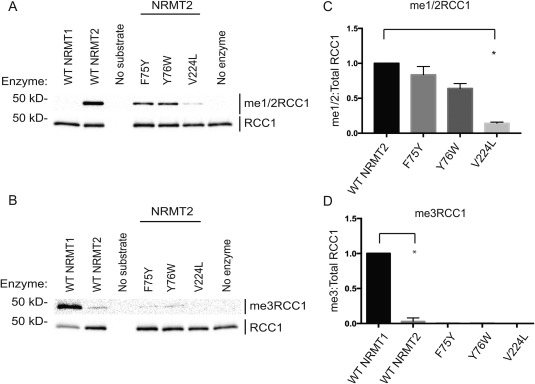
N‐terminal methylation patterns of wild type and mutant NRMT2. The directed mutation of aromatic residues F75 and Y76 in NRMT2 showed no effect on its (A) monomethylase/dimethylase (me1/2RCC1) or (B) trimethylase (me3RCC1) activities. The NRMT2 breast cancer mutant V224L exhibits (A) decreased monomethylase/dimethylase activity (B) but no corresponding increase in trimethylase activity. Total RCC1 is shown as loading control. (C) Densitometry analysis of panel A. Ratio of me1/2RCC1:Total RCC1 band intensity, normalized to WT NRMT2. (D) Densitometry analysis of panel B. Ratio of me3RCC1:Total RCC1 band intensity, normalized to WT NRMT1. As previously shown, trimethylation levels are significantly different between WT NRMT1 and WT NRMT2,19 but none of the NRMT2 mutants were significantly different from WT NRMT2. Low levels of trimethylation signal seen with WT NRMT2 are not due to trimethylation activity but cross‐reactivity of me3RCC1 antibody with lower levels of methylation when no trimethylation is present.19, 20 Each data point represents the mean ± SEM of three independent experiments. * denotes P < 0.05, determined by an unpaired two‐tailed Student's t‐test. Bands were quantified using ImageJ software (NIH).
In addition to an inability to switch catalytic activities, these mutations also did not significantly alter total methylation levels [Figs. 1(C,D) and 2(C,D)]. This is contrary to published data showing that the NRMT1 Y19F mutation significantly inhibits the ability of WT recombinant NRMT1 to methylate an N‐terminal peptide of CENP‐A.26 As our assay was done with full‐length recombinant RCC1 as substrate, it may be that enzyme/substrate binding is enhanced by interactions with the full protein and the interaction of Y19 with substrate is more imperative for peptide substrates. It may also be consensus sequence dependent. The CENP‐A N‐terminal sequence is Gly‐Pro‐Arg (GPR), while the RCC1 N‐terminal sequence is Ser‐Pro‐Lys (SPK). The recently solved crystal structure of human NRMT1 bound to CENP‐A N‐terminal peptide indicates the Arg residue in the CENP‐A consensus sequence forms hydrogen bonds and electrostatic interactions with Y19.26 These interactions may differ with a lysine residue and make Y19 less crucial for catalytic function.
As the mutations in Y19 and F75 did not significantly alter the methyltransferase activities of NRMT1 and NRMT2, we next mutated the other aromatic residue that differs between their active sites. W20 in NRMT1 is replaced with Y76 in NRMT2.19 Full‐length recombinant proteins were made for NRMT1 W20Y and NRMT2 Y76W, and their ability to mono/dimethylate or trimethylate full‐length recombinant RCC1 was assayed by Western blot. Again, neither mutation significantly affected methyltransferase activity [Figs. 1(A,B) and 2(A,B)]. This is also contrary to previous published data showing the W20Y mutation of NRMT1 significantly diminishes its ability to methylate the N‐terminal peptide of CENP‐A,26 and further indicates the most important catalytic residues may differ depending on substrate length or consensus sequence.
To differentiate between these two possibilities, we performed site directed mutagenesis on our plasmid used to make full‐length recombinant RCC1. We made GPR‐RCC1 to mimic the CENP‐A consensus sequence on a full‐length protein and Gly‐Pro‐Lys (GPK) and Ser‐Pro‐Arg (SPR)‐RCC1 to assess if the first or third amino acid is more important. Wild type SPK‐RCC1 and all three full‐length RCC1 consensus sequence mutants (GPK, SPR, and GPR) could be in vitro methylated by WT, Y19F, and W20Y NRMT1 [Supporting Information Fig. S1(A–D)], indicating that the impaired Y19F and W20Y activity seen by Wu et al.26 is not due to a difference in the three amino acid consensus sequence. To determine if the impaired activity resulted from substrate length, we tested the activity of Y19F and W20Y NRMT1 on a peptide containing the first 15 amino acids of WT RCC1 (SPKRIAKRRSPPADA). Interestingly, Y19F was still able to trimethylate the peptide, while W20Y was not [Supporting Information Fig. S1(E)]. These data show it is possible for a recombinant enzyme to have different activities toward full‐length and peptide substrates with the same consensus sequence.
Mutations found in human cancers alter NRMT1 and NRMT2 activities
Human cancer mutations of NRMT1 and NRMT2 were selected from the Cosmic Catalogue of Somatic Mutations in Cancer database based on their proximity to the active site or peptide‐binding channel. For NRMT1, we selected Q144H (lung cancer), N209I (endometrial cancer), and P211S (lung cancer). Both the N209I and P211S mutations are in the peptide binding channel of NRMT1,26 while the Q144H mutation is adjacent to H140, a third aromatic residue in the active site.19 Unlike Y19 and W20, this histidine is conserved between NRMT1 and NRMT2.19 For NRMT2, we selected V224L (breast cancer). This valine is analogous to M169 in NRMT1,19 which is directly adjacent to N168, an amino acid that forms both hydrogen bonds and electrostatic interactions with substrate.26, 27
In vitro methylation assays with the full‐length recombinant mutants and full‐length recombinant RCC1 as a substrate, showed the Q144H lung cancer mutation exhibited similar levels of monomethylation/dimethylation [Fig. 1(A)] and trimethylation [Fig. 1(B)] as WT NRMT1. In contrast, the N209I endometrial and P211S lung cancer mutants displayed significantly increased levels of monomethylation/dimethylation [Fig. 1(A,C)] and significantly decreased levels of trimethylation [Fig. 1(B,D) compared to WT NRMT1, indicating these mutations in patients could decrease global N‐terminal methylation levels in favor of monomethylation/dimethylation. As seen with WT NRMT2, the NRMT2 V224L breast cancer mutation exhibited no trimethylase activity [Fig. 2(B,D)], but it also exhibited significantly decreased monomethylase activity as compared to control [Fig. 2(A,C)]. This indicates patients harboring this mutation would have lower levels of priming activity by NRMT2 and potentially less trimethylation by NRMT1 as a consequence.19
Mass spectrometry verification of N209I and P211S shifted methylation activity
As the N‐terminal monomethyl/dimethyl RCC1 antibody (me1/2RCC1) that we created cannot discriminate between monomethylation and dimethylation, mass spectrometry (MS) analysis was used to determine if the N209I and P211S NRMT1 mutants were capable of monomethylation, dimethylation, or both. The results from the MS analysis [Fig. 3 and Supporting Information Fig. S2(A–J)] showed that, of the recombinant RCC1 that underwent successful cleavage of the initiating methionine (a portion of recombinant RCC1 fails to undergo this cleavage and is unable to be methylated in vitro), 63% was trimethylated by WT NRMT1 and the remaining 37% remained unmethylated.19 This is consistent with previous results showing WT NRMT1 will almost completely trimethylate RCC1 after 1 h in vitro.19 With the N209I mutation, unmethylated RCC1 levels increased to 73%, while RCC1 trimethylation levels dropped to 13%, dimethylation levels increased to 7%, and monomethylation levels increased to 6%. With the P211S mutant, unmethylated RCC1 levels also increased to 73%, trimethylation was further decreased to 5%, dimethylation increased to 13%, and monomethylation increased to 9%. The MS analysis is consistent with the Western blot results, indicating these mutants exhibit decreased trimethylase activity and increased monomethylase and dimethylase activity. They also indicate, that unlike the EZH2 mutants which switch catalytic activity from monomethylase to trimethylase, the NRMT1 mutations are decreasing the overall efficiency of the enzyme and preventing it from both converting unmodified substrate to monomethylated and monomethylated substrate to trimethylated.
Figure 3.
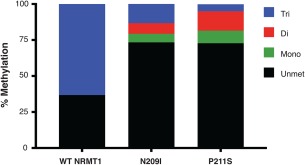
Mass spectrometry analysis of WT NRMT1 and NRMT1 mutants. Of the recombinant RCC1 with cleavage of the initiating methionine, 63% was N‐terminally trimethylated by recombinant WT NRMT1. The remaining 37% remained unmethylated. With the N209I mutation, unmethylated RCC1 levels increased to 73%, while RCC1 trimethylation levels dropped to 13%, dimethylation levels increased to 7%, and monomethylation levels increased to 6%. With the P211S mutant, unmethylated RCC1 levels also increased to 73%, trimethylation was further decreased to 5%, dimethylation increased to 13%, and monomethylation increased to 9%.
NRMT1 cancer mutants remain distributive methyltransferases
We have previously shown that NRMT1 works as a distributive enzyme, first monomethylating its substrate, then dissociating and reattaching for each subsequent methylation step.19 Richardson et al. confirmed this distributive nature of NRMT1 and additionally showed it is working through a random sequential bi‐bi mechanism.28 We have also shown that for the human RCC1 consensus sequence (Ser‐Pro‐Lys), affinity of NRMT1 for substrate increases with increasing substrate methylation levels,19 and hypothesize this helps the enzyme to quickly raise trimethylation levels without the accumulation of monomethylated or dimethylated substrate.
To monitor if the N209I and P211S mutants were impaired in the conversion of monomethylation/dimethylation to trimethylation, we held enzyme and substrate concentrations constant and varied the time of the in vitro methylation reactions. Western blot analysis showed that WT NRMT1, even at just 30 min, converted all monomethylation/dimethylation to trimethylation [Fig. 4(A)]. While low levels of trimethylation can be seen at 30 min with both the N209I and P211S mutants, monomethylation/dimethylation levels are higher and stay steady (N209I) or continue to increase (P211S) up to 2 h [Fig. 4(B,C)]. Finally after 2 h, monomethylation/dimethylation levels begin to decrease with a corresponding increase in trimethylation, indicating conversion of one to the other [Fig. 4(B,C)]. These data mirror the MS results (Fig. 3) and indicate that while N209I and P211S are still distributive enzymes capable of trimethylation, they are significantly slower at converting monomethylation and dimethylation into trimethylation.
Figure 4.
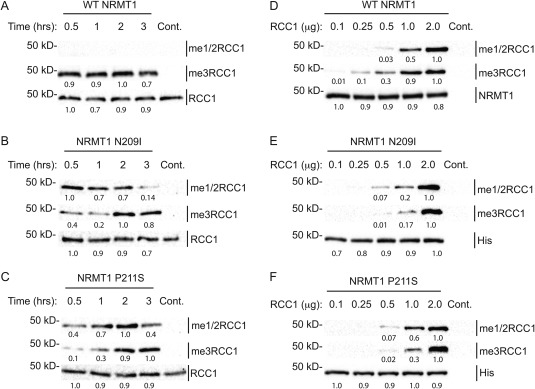
Catalytic studies of WT and mutant NRMT1. (A) WT NRMT1 fully trimethylates RCC1 (me3RCC1) in less than 30 min, compared to (B) N209I and (C) P211S, which exhibit primarily monomethylation/dimethylation (me1/2RCC1) until 2 h, where trimethylation levels begin to rise. The corresponding decrease in monomethylation/dimethylation is evident only after 3 h. These data indicate N209I and P211S are still distributive enzymes capable of trimethylation, but they are slower at converting monomethylation to trimethylation. Total RCC1 is shown as a loading control. Control (Cont.) reactions done without enzyme. (D) At low substrate levels, WT NRMT1 proceeds almost completely to trimethylation. As substrate concentration increases, the levels of monomethylation/dimethylation by NRMT1 increase because the ratio of unmodified substrate to previously methylated substrate is higher. (E–F) At low substrate levels, the NRMT1 N209I and P211S mutants show no methyltransferase activity. As substrate concentration increases, trimethylation begins to appear but does not reach WT levels until a 1:1 molar ratio of enzyme to substrate, indicating a higher substrate concentration is needed for optimum trimethylase activity. Anti‐NRMT1 is shown as a loading control for WT. Anti‐His is shown as a loading control for mutant NRMT1, as our NRMT1 antibody recognizes an epitope containing N209 and P211.20 Blots are representative images of three independent experiments. Bands were quantified using ImageJ software (NIH) and internally normalized to brightest band of each set, which was set at 1.0.
To assay whether the activity of the mutants could be restored by a significant increase in substrate concentration, we monitored the ability of the mutants to monomethylate/dimethylate or trimethylate RCC1 at varying substrate concentrations. Western blot analysis of the in vitro methylation assays revealed that N209I and P211S require a higher substrate concentration to reach the trimethylation levels seen with WT [Fig. 4(D–F)]. At low substrate levels (0.25 μg), WT NRMT1 shows only trimethylated substrate [Fig. 4(D)], while neither mutation exhibits any methyltransferase activity [Fig. 4(E,F)]. At 0.5 μg substrate, WT NRMT1 predominantly shows trimethylation [Fig. 4(D)], while both mutants are just beginning to exhibit monomethylase/dimethylase activity [Fig. 4(E,F)]. At 1.0 μg substrate, WT NRMT1 still favors trimethylated product, while both N209I and P211S still favor monomethylation/dimethylation [Fig. 4(D–F)]. It is not until the molar amount of mutant enzyme equals the molar amount of substrate (1 μg of NRMT = 40 pmol; 2 μg of RCC1 = 40 pmol) that the mutants have monomethylation/dimethylation and trimethylation levels comparable to WT NRMT1 [Fig. 4(D–F)]. This indicates the mutants require a higher substrate concentration to reach maximal activity.
Molecular modeling of N209I and P211S
Our examination of the NRMT1 crystal structure20 showed N209I and P211S to be in the peptide‐binding channel near the aromatic residues (Y19, W20, H140) of the active site.27 NRMT1 is a class I methyltransferase consisting of a seven‐stranded β sheet surrounded by five α‐helices.20 In addition, there are three helices in the N‐terminus segment, a pair of β hairpins, and a series of loops connecting the structural elements.26 It has been determined that the helices in the N‐terminal segment cluster with loop 4 (L4) and loop 67 (L67) to create the peptide‐binding domain, which is integrated by residues L31, Y34, I37, W136, L210, P211, I214, V217, Y215, and E213.26 While both N209 and P211 are in L67 [Fig. 5(A)], neither was previously predicted to directly interact with substrate.26
Figure 5.
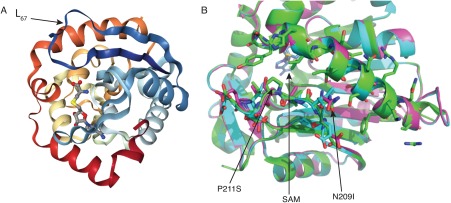
Molecular modeling of NRMT1 mutants. (A) Full crystal structure of NRMT1. Arrow denotes loop67 (L67, navy blue) where N209 and P211 are located. The methyltransferase co‐factor S‐adenosyl‐methionine (SAM) bound to the active site is indicated in gray. (B) Model comparing wild type NRMT1 (green, PDB code 2EX4) to mutated NRMT1 (pink), as calculated by the Robetta server.29 Amino acids N209 and P211 are green and indicated by arrows, the N209I and P211S mutations are adjacent in pink. SAM bound to the active site is indicated in blue.
To determine how the N209I and P211S mutations might otherwise affect the peptide‐binding channel, we performed molecular modeling.29 The modeling revealed that P211 is oriented toward the peptide‐binding channel, and its mutation to serine could alter the shape of the cavity itself [Fig. 5(B)]. Alternatively, prolines confer distinct shapes to unstructured regions, so its mutation to serine could change the configuration of L67 in an unpredictable manner. Mutation of N209 to isoleucine does not make any visually obvious changes to the structure of the peptide‐binding channel [Fig. 5(B)]. However, asparagine to isoleucine mutations have previously been shown to affect protein characteristics.30 The amide group of asparagine can hydrogen bond, while the isoleucine side chain is hydrophobic and does not. While these hydrogen bonds might not be directly formed with substrate, they may be necessary for proper orientation of L67. Taken together, we hypothesize that residues in the peptide‐binding channel that do not directly interact with the substrate can still regulate substrate binding by altering the overall orientation of L67.
NRMT1 mutations do not act as dominant negatives in cancer cells
It was determined that the EZH2 Y641 mutation acted in a dominant manner by exogenously expressing both WT EZH2 and EZH2Y641F in HEK293T cells, which already harbor WT EZH2 activity, and monitoring H3K27me3 levels.15 While expression of WT EZH2 produced a barely detectable increase in H3K27me3, expression of EZH2Y641F resulted in a significant increase in H3K27me3,15 indicating even with WT EZH2 present, the Y641F can change H3K27 methylation levels. HEK293T cells expressing EZH2Y641F were also more resistant to a small‐molecule inhibitor of single‐carbon transfer methyltransferases.15
To monitor if the P211S and N209I NRMT1 mutations worked in a similar dominant fashion, both were exogenously expressed using lentivirus at an MOI of 1 in A549 human lung carcinoma cells (as P211S was originally found in a lung cancer sample).31 Though expression levels of WT, N209I, and P211S NRMT1 were similar, only WT NRMT1 showed a slight increase in N‐terminal trimethylation levels [Fig. 6(A)]. Neither N209I nor P211S significantly changed either monomethylation/dimethylation or trimethylation levels of endogenous RCC1 [Fig. 6(A)], indicating they are not acting in a dominant negative manner. Expression of the mutants also did not significantly affect cellular proliferation [Fig. 6(B)].
Figure 6.
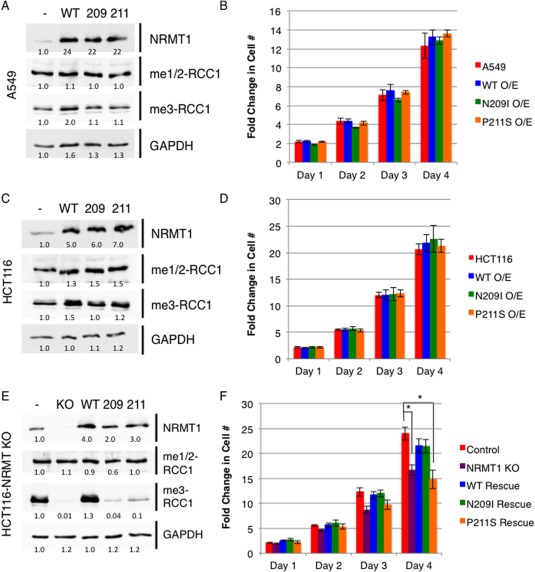
NRMT1 mutants are not acting as dominant negatives but reduce N‐terminal trimethylation levels when homozygous. (A–D) When overexpressed in (A) A549 or (C) HCT116, neither the 209I nor the P211S NRMT1 mutants (A,C) alter the level of RCC1 N‐terminal monomethylation/dimethylation (me1/2RCC1) or trimethylation (me3RCC1) or (B, D) cellular growth rates as compared to control cells expressing empty vector (‐) or cells overexpressing wild type (WT) NRMT1. When expressed in HCT116 cells where NRMT1 expression has been knocked out (KO) through CRISPR/Cas9 genome editing, (E) neither the N209I nor P211S mutant can restore N‐terminal trimethylation levels, and (F) P211S is also unable to rescue the growth defect seen with NRMT1 knockout. Each data point represents the mean ± SEM of three independent experiments. * denotes P < 0.05, determined by a paired two‐tailed Student's t‐test. GAPDH is shown as a loading control. Anti‐Mettl11a/NRMT1 (Abcam) used to determine WT and mutant NRMT1 expression levels. Blots are representative images of three independent experiments. Bands were quantified using ImageJ software (NIH) and internally normalized to wild type untransfected bands, which were set at 1.0.
The role of NRMT1 in lung cancer remains unclear, though it has been found to have slightly decreased expression in non‐small cell lung carcinoma.32 NRMT1 is most commonly found under‐expressed in breast cancer, glioblastoma, and leukemia.33, 34, 35, 36 We have correspondingly shown that in breast cancer it is acting as a tumor suppressor, and its loss promotes oncogenic growth.24 Conversely, NRMT1 has shown to be robustly overexpressed in a variety of colon cancer samples,37, 38, 39 where we predict NRMT1 may be acting as an oncogene. To monitor if the P211S and N209I mutations have a differential effect in a cancer type that typically overexpresses NRMT1, we performed the same overexpression experiments in HCT116 human colorectal carcinoma cells. As in A549 cells, neither expression of P211S nor N209I was able to change monomethylation/dimethylation or trimethylation levels of endogenous RCC1 [Fig. 6(C)] or alter cellular proliferation levels [Fig. 6(D)].
We had also recently made a CRISPR/Cas9 HCT116 NRMT1 knockout strain, which completely lacks NRMT1 expression and N‐terminal trimethylation, while maintaining wild type monomethylation/dimethylation levels [Supporting Information Fig. S3 and Fig. 6(E)]. To test if the P211S and N209I mutations could alter cellular phenotypes as homozygous mutations, we transduced the NRMT1 knockout cell lines with both mutations at an MOI of 1. As opposed to rescue with WT NRMT1, neither mutation could rescue N‐terminal trimethylation levels, though they were expressed at similar levels for over 72 h [Fig. 6(E)]. These data confirm the impaired biochemical activities of these mutants cannot be overcome in cells with endogenous substrate levels, even after prolonged exposure.
As loss of NRMT1 function has been shown to alter cellular growth rates, we also monitored the ability of N209I and P211S to rescue cellular proliferation rates in the HCT116 NRMT1 knockout line. As compared to control pSpCas9 transfected cells, the NRMT1 knockout strain grows significantly slower [Fig. 6(F)]. This would be expected if NRMT1 acts as an oncogene in this cell type. Rescue with transduction of WT NRMT1 restores proliferation rates [Fig. 6(F)]. Surprisingly, expression of the N209I mutant also restores proliferation rates, though the P211S mutation does not [Fig. 6(F)]. Why one mutation can restore proliferation and the other cannot, though neither restores N‐terminal trimethylation levels, now remains to be determined. These data indicate the NRMT1 mutations are loss‐of‐function mutations and not neomorphic gain‐of‐function alleles, like the EZH2 mutations, and will need to become homozygous or combined with other NRMT1 loss of function mutations before effects on proliferation and other oncogenic phenotypes will be seen.
Discussion
Our work is the first to describe a biochemical alteration in NRMT1 and NRMT2 methyltransferase activities in mutations identified in human cancer samples. Here, we demonstrated that NRMT1 mutations in the conserved aromatic residues of the active site did not result in switched catalytic specificities or altered levels of substrate methylation. This is contrary to what is seen in the SET domain histone methyltransferase EZH2, where mutation of its Y641 residue to either a phenylalanine or asparagine changes its catalytic specificity from a monomethylase to a trimethylase.15 There are a few possible explanations for this discrepancy. First, while EZH2 is a SET domain methyltransferase, NRMT1 and NRMT2 are seven‐β‐strand methyltransferases.19 Though both types of methyltransferases contain a series of aromatic residues in their active site that are reminiscent of the aromatic cages found in methyllysine‐binding proteins and likely contribute to substrate specificity, they are structurally distinct methyltransferases that may have different modes of substrate recognition.15, 19
Alternatively, in addition to the aromatic residue composition, there is a second structural feature of NRMT1 and NRMT2 that could dictate catalytic specificity. Despite the high sequence conservation between NRMT1 and NRMT2, NRMT2 possesses an extra 60 amino acid N‐terminal domain “tail” which is not found in NRMT1.19 Given the apparent flexibility of this tail, it is possible it could partially fold over the active site and limit substrate entrance. This would then take precedence over the aromatic residues in substrate selection and binding. A similar regulatory mechanism is seen in the human arginine methyltransferase PRMT1.40 PRMT1 has seven alternative splice variants that differ in their N‐terminal composition, and these unique sequences influence both catalytic activity and substrate specificity.40 To address this possibility for NRMT1 and NRMT2, we attempted to make NRMT2 with the tail domain deleted and NRMT1 with the tail domain added. However, only the NRMT2 without the tail expressed as soluble recombinant protein, and it showed no methyltransferase activity in 1‐h methyltransferase assays (data not shown).
Although the NRMT1 aromatic cage mutants showed no alteration in catalytic specificity, the cancer mutations N209I and P211S (endometrial and lung, respectively) showed a significant decrease in trimethylase activity and a significant increase in monomethylase/dimethylase activity. The NRMT2 breast cancer mutation V224L also showed a significant decrease in monomethylase activity but lacked a reciprocal gain in trimethylation activity. The recently solved crystal structures of NRMT1 complexed with substrate peptides illustrates that N209I and P211S are in the peptide binding channel,26 and V224 is adjacent to an asparagine that forms both hydrogen bonds and electrostatic interactions with substrate,26 indicating the mutations do not directly change catalytic specificity but alter substrate preference. This is validated by our kinetic experiments that showed N209I and P211S are still distributive enzymes capable of trimethylation. However, they are less efficient at methylating unmodified substrate and converting monomethylated/dimethylated substrate into trimethylated.
Whether these mutations can act as drivers of oncogenesis or promote further oncogenic transformation remains to be elucidated. Our data indicate it may depend on in which type of cancer it is found. As seen in the HCT116 NRMT1 knockout line, cancers that typically overexpress NRMT1 may find mutants with decreased trimethylase activity detrimental to their growth. In addition, loss of N‐terminal trimethylation has been shown to impair DNA repair,18, 24 so it may also make these tumors more sensitive to DNA damaging chemotherapeutics or γ‐irradiation. Cancers, such as breast, that become more oncogenic with loss of NRMT124 may find these mutations as helpful drivers of oncogenesis, though the potential for increased sensitivity to DNA damaging agents would remain.
In the case of NRMT1, we propose its ability to work both as an oncogene and a tumor suppressor is likely dependent on which pathways are driving oncogenesis in specific tissues. For example, one well‐studied NRMT1 target is the tumor suppressor retinoblastoma protein (Rb). We have previously shown that NRMT1 is acting as a tumor suppressor in estrogen receptor (ER) positive MCF‐7 breast cancer cells, as its loss promotes DNA damage accumulation, proliferation, migration, and xenograft tumor formation.24 Patients with ER+ tumors have poorer disease outcomes if they have an Rb mutation.41 If methylation by NRMT1 activates Rb function, loss of NRMT1 could mimic an Rb mutation, contributing to the increased oncogenicity seen with NRMT1 loss. In contrast, NRMT1 is found overexpressed in colon cancers,37, 38, 39 indicating it may be acting as an oncogene in this tissue. One difference between breast cancers and colon cancers is that colon cancer cells harboring activating K‐Ras mutations require wild type Rb for oncogenic transformation and prevention of apoptosis.42, 43 Thus, in this particular tumor type, overexpression of NRMT1 could be beneficial.
Little is known about NRMT1 expression levels in human lung cancer samples, though one study found a 1.5 fold decrease in NRMT1 expression in non‐small cell lung carcinomas (NSCLC).32 Unlike small cell lung carcinomas (SCLC), which frequently harbor Rb mutations, NSCLC tumors favor mutation in CDKN2A.44 As an inhibitor of MDM2 activity, CDKN2A indirectly controls both p53 and Rb protein levels, so NSCLC cancer harboring both a CDKN2A and NRMT1 mutation would have reduced levels of Rb with potentially reduced activity. In fact, the NRMT1 P211S mutation was found in a cell line derived from a metastatic lymph node of a patient with NSCLC,31 and Functional Analysis through Hidden Markov Models (FATHMM), which predicts the functional consequences of single nucleotide variants, rates it as strongly pathogenic (Cosmic Catalogue of Somatic Mutations in Cancer). The NRMT1 N209I mutation was identified as a somatic mutation in an endometrial tumor sample and also has a strongly pathogenic FATHMM prediction (Cosmic Catalogue of Somatic Mutations in Cancer). Of the 48 currently reported NRMT1 cancer mutations, eight are missense mutations in the L4 and L67 loop regions that help create the peptide‐binding channel, and seven of these eight mutations have strongly pathogenic FATHMM predictions (Cosmic Catalogue of Somatic Mutations in Cancer).
Why loss of N‐terminal trimethylation by NRMT1 would result in phenotypes despite the continued presence of monomethylation by NRMT2 also remains to be elucidated. As with lysine methylation, we predict the different levels of N‐terminal methylation promote different functional outcomes. For example, in the case of histone H4 lysine 20 (H4K20) methylation, monomethylation promotes transcriptional elongation by recruiting the MSL complex, increasing local histone H4 lysine 16 acetylation, and releasing RNA polymerase II (Pol II) into active elongation.4 In contrast, H4K20 trimethylation promotes Pol II pausing by inhibiting MSL recruitment.4 These distinct functional outcomes are driven by the specificity of the MSL chromodomain for H4K20 monomethylation and dimethylation and its inability to bind trimethylation.10 Whether the different levels of N‐terminal methylation also have readers with distinct structural domains or monomethylation simply is unable to promote strong DNA–protein interactions are currently under investigation.
The discovery of the Y641 EZH2 mutations as drivers of B cell lymphoma has lead to the development of many new EZH2 inhibitors. Two of these inhibitors, GSK126 and EPZ‐6438, both highly selective S‐adenosyl‐methionine‐competitive small molecule inhibitors, have been respectively shown to inhibit the proliferation of EZH2 diffuse large B‐cell lymphoma cell lines and mouse xenografts expressing the Y641 mutation.45, 46, 47 However, unlike the EZH2 Y641 mutations, the identified NRMT1 mutations are not gain‐of‐function, and therapeutic use of NRMT1 inhibitors would have to be context specific. In tumors such as colorectal, that significantly overexpress NRMT1, NRMT1 inhibitors could be a viable therapeutic option.37, 38, 39 In breast cancers, however, use of NRMT1 inhibitors alone could be detrimental but beneficial in combination with DNA‐damaging chemotherapeutics or γ‐irradiation. As we have shown that neither the N209I nor P211S NRMT1 mutations can restore N‐terminal trimethylation after loss of NRMT1, homozygosity for these mutations may also be a useful marker for tumors especially susceptible to chemo and irradiation therapies. Novel bisubstrate analogues and potent inhibitors of NRMT1 have recently been designed and continue to be optimized,48, 49 and it will be interesting to see if any of the derivatives affect cancer cell proliferation and/or sensitivity to chemo and radiation therapy in a tissue‐specific manner.
Materials and Methods
Constructs and antibodies
To make His6‐tagged recombinant protein, the human NRMT1 and NRMT2 ORFs (GE Dharmacon, Marlborough, MA) were amplified to introduce a 5′ NdeI restriction site and a 3′ XhoI restriction site, and subcloned into pET15b vector (EMD Millipore, Billerica, MA). These were used as templates for constructing all subsequent NRMT1 and NRMT2 mutants using the Quikchange site‐directed mutagenesis protocol (Agilent Technologies, Santa Clara, CA). The following forward primers and their reverse complements were used:
Y19F: 5′‐CCAAGGCCAAGACCTTCTGGAAACAAATCCCAC‐3′
W20Y: 5′‐CCAAGGCCAAGACCTACTACAAACAAATCCCACCC‐3′
Q144H: 5′‐GCCACCTCACCGATCACCACCTGGCCGAGTTC‐3′
N209I: 5′‐GAGGAGAGGCAGGAGATCCTCCCCGATGAGATC‐3′
P211S: 5′‐GGCAGGAGAACCTCTCCGATGAGATCTACC‐3′
F75Y: 5′‐GCCAGAGCTAAACTTTACTACCAAGAAGTACCAGC‐3′
Y76W: 5′‐GCCAGAGCTAAACTTTTCTGGCAAGAAGTACCAGCCAC‐3′
V224L: 5′‐CATATTGAAGGACAATCTGGCCCGGGAGGGCTGTATC‐3′
All His6 proteins were purified as previously described.50
Primary antibodies used for Western and dot blots are as follows: 1:5000 rabbit anti‐me1/2RCC1 (monomethylated/dimethylated SPK‐RCC1),17 1:10,000 rabbit anti‐me3RCC1 (trimethylated SPK‐RCC1),17 1:1000 goat anti‐RCC1 (Santa Cruz Biotechnology, sc‐1162, Santa Cruz, CA), 1:2000 rabbit anti‐NRMT1,20 1:3000 rabbit anti‐GAPDH (Trevigen, Gaithersburg, MD), and 1:1000 mouse anti‐polyHistidine (Sigma Aldrich, St. Louis, MO). 1:1000 rabbit anti‐Mettl11a/NRMT1 (Abcam, Cambridge, United Kingdom) was used to detect WT and mutant NRMT1 for lentiviral expression assays (Fig. 6).
In vitro methylation assays
All methylation assays were conducted at 30°C using 1 μg recombinant enzyme, 0.5 μg recombinant RCC1 substrate, and 100 μM AdoMet unless otherwise noted. The reaction volume was adjusted to 50 μL with methyltransferase buffer (50 mM potassium acetate, 50 mM Tris/HCl, pH 8), and reactions were run for 1 h. Methylation assays using varied RCC1 concentration were conducted using 1 μg recombinant enzyme, 0.1–2 μg recombinant RCC1 substrate, and 100 μM AdoMet. The reaction volume and time were unchanged. Methylation assays conducted at varying times used 1 μg recombinant enzyme, 0.5 μg recombinant RCC1 substrate, and 100 μM AdoMet. The reaction volume was unchanged, but reactions were run for 30 min to 3 h. Samples were prepared for MS analysis by performing a methyltransferase assay using 1 μg recombinant enzyme, 0.5 μg recombinant RCC1 substrate, and 100 μM AdoMet. Reaction volume was adjusted to 20 μL with methyltransferase buffer, and reactions were run for 1 h at 30°C. Reactions were run on an SDS/PAGE gel, and bands visualized by Coomassie Blue stain. The analysis for the presence and extent of RCC1 N‐terminal methylation by MS was conducted as previously described.19 For peptide in vitro methylation assays, 1 μg recombinant enzyme was mixed with 1.5 μg unmethylated RCC1 N‐terminal peptide (SPKRIAKRRSPPADA), 100 μM AdoMet, and methyltransferase buffer to 15 μL. After 1 h at 30°C, one third of the reaction was spotted on nitrocellulose and trimethylation of peptide was assessed using the rabbit anti‐me3RCC1 antibody as described above. Another third of the reaction was spotted on separate nitrocellulose and used to assess peptide monomethylation/dimethylation using the rabbit anti‐me1/2RCC1 as described above.
Cell culture
A549 human lung carcinoma cells (ATCC) were maintained in Dulbecco's Modified Eagle Medium (DMEM; Life Technologies, Grand Island, NY) with 10% fetal bovine serum (FBS; Atlanta Biologicals, Atlanta, GA) and 1% penicillin‐streptomycin (P/S; Life Technologies). HCT116 human colorectal carcinoma cells lines (a generous gift from Dr. Ian Macara, Vanderbilt University) were maintained in McCoy's 5a Modified Medium with 10% FBS and 1% P/S. HEK293LT human embryonic kidney cells (also a generous gift from Dr. Ian Macara) were grown in DMEM with 10% FBS and 1% P/S.
Generation of NRMT1 CRISPR/Cas9 knockout cell line
Suitable CRISPR/Cas9 target sites in the human NRMT1 gene were identified using an online CRISPR Design Tool (http://tools.genome-engineering.org).51 A target site in the first exon [Supporting Information Fig. S3(A)] was chosen and the following oligos designed and ordered from Integrated DNA Technologies (IDT, Coralville, IA).
Top: 5′‐ CACCGACGGTGGACGGCATGCTTGG ‐ 3′
Bottom: 5′‐ AAACCCAAGCATGCCGTCCACCGTC‐ 3′
The oligos were annealed, phosphorylated, and subcloned into BbsI‐digested pSpCas9(BB)‐2A‐Puro (Addgene, Cambridge, MA) as previously described.51 Resulting clones were verified by DNA sequencing. 6 × 105 HCT116 cells were transfected with 250 ng either empty pSpCas9(BB)‐2A‐Puro or the same vector containing the NRMT1 target sequence. Forty eight hours post‐transfections, cell were treated with 2 μg/mL puromycin. Surviving cells were transferred to individual wells in a 96‐well plate. Cells were expanded and passaged. Half were used to make carry plates the other half were used to isolate genomic DNA. For the first six wells, the genomic DNA was PCR amplified with primers flanking the target sequence. The resultant PCR products were sequenced at the University of Louisville Genomics Core. All six clones contained frameshift mutations and were selected for expansion and analyzed for NRMT1 expression and N‐terminal methylation by Western blot (Supporting Information Fig. S3). Subclone #6 was used in all subsequent experiments.
Lentivirus production
Wild type (WT), N209I, or P211S human NRMT1 were amplified from the pet15b vector to introduce a 5′ PmeI restriction site and a 3′ PmeI restriction site, and subcloned into pWPI lentiviral expression vector (Addgene). GFP‐tagged lentivirus expressing WT NRMT1, N209I NRMT1, or P211S NRMT1 were made by cotransfecting HEK293LT cells with 50 μg pWPI containing the appropriate NRMT1 cDNA, 37.5 μg psPAX2 packaging vector, and 15 μg pMD2.G envelope plasmid using calcium phosphate transfection. Forty eight hours post‐transfection, viral supernatants were collected, concentrated with 100K ultrafilters (EMD Millipore), and titered in the HEK293LT cells. A549, HCT116, or HCT116 NRMT1 KO cells were infected with virus to a multiplicity of infection (MOI) of 1. Three days post‐transduction, cells were counted and used in cell growth assays; remaining cells were used for Western blot analysis.
Cell growth assays
One thousand control A549, HCT116, HCT116 NRMT1 KO, or HCT116 pSpCas9 cells were plated in triplicate in a 96‐well plate in 100 μL of the appropriate cell culture media. Concurrently, A549, HCT116, or HCT116 NRMT1 KO cells transduced with WT, N209I, or P211S NRMT1‐expressing virus were plated in triplicate in 96‐well plates. Five sets of triplicates for each condition were made. On the day of plating (Day 0), 20 μL of Aqueous One Solution (Promega, Madison, WI) was added to the first set of triplicates for each condition and the absorbance at 490 nm was read after 2 h. Readings were taken on Day 0 and daily for four additional days. Relative fold increase was calculated by dividing average absorbance on each day by average absorbance at Day 0.
Conflict of Interest
The authors declare no competing interests.
Supporting information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Impact Statement This work presents the biochemical characterization of NRMT1 mutations found in human cancers. All but one has altered methyltransferase activity, and the most severe fails to rescue growth defects seen with NRMT1 knockout. The study of these mutations will help clarify their roles as potential drivers of oncogenesis or markers of cells sensitive to DNA damaging agents.
References
- 1. Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705. [DOI] [PubMed] [Google Scholar]
- 2. Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45. [DOI] [PubMed] [Google Scholar]
- 3. Greeson NT, Sengupta R, Arida AR, Jenuwein T, Sanders SL (2008) Di‐methyl H4 lysine 20 targets the checkpoint protein Crb2 to sites of DNA damage. J Biol Chem 283:33168–33174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kapoor‐Vazirani P, Vertino PM (2014) A dual role for the histone methyltransferase PR‐SET7/SETD8 and histone H4 lysine 20 monomethylation in the local regulation of RNA polymerase II pausing. J Biol Chem 289:7425–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richards EJ, Elgin SCR (2002) Epigenetic codes for heterochromatin formation and silencing. Cell 108:489–500. [DOI] [PubMed] [Google Scholar]
- 6. Santos‐Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NCT, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri‐methylated at K4 of histone H3. Nature 419:407–411. [DOI] [PubMed] [Google Scholar]
- 7. Smith BC, Denu JM (2009) Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta 1789:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao C, Herold JM, Kireev D, Wigle T, Norris JL, Frye S (2011) Biophysical probes reveal a “compromise” nature of the methyl‐lysine binding pocket in L3MBTL1. J Am Chem Soc 133:5357–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garske AL, Craciun G, Denu JM (2008) A combinatorial H4 tail library to explore the histone code. Biochemistry 47:8094–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moore SA, Ferhatoglu Y, Jia Y, Al‐Jiab RA, Scott MJ (2010) Structural and biochemical studies on the chromo‐barrel domain of Male Specific Lethal 3 (MSL3) reveal a binding preference for mono‐ or dimethyllysine 20 on histone H4. J Biol Chem 285:40879–40890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blus BJ, Wiggins K, Khorasanizadeh S (2011) Epigenetic virtues of chromodomains. Crit Rev Biochem Mol Biol 46:507–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M (2010) Quantitative interaction proteomics and genome‐wide profiling of epigenetic histone marks and their readers. Cell 142:967–980. [DOI] [PubMed] [Google Scholar]
- 13. Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callén E, Celeste A, Pagani M, Opravil S, De La Rosa‐Velazquez IA, Espejo A, Bedford MT, Nussenzweig A, Busslinger M, Jenuwein T (2008) A chromatin‐wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Devel 22:2048–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E (2012) Molecular architecture of human polycomb repressive complex 2. eLife 1:e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yap DB, Chu J, Berg T, Schapira M, Cheng SWG, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, Marquez VE, Marra MA, Gascoyne RD, Humphries RK, Arrowsmith CH, Morin GB, Aparicio SA. (2011) Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117:2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, Martinez‐Garcia E, Zhang H, Zheng Y, Verma SK, McCabe MT, Ott HM, Van Aller GS, Kruger RG, Liu Y, McHugh CF, Scott DW, Chung YR, Kelleher N, Skaknovich R, Creasy CL, Gascoyne RD, Wong KK, Cerchietti L, Levine RL, Abdel‐Wahab O, Licht JD, Elemento O, Melnick AM (2013) EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23:677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen T, Muratore TL, Schaner‐Tooley CE, Shabanowitz J, Hunt DF, Macara IG (2007) N‐terminal [alpha]‐methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat Cell Biol 9:596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cai Q, Fu L, Wang Z, Gan N, Dai X, Wang Y (2014) α‐N‐methylation of damaged DNA‐binding protein 2 (DDB2) and its function in nucleotide excision repair. J Biol Chem 289:16046–16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petkowski JJ, Bonsignore LA, Tooley JG, Wilkey DW, Merchant ML, Macara IG, Schaner Tooley CE (2013) NRMT2 is an N‐terminal monomethylase that primes for its homologue NRMT1. Biochem J 456:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tooley CE, Petkowski JJ, Muratore‐Schroeder TL, Balsbaugh JL, Shabanowitz J, Sabat M, Minor W, Hunt DF, Macara IG (2010) NRMT is an alpha‐N‐methyltransferase that methylates RCC1 and retinoblastoma protein. Nature 466:1125–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petkowski JJ, Schaner Tooley CE, Anderson LC, Shumilin IA, Balsbaugh JL, Shabanowitz J, Hunt DF, Minor W, Macara IG (2012) Substrate specificity of mammalian N‐terminal α‐amino methyltransferase NRMT. Biochemistry 51:5942–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, Allali‐Hassani A, Campagna‐Slater V, Vedadi M, Arrowsmith CH, Plotnikov AN, Schapira M (2010) Structural biology of human H3K9 methyltransferases. PLoS One 5:e8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ (2003) Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 421:652–656. [DOI] [PubMed] [Google Scholar]
- 24. Bonsignore LA, Butler JS, Klinge CM, Schaner Tooley CE (2015) Loss of the N‐terminal methyltransferase NRMT1 increases sensitivity to DNA damage and promotes mammary oncogenesis. Oncotarget 6:12248–12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonsignore LA, Tooley JG, Van Hoose PM, Wang E, Cheng A, Cole MP, Schaner Tooley CE (2015) NRMT1 knockout mice exhibit phenotypes associated with impaired DNA repair and premature aging. Mechanisms Ageing Devel 146–148:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu R, Yue Y, Zheng X, Li H (2015) Molecular basis for histone N‐terminal methylation by NRMT1. Genes Dev 29:2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dong C, Mao Y, Tempel W, Qin S, Li L, Loppnau P, Huang R, Min J (2015) Structural basis for substrate recognition by the human N‐terminal methyltransferase 1. Genes Dev 29:2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Richardson SL, Mao Y, Zhang G, Hanjra P, Peterson DL, Huang R (2015) Kinetic mechanism of protein N‐terminal methyltransferase 1. J Biol Chem 290:11601–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Das G, Hickey DR, McLendon D, McLendon G, Sherman F (1989) Dramatic thermostabilization of yeast iso‐1‐cytochrome c by an asparagine—isoleucine replacement at position 57. Proc Natl Acad Sci USA 86:496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu J, Lee W, Jiang Z, Chen Z, Jhunjhunwala S, Haverty PM, Gnad F, Guan Y, Gilbert HN, Stinson J, Klijn C, Guillory J, Bhatt D, Vartanian S, Walter K, Chan J, Holcomb T, Dijkgraaf P, Johnson S, Koeman J, Minna JD, Gazdar AF, Stern HM, Hoeflich KP, Wu TD, Settleman J, de Sauvage FJ, Gentleman RC, Neve RM, Stokoe D, Modrusan Z, Seshagiri S, Shames DS, Zhang Z (2012) Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res 22:2315–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee E‐S, Son D‐S, Kim S‐H, Lee J, Jo J, Han J, Kim H, Lee HJ, Choi HY, Jung Y, Park M, Lim YS, Kim K, Shim Y, Kim BC, Lee K, Huh N, Ko C, Park K, Lee JW, Choi YS, Kim J (2008) Prediction of recurrence‐free survival in postoperative non–small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin Cancer Res 14:7397. [DOI] [PubMed] [Google Scholar]
- 33. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Meterissian S, Omeroglu A, Hallet M, Park M (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14:518–527. [DOI] [PubMed] [Google Scholar]
- 34. Lee S, Chen J, Zhou G, Shi RZ, Bouffard GG, Kocherginsky M, Ge X, Sun M, Jayathilaka N, Kim YC, Emmanuel N, Bohlander SK, Minden M, Kline J, Ozer O, Larson RA, LeBeau MM, Gree ED, Trent J, Karrison T, Liu PP, Wang SM, Rowley JD (2006) Gene expression profiles in acute myeloid leukemia with common translocations using SAGE. Proc Natl Acad Sci USA 103:1030–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neale G, Su X, Morton CL, Phelps D, Gorlick R, Lock RB, Reynolds CP, Maris JM, Friedman HS, Dome J, Khoury J, Triche TJ, Seeger RC, Gilbertson R, Khan J, Smith MA, Houghton PJ. (2008) Molecular characterization of the pediatric preclinical testing panel. Clin Cancer Res 14:4572–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nikolsky Y, Sviridov E, Yao J, Dosymbekov D, Ustyansky V, Kaznacheev V, Dezso Z, Mulvey L, Macconaill LE, Winckler W, Serebryiskaya T, Nikolskaya T, Polyak K (2008) Genome‐wide functional synergy between amplified and mutated genes in human breast cancer. Cancer Res 68:9532. [DOI] [PubMed] [Google Scholar]
- 37. Hong Y, Downey T, Eu KW, Koh PK, Cheah PY (2010) A ‘metastasis‐prone’ signature for early‐stage mismatch‐repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin Experim Metastasis 27:83–90. [DOI] [PubMed] [Google Scholar]
- 38. Kaiser S, Park Y‐K, Franklin JL, Halberg RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG, Kong S, Sakthivel B, Xu H, Reichling T, Azhar M, Boivin GP, Roberts RB, Bissahoyo AC, Gonzales F, Bloom GC, Eschrich S, Carter SL, Aronow JE, Kleimeyer J, Kleimeyer M, Ramaswamy V, Settle SH, Boone B, Levy S, Graff JM, Doetschman T, Groden J, Dove WF, Threadgill DW, Yeatman TJ, Coffey RJ Jr, Aronow BJ (2007) Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol 8:R131‐R131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sabates‐Bellver J, Van der Flier LG, de Palo M, Cattaneo E, Maake C, Rehrauer H, Laczko E, Kurowski MA, Bujnicki JM, Menigatti M, Luz J, Ranalli TV, Gomes V, Pastorelli A, Faggiani R, Anti M, Jiricny J, Clevers H, Marra G (2008) Transcriptome profile of human colorectal adenomas. Mol Cancer Res 5:1263. [DOI] [PubMed] [Google Scholar]
- 40. Goulet I, Gauvin G, Boisvenue S, Côté J (2007) Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J Biol Chem 282:33009–33021. [DOI] [PubMed] [Google Scholar]
- 41. Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, Knudsen ES (2010) RB‐pathway disruption in breast cancer: differential association with disease subtypes, disease‐specific prognosis and therapeutic response. Cell Cycle 9:4153–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Collard TJ, Urban BC, Patsos HA, Hague A, Townsend PA, Paraskeva C, Williams AC (2012) The retinoblastoma protein (Rb) as an anti‐apoptotic factor: expression of Rb is required for the anti‐apoptotic function of BAG‐1 protein in colorectal tumour cells. Cell Death Dis 3:e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williams JP, Stewart T, Li B, Mulloy R, Dimova D, Classon M (2006) The retinoblastoma protein is required for Ras‐induced oncogenic transformation. Mol Cell Biol 26:1170–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim NS, Mee K, Somin S, Yujeong K, Yonghwan Y Sukjoon (2016) Differential regulation and synthetic lethality of exclusive RB1 and CDKN2A mutations in lung cancer. Intl J Oncol 48:367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang K‐C, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter‐Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H (2014) Selective inhibition of EZH2 by EPZ‐6438 leads to potent antitumor activity in EZH2‐mutant non‐Hodgkin lymphoma. Mol Cancer Ther 13:842. [DOI] [PubMed] [Google Scholar]
- 46. Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter‐Scott M, Smith JJ, Moyer MP, Copeland RA, Pollock RM, Kuntz KW, Raimondi A, Keilhack H (2014) Synergistic anti‐tumor activity of EZH2 inhibitors and glucocorticoid receptor agonists in models of germinal center non‐Hodgkin lymphomas. PLoS One 9:e111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Iii ADP, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature 492:108–112. [DOI] [PubMed] [Google Scholar]
- 48. Zhang G, Richardson SL, Mao Y, Huang R (2015) Design, synthesis, and kinetic analysis of potent protein N‐terminal methyltransferase 1 inhibitors. Organ Biomol Chem 13:4149–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang G, Huang R (2016) Facile synthesis of SAM‐peptide conjugates through alkyl linkers targeting protein N‐terminal methyltransferase 1. RSC Adv 6:6768–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen T, Brownawell AM, Macara IG (2004) Nucleocytoplasmic shuttling of JAZ, a new cargo protein for exportin‐5. Mol Cell Biol 24:6608–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3