

Abstract
Nanovehicles can efficiently carry and deliver anticancer agents to tumour sites. Compared with normal tissue, the tumour microenvironment has some unique properties, such as vascular abnormalities, hypoxia and acidic pH. There are many types of cells including tumour cells, macrophages, immune and fibroblasts cells, fed by defective blood vessels in the solid tumour. Exploiting the tumour microenvironment can benefit the design of nanoparticles for enhanced therapeutic effectiveness. In this review article, we summarized the recent progress in various nanoformulations for cancer therapy, with special emphasis on tumour microenvironment stimuli-responsive ones. Numerous tumour microenvironment modulation strategies with promising cancer therapeutic efficacy have also been highlighted. Future challenges and opportunities of design consideration are also discussed in details. We believe that these tumour microenvironment modulation strategies offer a good chance for the practical translation of nanoparticle formulas into clinic.
Graphical abstract
Exploiting the tumour microenvironment can benefit the design of nanomaterials for enhanced therapeutic effectiveness.
1. Introduction
Cancer is one of the world’s leading cause of death.1 Many efforts from different fields have been made to explore and improve the strategies to treat the disease efficiently and safely. Various nanomaterials (including but not limited to polymeric, inorganic and biomolecular) attracted a great deal of attention for cancer treatment due to their unique physicochemical properties, which give rise to the desired diagnostic and/or therapeutic applications.2–7 For example, carbon based nanoparticles (NPs) are intrinsically photothermal therapeutic agents with high light absorption and photothermal conversion efficiency.8 Rare earth upconversion NPs can convert low-energy near-infrared (NIR) light to high-energy ultraviolet (UV) and visible light, and therefore, serve as inner transducers for effective UV-based phototherapy upon exposure to NIR laser irradiation.9 With the high surface to volume ratio, finely-tuneable morphologies and surface properties, NPs are also capable of carrying diverse theranostic functionalities into one vehicle via loading, chemical conjugation or integration for cancer imaging and treatment.10–12 Different drug delivery systems have different loading capacities. For the polymer micelles, the loading amount is typically within the range of 10–20 % (wt/wt).13 Inorganic NP delivery systems such as mesoporous silica nanoparticles (MSNs), however, offer much higher loading capacity. In particular, the hollow structured MSNs can load as much as 1 g of lipophilic drug molecules per gram of silica).14 Compared with single modality imaging or therapeutic agents, multifunctional NPs can show the desired synergistic properties to endow early-stage diagnosis, provide more comprehensive information of the tumour region and improve the treatment efficacy.15–23
A major concern of cancer treatment is the non-targeted distribution of theranostic agents throughout the body. Most conventional anticancer agents do not distinguish normal cells and cancer cells. 24, 25 NPs have preferential accumulation in the tumour area due to the enhanced permeation and retention (EPR) effect.26–29 The physical and biological barriers in the body can affect the accumulation of NPs in the tumour. However, most NPs can be sequestered by the reticuloendothelial system (RES) before they reach the tumour, which not only causes a decrease in tumour accumulation, but also leads to possible damage to RES-rich organs.30 Engineering the physicochemical properties of NPs can help minimize the RES sequestration of NPs. For example, protein resistant ligands such as poly(ethylene glycol) (PEG) and zwitterionic molecules, are widely used as antifouling surface ligands for NPs to avoid nonspecific protein adsorption and cell adhesion before reaching the tumour sites.69, 70 Despite these progresses, only a median of 0.7% of the administered NPs can reach tumours.31 Design of NPs that can specifically respond to tumour microenvironment is significant to reduce the side effect to healthy tissues. The tumour microenvironment is complicated and quite different from normal tissues.32–34 The abnormal structures increase the interstitial fluid pressure, leading to unevenness of blood flow, hypoxia and acidic pH. Owing to these unique characteristics, stimulus-responsive therapeutic NPs are developed to exploit or modulate the physiology of tumour for anti-tumour applications. This review summarizes the recent NP designs by exploiting tumour microenvironment to enhance the anticancer therapy effect. We aim to give a comprehensive view of the use of NPs for cancer therapy affected by tumour microenvironment.
2. Tumour microenvironment
The solid tumour consists of cancer cells and numerous other type of stromal cells including fibroblasts, macrophages, lymphocytes, adipocytes, etc.33, 35–41 Each cell type has its own functions. All the cells are embedded in the extracellular matrix composed of collagen and proteoglycans, which provides a hydrated matrix to support tumour growth (Fig. 1).42, 43 The tumour blood vessels have irregular diameters and leaky structures. Some focal regions even lack endothelial cells or basement membrane.44
Fig 1.

The main components of the tumour microenvironment, which include malignant cells, many non-malignant cells (for example T cells, tumour associated fibroblasts and dendritic cells), tumour extracellular matrix and blood vessels.
2.1 Cancer-associated fibroblasts
Cancer-associated fibroblasts (CAFs) are the main component in cancer stroma which can secret extracellular matrix components, growth factors, cytokines and hormones.45, 46 Compared with normal fibroblasts which produce collagen subtypes to connect tissues, CAFs are large, spindle-shaped mesenchymal cells, which can promote tumour initiation, progression and metastasis. The CAFs take part in a heterotypic cross-talk with the cancer cells lining the desmoplastic invasion front.47 They are effected by growth factors secreted by cancer cells, such as transforming growth factor-β1 (TGF-β1) and platelet derived growth factor (PDGF).47, 48 On the other hand, CAFs can secrete fibroblast growth factor (FGF), stromal cell-derived factor 1 (SDF1), matrix metalloproteinases (MMPs) and insulin-like growth factor 1 (IGF1), which further promote tumour growth, angiogenesis and metastasis.49
2.2 Immune cells
Immune cells are another important component in mouse and human tumours. Immune cells can secrete inflammatory mediators to affect tumour microenvironment. Tumour infiltrating lymphocytes (TIL), including T cells, B cells and NK cells, are mainly responsive for local stimulation. The role of immune response in tumour has been controversial, either inhibit or actively promote tumour growth.50
2.2.1 T cells
T cells play an important role in cell-mediated immunity. There are several kinds of T cells, including cytotoxic T cells, alpha beta T cells and gamma delta T cells, each of which has its unique functions.51 For example, cytotoxic CD8+ T cells can destroy tumour cells and eliminate large solid tumour.52, 53 CD4+ T cells play a major role in protecting body from infection and modulating immune responses to tumour cells.54 However, cancer cells and tumour stromal cells in tumour microenvironment can produce monocyte chemoattractant protein-1 (MCP-1), which inhibit the functions of T cells. Therefore, activation of the immune system is important for cancer therapy. Besides, some studies demonstrated that the immune recognition of CD4+ T cells might not be always advantageous.
2.2.2 Tumour-associated macrophages
Tumour-associated macrophages are particularly abundant and are present at all stages of most human and experimental murine tumours.37, 55 Macrophages are located in the stromal compartment of solid tumours and can participate either in the tumour progression (e.g. cell proliferation, metastasis and invasion) or in the anti-tumour processes, depending on the subtypes of macrophages.37, 56 The tumour-associated macrophages are the major immunoregulatory cells to immune response, interacting with a wide range of growth factors, cytokines and chemokines.57
2.2.3 Dendritic cells
Dendritic cells (DCs) are unique immune cells in the tumour. DCs can induce, maintain and launch the antitumor immunity, and therefore are employed for cancer immunotherapy.58 Some DCs can activate B cells, natural killer (NK) cells and natural killer T (NKT) cells.59–61 Some DCs have been found to suppress T cell responses in tumour tissues, and the DCs can be employed as the sensor by capturing invading microbes and transmitting the resulting information to lymphocytes. Furthermore, DCs also play an important role in tumour-specific effector T cell generation.62
2.2.4 B cells
B cells are one type of lymphocytes, located in the draining lymph nodes and lymphoid structures adjacent to the tumour microenvironment.63 There are heterogeneous populations of B cells with distinct functionalities in the tumour.64, 65 B cell-associated autoimmune responses are frequently found in different tumours. B cells can secret cytokines and influence the functions of other cells in the cancer development.66 For instance, B cells can secret IL10 and Ig G to gain antigen IgG antibody complexes to realize immunosuppression or facilitate tumour progression.67 In addition, B cells can recognize specific antigens, regulate antigen processing and modulate T cell immune responses.64
2.3 Myeloid-derived suppressor cells (MDSCs)
MDSCs are a group of heterogenous immune cells raised from bone marrow progenitor cells.68 There are two major types of MDSCs: monocytic MDSCs and polymorphonuclear MDSCs. The former are similar to monocytes, and the latter are similar to neutrophils.69 MDSCs mainly accumulate in tumour tissues and peripheral lymphoid organs, and can suppress tumour progression by inhibiting the functions of T cells and NK cells.70 Therefore, one promising cancer therapeutic strategy is the expansion of MDSCs. Moreover, MDSCs can be used as target to enhance the accumulation of chemotherapeutic and immunotherapeutic agents.
2.4 Tumour endothelial cells (TECs)
Compared with normal endothelial cells, TECs are heterogeneous and can respond to growth factors and different drug molecules. Many factors including VEGF and FGF in the tumour can stimulate TECs for cancer growth. It is well documented that tumour blood vessels with irregular shapes and leaky structure play critical roles in tumour progression and metastasis. TECs are phenotypically similar among almost all cancers, therefore are a promising target for most cancer types.71 Several TEC targeting systems have been reported. For example, arginine-glycine-aspartic acid (RGD) peptide has been used to recognize integrin αvβ3, which is overexpressed in TECs.72
2.5 Natural killer (NK) cells and natural killer T (NKT) cells
NK and NKT cells are key players in regulating antitumor immunity, sharing similar phenotypes and functions.73 NK cells are located in lymphocytic infiltrates among and around the tumour tissue, not in direct contact with tumour cells,73 but can cause a fast immune response in the tumour stroma. NKT cells are a heterogeneous group of T cells which have properties of both T cells and NK cells. NKT cells express αβ T-cell receptors and a variety of molecular markers that are typically associated with NK cells. NKT cells can actively stimulate NK cells by secreting IFNγ, which then participate in shaping the adaptive immune response.74 They can also directly stimulate immune response, such as recognizing glycolipid antigen and producing pro-inflammatory T helper 1 (TH1) cytokines and anti-inflammatory TH2 cytokines.75
2.6 Pericytes
Pericytes are contractile cells that wrap around the endothelial cells of blood capillaries throughout the body.76 In tumour vasculature, pericytes provide support structure for blood vessels.77 They are important regulators of angiogenesis and vascular stability,78 and can be employed as a stromal target for cancer therapy. For example, Sood group found that dual targeting of endothelial cells and pericytes by anti-VEGF therapy and PDGF-β aptamer is more effective than monotherapy in human ovarian carcinoma models.79 However, the pericyte coverage of tumour vasculature is low due to the high level of pro-angiogenic factors, which also correlates with poor prognosis and unusual metastases.80 Hence, increase of pericyte coverage has also been considered as a promising strategy for cancer therapy.
2.7 Tumour-associated neutrophils (TANs)
TANs play an essential role in enhancing angiogenesis and immunosuppression at the tumour site via secreting cytokines and chemokines. Many studies found that TANs can release matrix metalloprotease 9 (MMP9) and VEGF,81 and also have great influence in tumour motility, migration and invasion.82 Another important function of TANs is to produce reactive oxygen species (ROS), the level of which is highly related to carcinogenesis. Stossel group demonstrated that neutrophils can induce mutation in Chinese hamster ovary (CHO) cells.83 Under physiological conditions, TANs rapidly undergo apoptosis and have a short blood circulation half-life of around 6–8 h.81, 84
3 Tumour vascular abnormalities
Although tumour blood vessels, like normal blood vessels, are composed of endothelial cells, mural cells and basement membrane, the blood vessels of cancerous tumours have multiple structural and functional abnormalities.85, 86 The vessels are leaky, poorly organized, irregularly shaped and tortuous.
Most chemotherapeutic agents face the challenge of lacking tumour selectivity. Nanosized vehicles are excellent candidates for efficient tumour delivery.87, 88 Exploiting the tumour microenvironment can help design nanoparticles with high tumour accumulation by passive or active targeting, and thus, enhance their cancer therapy efficacy. 85, 86, 89
3.1 Passive targeting to tumour by enhanced permeability and retention (EPR) effect
The EPR effect is the property by which NPs of suitable sizes tend to accumulate in tumour tissue much more than they do in normal tissues and prolong the retention time of NPs in the tumour region (Fig. 2).26–29 The reason for this phenomenon is that the abnormal tumour blood vessels promote vascular permeability.29 NPs in the size range of 20–200 nm tend to penetrate inside the interstitial space due to the poorly aligned defective endothelial cells.86 Moreover, the clearance of NPs from the interstitial space of tumour tissue tends to be slow owing to shortage of lymphatic drainage in tumour tissue.90 The EPR effect can be optimized by evading immune surveillance and enhancing circulation of NPs.87 The properties of NPs strongly influence the EPR effect.
Fig 2.

Schematic representation of the enhanced permeability and retention (EPR) effect. Angiogenic vessels in the tumour site is abnormal in form with large vascular fenestrae.
3.1.1 The effect of size and shape of NPs
The size and shape of NPs can significantly affect their biodistribution and accumulation in the tumour tissue.91–94 The size should be larger than 4–5 nm to avoid kidney filtration for a longer blood circulation. Meanwhile, they should be less than 200 nm so that they could extravasate the leaky vasculature.95 Meng et al. demonstrated that 50 nm MSNs coated with PEI-PEG copolymer achieved better passive tumour accumulation than 100 nm ones in KB-31 tumour model.92 MSNs are emerging as a drug carrier for cancer therapy. Tang’s group has investigated the distribution, absorption, excretion and toxicity of 110 nm MSNs.96 They found that the MSNs were difficult to absorb after hypodermic and intramuscular administration. Furthermore, Kim et al. systematically investigated particle size-dependent tumour accumulation using PEGylated MSNs in a U87MG tumour model. They found that the 100–150 nm-sized particles had an 4–6.5 fold higher tumour uptake than the ones with diameter < 30 or >300 nm.97 Decuzzi et al. also studied the MDA-MB-231 breast tumour uptake of spherical silica beads with sizes ranging from 700 nm to 3 μm.94 They found that the number of beads accumulated in the tumour site increased monotonically as the diameter decreased from 3 μm to 700 nm. Even through all the above works employed spherical silica as the model to investigate EPR effect, the EPR effect is also highly dependent on the tumour type and tumour size due to the differences of tumour vascularization and angiogenesis.28, 88
The biodistribution pattern of NPs is also highly dependent on their shape and rigidity.98–102 Non-spherical NPs have different in vivo behaviours from spherical ones.103 For example, spherical particles with a diameter larger than 200 nm would not pass through the spleen, but concave-shaped red blood cells (~10 μm in diameter) can infiltrate the splenic tissue rather easily.98 Huang et al. found short-rod MSNs (aspect ratio of 1.5) had a more rapid clearance rate than long-rod MSNs (aspect ratio of 5).99 Rapid clearance of NPs from blood is a main issue for their clinical application. Keeping NPs in the blood can help them reach tumour tissue by EPR effect. The long-rod MSNs could improve blood residence of NPs and enhance EPR effect. Alexis et al. found that compared with spherical micelles, the wormlike micelles intend to remain in the circulation for longer time due to a slower clearance by the mononuclear phagocyte system.104 It’s worth mentioning that the understanding of shape effect on in vivo behaviour is still limited due to the lack of proper synthetic methods to prepare uniform NPs with finely tuned morphology.
3.1.2 The effect of surface properties
Modification of surface property is also widely used to improve the blood circulation and tumour accumulation of NPs.105 The blood vessels have a negatively charged surface, and therefore, positively charged NPs can bind to the luminal surface of vascular walls and be cleared rapidly from the blood circulation.105 Negatively charged particles, on the other hand, show a higher accumulation in the liver due to charge-selective filtration.106 A promising strategy to prevent NPs from clearance is to camouflage them with antibiofouling synthetic polymers or cell membranes.107 It has been reported that neutral synthetic polymers can reduce undesirable adhesion of protein.108 Poly(ethylene glycol) (PEG), an FDA approved polymer is well-known for its ability to enhance surface hydrophilicity of NPs and reduce protein association.109 PEG is generally regarded as safe (GRAS). An early report by Abuchowsky et al. found that the modification of albumin and catalase by PEGylation can enhance blood circulation in mice.110, 111 In fact, PEGylation strategy has been successfully applied to various agents including nanovehicles. Recently, Chan’s group reported that molecular weight and surface density of PEG greatly influence the ability of NPs to resist protein adsorption with longer PEG chains and higher PEG surface density being better.112, 113 Zwitterionic materials which contain both positively and negatively charged groups and remain an overall neutral charge are promising alternatives that can effectively resist nonspecific protein adsorption.114, 115 Zwitterionic polymers such as poly(phosphorylcholine),116, 117 poly(sulfobetaine)118 and poly(carboxybetaine) demonstrated comparable performance with PEG to resist protein adsorption from plasma.117, 119 Yang et al. demonstrated that zwitterionic poly(carboxybetaine) (PCB)-coated gold NPs (PCB-GNP) exhibited much longer circulation half-life (t1/2 = 55.8 h) than PEG-coated GNPs (8.7 h). Furthermore, while the second dose of PEG-G NPs suffered from a dramatic decrease of t1/2 to 5.2 h, the PCB-G NPs retained a similar blood circulation half-life (t1/2 = 55.6 h for the second dose).119
Another effective approach is to use cell-membrane to camouflage NPs.120–124 The NPs have been entrapped in various types of cell membrane, such as human platelets, red blood cells, leukocytes and cancer cells. The membrane-cloaked NPs could mimic cells in order to escape from the macrophage uptake. Hu et al. reported the preparation of polymeric NPs enclosed in the plasma membrane of human platelets. These NPs displayed platelet-mimicking properties and a reduction in macrophage uptake by macrophage-like cells in autologous human plasma.120 Red blood cells were also employed as a masquerade for gold NPs (RBC-Au NPs).122 The resulting RBC-AuNPs could avoid macrophage uptake by bestowing immunosuppressive functionalities and shield the particles from interacting with thiolated compounds on the AuNPs. Leukocyte and cancer-cell membranes have also been investigated to enhance the blood circulation and tumour accumulation.125, 126
3.2 Active targeting of NPs
Although NPs intend to accumulate in the tumour tissue due to the EPR effect, passive tumour targeting is dependent on the tumour vascularization and angiogenesis, and therefore lacks specificity and consistency. Both tumour model type and tumour condition can seriously affect the passive targeting effectiveness. 28, 88 Ligands which could specifically bind to the corresponding receptors overexpressed in the tumour area (either the tumour cells or the tumour microenvironment) have been attached to the NPs for active targeting.127–131 In this review, we mainly focus on targeting to tumour endothelium, including tumour vascular endothelial growth factor receptor (VEGFR),132, 133 integrin αvβ3 134, 135 and vascular cell adhesion molecule-1 (VCAM-1).136
3.2.1 Vascular endothelial growth factor (VEGF)
Angiogenesis is essential for tumour growth and metastasis,. VEGF receptor (VEFR) is an important signalling protein inducing tumour angiogenesis overexpressed in the endothelium of most solid tumours.137 Senger et al. first identified vascular permeability factor (VPF), a protein inducing vascular leakage in guinea pigs, hamsters, and mice in 1983.138 Since then, anti-VEGF monoclonal antibodies and other VEGF blockers have been reported to successfully inhibit tumour growth. Goel et al. proposed the use of MSNs for VEGFR specific targeting. Compared with non-targeted NPs, the targeted NPs showed almost three times enhancement of tumour accumulation in a U87MG human glioblastoma xenograft model. The MSNs were also modified with macrocyclic chelator 1,4,7-triazacyclononane-triacetic acid (NOTA) and labelled with 64Cu for positron emission tomography (PET) imaging and loaded with anti-VEGFR drug (sunitinib) for therapy (Fig. 3A).139 VEGF has also been applied to a variety of NPs for active targeting. For example, Chen et al. conjugated VEGF and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, 64Cu chelator) onto quantum dots (QDs) for VEGFR-targeted PET/near-infrared fluorescence (NIRF) imaging.140 Chekhonin group developed a magnetic system that monoclonal antibodies against vascular endothelial growth factor (mAbVEGF) was coupled to BSA coated magnetic NPs (Fe3O4).141 VEGF has also been coupled to organic NPs like boronated polyamidoamine dendrimers. Fluorescence imaging confirmed selective accumulation of the dendrimer at the periphery of 4T1 breast carcinoma tumour. These dendrimers were further employed for boron neutron capture therapy (BNCT).142
Fig 3.
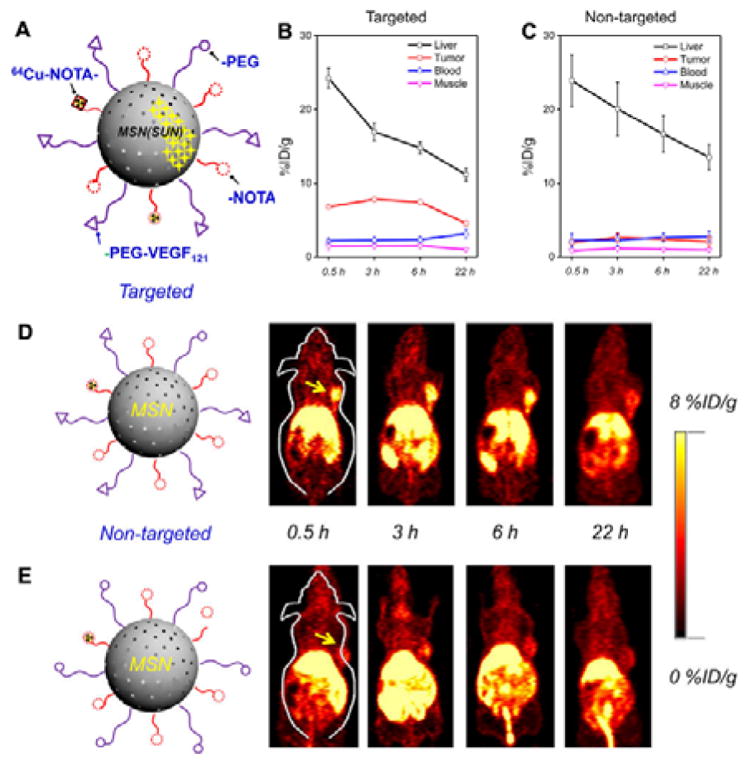
(A) Schematic illustration of radioisotope 64Cu-labelled VEGF121 conjugated mesoporous silica NPs (64Cu-NOTA-MSN-VEGF121). (B, C) Region-of-interest (ROI) quantification of liver, U87MG tumour, blood, and muscle upon intravenous injection of (B) 64Cu-NOTA-MSN-PEG-VEGF121 (targeted group), and (C) 64Cu-NOTA-MSN-PEG (non-targeted group) after various time points. (D, E) VEGFR targeted PET imaging in U87MG tumour-bearing mice. Coronal PET images of 64Cu-NOTA-MSN-PEGVEGF121 (D) and 64Cu-NOTA-MSN-PEG (E) injected intravenously in U87MG tumour mice at various time points. The location of the tumour is indicated by the yellow arrows. (Reprinted with permission from ref. 139. Copyright 2014, American Chemical Society.)
3.2.2 αvβ3 integrin
αvβ3 integrin is another important endothelial cell receptor, which is expressed in tumour-associated endothelial cells of various fast growing tumours.143 The αvβ3 integrin is important for mediating cell-to-cell and cell-to-matrix interactions.144 Researchers have paid much attention to develop αvβ3-targeted NPs for cancer diagnosis and therapy. Cai et al. reported the development of cyclic RGD peptide-labelled QDs for tumour vasculature targeted NIR fluorescence imaging.145 Graf et al. described the cyclic pentapeptide c(RGDyK) modified poly(D,L-lactic-co-glycolic acid)-block-polyethylene glycol (PLGA-PEG) NPs for platinum prodrug delivery.146 The PLGA is a FDA approved polymer due to its non-toxicity, good bioavailability and biocompatibility.147 PLGA can undergo hydrolysis in the body to produce the original monomers, lactic acid and glycolic acid. Zhen et al. used RGD modified ferritin (RFRT) as a carrier to selectively deliver zinc hexadecafluorophthalocyanine (ZnF16Pc, a photosensitizer) to the tumour endothelium.148 The RFRT has a strong binding affinity for integrin αvβ3. The 1O2 produced by ZnF16Pc increases tumour vascular permeability and leads to a higher tumour uptake of the second dose of therapeutic NPs (Fig. 4A–C). Other NPs including but not limited to dendrimers, copolymer NPs, magnetic NPs and upconversion NPs have also been modified with RGD for enhancing tumour accumulation.149–155
Fig 4.
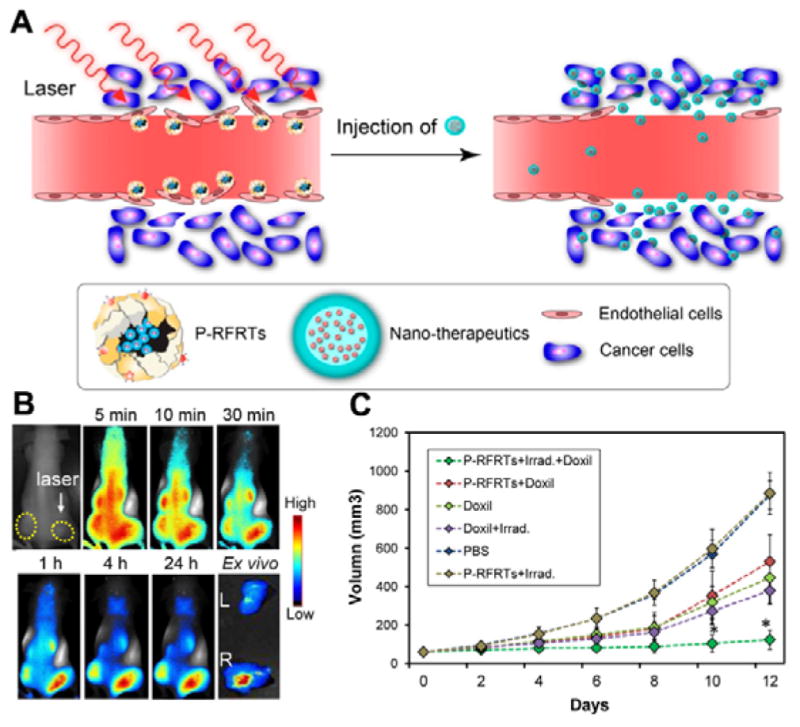
(A) Working mechanism of ZnF16Pc-loaded RGD peptide modified ferritin (P-RFRTs) mediated photodynamic therapy (PDT) was performed by accumulation of P-RFRTs in the tumour after intravenous injection and irradiation with a 671 nm laser. (B) EPR enhancement by PDT-induced P-RFRTs. At 24 h post-injection of P-RFRT, the right 4T1 tumour was irradiated by a 671 nm laser for 5 min followed by IRDye800-HSA injection. The right tumour was enhanced EPR effect showed significantly higher albumin accumulation. (C) EPR enhancement led to improved tumour therapy outcome. Doxil was injected 5 min after the end of P-RFRT-mediated PDT. Control groups include animals receiving P-RFRTs and Doxil but no irradiation, Doxil only, irradiation only, P-RFRTs and irradiation but no Doxil, and PBS only. Compared to the control groups, animals receiving the PDT and Doxil combination showed much more significant tumor growth suppression. (Reprinted with permission from ref. 148. Copyright 2014, American Chemical Society.)
3.2.3 Vascular cell adhesion molecule-1 (VCAM-1)
VCAM-1 is expressed on both luminal and lateral sides of endothelium during inflammation and cancer, mediating cell adhesion and spreading.136, 156, 157 Molecules that bind specifically to VCAM-1 are promising ligands for NPs to improve tumour accumulation. Gosk et al. prepared PEG modified immunoliposomes (IL) directed against VCAM-1 and investigated the targetability to tumour vessels.158 Since VCAM-1 is also aberrantly overexpressed on breast cancer cells, it has been reported as a potential therapeutic target against lung metastasis of breast cancer.159 Cao et al. encapsulated water-insoluble VCAM-1 inhibitor, succinobucol (SCB) in triblock polymer NPs.160 These SCB loaded NPs significantly reduced VCAM-1 expression on 4T1 cells. After oral administration, these NPs could efficiently inhibit the lung metastasis of breast cancer. The same group further developed SCB-loaded pH-responsive wormlike micelles for lung metastasis treatment.161
4 Tumour microenvironment mediated nanotherapeutics
4.1 Tumour hypoxia
Tumour hypoxia means that oxygen level in the tumour tissue is lower than physiological level.162, 163 Rapid tumour cell proliferation and tumour structural and functional abnormalities limit oxygen diffusion.164 The oxygen pressure can be near zero mmHg in some solid tumours, while it is about 30 mmHg in normal tissue.165 Oxygen availability also decreases as the distance from the blood vessel increases. Therefore, the hypoxic cells are far away from blood vessels and cannot be exposed to anticancer drugs effectively during the treatment.166, 167 As a result, the hypoxic cells in solid tumours cannot be effectively killed by chemotherapy and radiotherapy. To enhance the tumour therapy efficacy, many efforts have been devoted to modulate tumour oxygen level.168–173
4.1.1 Modulating tumour oxygen level
Photodynamic therapy (PDT) is a powerful treatment, which involves light and photosensitizer (PS).174, 175 The PS can generate reactive oxygen species (ROS) from oxygen to kill cells under light activation at specific wavelength.176 However, the application of PDT is limited by the hypoxia condition in tumours. To enhance the PDT efficacy, it is highly desirable to selectively heighten the local oxygen level in the tumour area. Cheng et al. reported the oxygen self-enriching photodynamic therapy (Oxy-PDT) by loading a near-infrared photosensitizer IR780 into perfluorocarbon nanodroplets.177 Due to the high solubility of respiratory gases in perfluorocarbon, the perfluorocarbon is a good candidate as oxygen carrier.178, 179 The NIR photosensitizer IR780 and perfluorohexane (PFH) were employed to prepare lipid nanodroplets with PEG on the surface (Fig. 5A). After PEGylation, the size of nanodroplets is around 200 nm, which is suitable for passive targeting to tumour. When the Oxy-PDT was irradiated with 808 nm laser, oxygen enriched in the PFH was activated to generate cytotoxic singlet oxygen by IR780. In addition, they found that the lifetime of 1O2 in PFH (5×10−2 s) is much longer than that in water (5×10−6 s) and intracellular environment (6×10−7 s). Oxy-PDT treatment with NIR laser irradiation showed the maximum amount of 1O2, and therefore the highest cell mortality (Fig. 5B and C). The in vivo experiment further proved that Oxy-PDT had a favourable tumour targeting due to the EPR effect (Fig. 5E) and improved tumour inhibition compared with traditional PDT (Fig. 5F). In addition, hollow Bi2Se3 NPs also displayed their ability of delivering perfluorocarbon for enhanced radiotherapy.180 Haemoglobin (Hb) which can bind with oxygen to form oxygenated Hb has also been employed as an oxygen carrier to promote cancer therapy.181–183
Fig 5.

(A) Schematic drawing of the oxygen self-enriched photodynamic therapy (Oxy-PDT) NPs. (B, C) H2DCFDA were employed to detect ROS generation by treating cells with various agents. Nuclei were stained with Hoechst 33342. Scale bar, 50 μm (B) and 10 μm (C). (D) Cell viability of CT-26 cells by the CCK-8 assay under hypoxic conditions. (E) Near-infrared fluorescence images of mice at different time points after intravenous injection of Oxy-PDT NPs. (F) Tumour therapy by single injection of Oxy-PDT NPs intravenously. (Reprinted with permission from ref. 177. Copyright 2015, Nature Publishing Group.)
Besides as oxygen carrier, NPs can also serve as oxygen generator under the stimulation of tumour microenvironment. Guo group developed H2O2-activatable, and O2-evolving PDT NPs (HAOP NPs).184 As shown in Fig. 6A, the photosensitizer methylene blue (MB) and catalase were encapsulated in the HAOP NPs and black hole quencher (BHQ) was doped in the poly(D,L-lactic-co-glycolic acid) (PLGA) polymer shell to quench the energy of excited photosensitizer. Compared with normal cells, cancer cells produce excessive amount of H2O2 (up to 0.5 nmol/104 cells/h).185 The H2O2 can be catalysed by catalase to generate oxygen, which then ruptures the PLGA, releases MB and decreases the Förster resonance energy transfer (FRET) and improves the PDT efficacy of MB (Fig. 6B). The tumour inhibition results showed that HAOP NPs had significantly higher in vivo PDT therapeutic effect than the group without catalase or without laser (Fig. 6C). Catalase-loaded tantalum oxide (Ta2O5) nanoshells were reported by Liu’s group. The catalase can enhance oxygen level in the tumour, benefiting radiotherapy performance of Ta2O5.186 Some inorganic materials can also be employed to generate oxygen. Manganese dioxide (MnO2) has high catalytic reactivity for H2O2 decomposition to produce oxygen under acidic pH. The MnO2 can also release Mn2+ ion in the body. The Mn2+ is a necessary element for humans to survive with low concentration. Liu et al. designed a multifunctional pH-responsive human serum albumin (HSA)-coated MnO2 NPs (Fig. 6D)187 that contain chlorin e6 (Ce6, photosensitizer), c,t,c-[Pt(NH3)2-(O2CCH2CH2COOH)(OH)Cl2] (cis-Pt(IV)SA, Pt prodrug) and BSA. At acidic pH, H2O2 reacts with MnO2 to generate oxygen promoting both chemo- and photodynamic therapy. The immunostaining indicated that these NPs could overcome tumour hypoxia-associated resistance (Fig. 6E). Furthermore, the chemo- and photodynamic dual therapy synergistically inhibited the tumour growth (Fig. 6F). Prasad et al. reported the polyelectrolyte albumin with MnO2 (A-MnO2) nanocomplex.188 The nanocomplex can generate oxygen and increase tumour pH to enhance radiotherapy efficacy. MnO2 has also been used to combine with upconversion NPs for pH/H2O2 responsive upconversion imaging and synergetic therapy.189 The oxygen-elevated synergetic radio/PDT was achieved by generating oxygen due to the MnO2-H2O2 reaction in tumour acidic environment.
Fig 6.

(A) Work mechanism of H2O2-controllable release of photosensitizer and O2 generation to enhance PDT by encapsulating catalase in the core and black hole quencher (BHQ) in the PLGA shell and modification with poly(vinylalcohol) (PVP) and cRGD to form a cell specific, H2O2-activatable, and O2-evolving PDT NP (HAOP NP). (B) In vitro release profiles of methylene blue (MB) under various conditions. (C) Relative tumour volume (V/V0) change with HAOP NPs under various treatments. (D) Schematic illustration for the synthesis of HSA-MnO2-Ce6&Pt (HMCP) NPs. (E) Immunofluorescence staining of tumour blood vessel (CD31) and hypoxia (pimonidazole) with and without HMCP treatment. (F) 4T1 tumour growth curves of mice after various treatments. Light irradiation was conducted at 24 h p.i. by a 661 nm laser at the power density of 5 mW cm−2 for 1 h (n = 5/group). (Fig.6A–C was reprinted with permission from ref. 184. Copyright 2015, American Chemical Society. Fig.6 D–F was reprinted with permission from ref. 187. Copyright 2016, John Wiley & Sons, Inc.)
4.1.2 Hypoxia-responsive drug release
The hypoxia in tumour can also be used to control drug release or activate prodrug in the tumour tissue. 169, 190,185 Gu’s group reported hypoxia-responsive conjugated polymer-based doxorubicin hydrochloride (DOX) release nanocarrier capable of light triggered ROS generation (Fig. 7A).191 The dithiophene-benzotriazole (DB), 2-nitroimidazole (NI) and polyvinyl alcohol (PVA) were the main parts of the polymer. The DB part can generate ROS under visible/near-infrared (Vis/NIR) light activation. NI, a hydrophobic molecule, can be converted to hydrophilic 2-aminoimidazole under a hypoxic environment. The hydrophilic PVA can prolong the nanocarrier circulation time in bloodstream. DOX was encapsulated in the polymer nanocarrier. As given in Fig. 7B, the TEM results indicated that these nanocarriers disassemble gradually. Under 532 nm light irradiation, this nanoformula completely inhibited tumour growth (Fig. 7C). However, living tissues have a strong absorption of light in the visible range below 600–700 nm, thus the penetration of 532 nm light is limited. Future studies might require the use of NIR light for PDT. The N=N double bond of azobenzene (AZO) is another widely used hypoxia-sensitive moiety to control cargo release or detect hypoxia.170, 190, 192–194 Torchilin group demonstrated a hypoxia-targeted siRNA delivery system based on AZO.170 Polyethyleneimine (PEI) and 1,2-dioleyl-sn-glycero-3-phosphoethanolamine (DOPE) were employed as the siRNA carriers (PAPD). AZO was used as a hypoxia-responsive bioreductive linker (Fig. 7D). Under hypoxic tumour environment, the PEG group can be detached due to the degradation of the AZO linker. In vitro experiment showed that these hypoxia-responsive NPs can penetrate multicellular spheroids (Fig. 7E). Further animal experiments confirmed that these PAPD/siRNA NPs can downregulate the target gene (Fig. 7F). As the hypoxia responsive drug release system only releases the drugs in the tumour microenvironment, it reduces undesired side effects to normoxic tissues.
Fig 7.

(A) Formation of light-activated hypoxia-responsive drug-delivery system by hypoxia-sensitive 2-nitroimidazole-grafted conjugated polymer (CP-NI) and polyvinyl alcohol (PVA)-based surface coatings and encapsulated DOX. (B) TEM images of DOX/CP-NI NPs at various time points after 5 min light irradiation (scale bar, 200 nm). (C) The HeLa tumour growth curves upon different treatments (2.0 mg kg−1 DOX, 3.6 mg kg−1 CP-NI). *p < 0.05, **p < 0.01 (n = 5/group). (D) Schematic representation of the synthesized PEG-Azo-PEI-DOPE and the work mechanism under hypoxic condition. (E) Images of siRNA distribution in various sized multicellular spheroids after 4 h incubation under normaxia and hypoxia conditions. (F) Ex vivo fluorescence optical imaging of tumours after intravenous injection of PBS, PEG-Azo-PEI-DOPE/anti-GFP siRNA complexes (PAPD/siGFP), and PEG-Azo-PEI-DOPE/negative siRNA complexes (PAPD/siNeg). ((Fig.7A–C was reprinted with permission from ref. 191. Copyright 2016, John Wiley & Sons, Inc. Fig.7 D–F was reprinted with permission from ref. 170. Copyright 2014, John Wiley & Sons, Inc.)
4.2 Acidic pH in tumour microenvironment
Acidic extracellular pH is an important character of solid tumour microenvironment.195 In the 1930s, Warburg first found that due to a lack of full capacity of glucose oxidation to produce energy of proliferating tumour cells, the extracellular tumour pH is within the range of 6.0–7.0, whereas the extracellular pH of normal tissue and blood is maintained at 7.4.196 The acidic pH microenvironment has been widely utilized to selectively trigger nanovehicles for enhanced cancer therapy efficacy. On the other hand, the acidic extracellular pH can activate some lysosomal enzymes to balance pH. Lactic acid is the main metabolite by anaerobic glycolysis in hypoxia.196 Moreover, acidic microenvironment can increase drug resistance and affect tumour metastasis.197, 198 To combat these effects, it is important to develop nanosystems to modulate the tumour extracellular pH. We classified the pH responsive NPs into two categories: inorganic ones and organic ones.
4.2.1 pH sensitive inorganic NPs
Inorganic NPs have received increasing attention in cancer treatment due to their feasible and finely tuneable synthesis, as well as ease of functionalization. Acid soluble inorganic NPs such as calcium carbonate (CaCO3),199–201 calcium phosphate (CaP)202, 203 and manganese dioxide (MnO2),188, 189 have emerged as excellent candidates for pH-responsive cancer therapy. Achilefu group synthesized CaCO3 NPs to modulate the tumour environment.200 CaCO3 can decompose gradually under mild acidic environment (pH ~6.8) into Ca2+ and CO2, and the pH can be modulated by consuming protons at the same time. The animal experiment further confirmed the tumour inhibition effect of these NPs. Furthermore, Liu’s group loaded Mn2+-chelated Ce6 (Ce6(Mn)) and DOX into CaCO3 by a co-precipitation method, and then modified the monodisperse CaCO3 NPs with PEG (Fig. 7A).199 The 100 nm sized Ce6(Mn)@CaCO3-PEG gradually decomposed at pH 6.5 and dissociated rapidly at pH 5.5. As the Ce6 (Mn) was released from the CaCO3, the Mn2+ T1-weighted magnetic resonance (MR) signal significantly enhanced (Fig. 8C). These NPs were successfully used for imaging guided therapy. CaP is another pH responsive biomaterial widely used for siRNA and drug delivery.203–205 Compared with CaCO3, CaP is nontoxic, biocompatible and degradable in the early lysosome.203–205 However, the highly charged CaP surface is easy to bind with proteins, which shortens the blood circulation life of CaP. MnO2 is also pH sensitive. It can react with H2O2 to generate oxygen and relieve tumour hypoxia. The detail has been discussed in Section 4.1.1.
Fig 8.

(A) Schemes showing the synthesis and structure of Ce6(Mn)@CaCO3-PEG NPs. (B) TEM images of Ce6(Mn)@CaCO3-PEG immersed with different pH values (5.5, 7.4 and 6.5) PBS buffers after various times. (C) T1-weighted MR images of Ce6(Mn)@CaCO3-PEG with different concentrations and different pH values after 4 h incubation. (Reprinted with permission from ref. 199. Copyright 2016, Elsevier Ltd.)
4.2.2 pH sensitive polymer
Various pH-sensitive polymers have been developed in the last few years for tumour targeted delivery. Most polymers are sensitive to relatively low acidic condition (pH 4.5–5.5) and only can respond in endo-/lysosome pH. Polymers which can be triggered by tumour extracellular environment (pH 6.5–7.0) are highly desired.195, 206–208 2-Propionic-3-methylmaleic anhydride (CDM) is a representative linker, which can be cleaved at a mild acidic environment (pH~6.8). Wang’s group conjugated platinum (Pt) prodrug onto poly(amidoamine) (PAMAM) via ester bond, and then used CDM to connect (PAMAM/Pt) and polycaprolactone (PCL).209 By assembling PCL-CDM-PAMAM/Pt, PEG-b-PCL and PCL, they constructed iCluster/Pt NPs with a diameter of around 100 nm (Fig. 9A). The iCluster/Pt NPs are stable in physiological environment and can be shattered to small PAMAM/Pt dendrimers in the acidic tumour microenvironment by cleaving DCM (Fig. 9B–D). Once the dendrimers enter the tumour cells, the Pt drug can be rapidly released in the redox environment. To monitor the penetration of these NPs in acidic tumour microenvironment, the PCL block of the hydrophobic core was labelled with rhodamine B (RhB, red) and PAMAM was labelled with fluorescein (Flu, green). As shown in Fig. 9E, there was nearly no green fluorescence detected in the internal area even after 24 h incubation with cluster/Pt NPs without CDM. However, for iCluster/Pt NPs, green signals can be clearly seen both in the internal and edge of spheroids after 24 h incubation. Furthermore, the iCluster/Pt NPs significantly delayed tumour growth and prolonged the median survival time of A549R cisplatin-resistant human lung tumour model in mice (Fig. 9F). Wang’s group also extended the pH sensitive assembly based on CDM for siRNA and docetaxel delivery.208, 210
Fig 9.

(A) Chemical structure of PCL-CDM-PAMAM/Pt and the mechanism of the self-assembly Pt-containing pH-instable clustered (iCluster/Pt) NPs in response to tumour environment. (B) Size distribution of iCluster/Pt measured by DLS. (C) TEM images of iCluster/Pt and Cluster/Pt immersed with PBS at pH 6.8 for various amounts of time. (D) In vitro release profiles of PAMAM (green line) and platinum drug (red line) from iCluster. (E) Multicellular spheroid model of BxPC-3 cells to confirm the penetration of RhBiClusterFlu and RhBClusterFlu at pH 6.8 after 4 h or 24 h incubation (Scale bar = 200 μm). (F) Inhibition of A549R cisplatin-resistant human lung cancer model by iCluster/Pt. Mice were i.v. administered an equivalent platinum dose of 1.5 mg/kg on days 0, 3, and 6. (G) Kaplan-Meier plots of the animal survival in 4T1 tumour mice (n = 10). Mice were treated at a platinum dose of 3 mg/kg via i.v. administration on days 10, 15, and 20 after tumor inoculation. (Reprinted with permission from ref. 209 with permission, Copyright 2016, National Academy of Sciences.)
Reversible protonation/deprotonation systems have also been exploited for the design of pH sensitive polymers.211 Zwitterionic-to-cationic charge conversion blocks, such as acylsulfonamide,212 carboxybetaine213 and phosphorylcholine,214 are excellent candidates. For example, Mizuhara et al. developed a pH-responsive zwitterionic ligand based on the alkoxyphenyl acylsulfonamide, which can keep neutrality under physiological condition and become positively charged at tumour microenvironment (pH < 6.5) with concomitant enhancement of cellular uptake.212 L-Histidine (His), an amino acid with a pKb of 6.5, is also a representative protonation/deprotonation ligand to respond to mild acidic tumour pH.215, 216 Bae’s group prepared a micelle from polyHis-b-PEG and poly(L-lactic acid) (pLLA)-b-PEG-b-polyHis-biotin.216 DOX was encapsulated in the micelle. As the pH decreased to below 7.0, most biotin molecules were exposed on the surface due to the ionization of polyHis and could interact with cells. When the pH was lower than 6.5, the micelles destabilized, resulting in enhanced drug release. They further conjugated polyHis with TAT peptide or PEI for cancer treatment.217, 218 Recently, an ultrasensitive pH-responsive fluorescent micellar structure containing tertiary amine substituents as pH detector has been reported by Gao’s group.211, 219 These NPs can rapidly respond to difference in less than 0.25 pH units in the tumour extracellular microenvironment.
4.2.3 pH Low Insertion Peptide (pHLIP)
pH low insertion peptide (pHLIP) is 36-aa peptide which can bind and insert across cell membrane to form an alpha-helix at low pH.220 The molecular mechanism of a pHLIP peptide is that the protonation of pHLIP’s residues such as Asp and Glu at acidic pH can increase peptide hydrophobicity and trigger peptide folding to insert into the cell membrane. The N terminus of pHLIP stays outside the cell membrane, and the C terminus can go inside a cell.221 Moreover, NPs can be modified easily by conjugating to the C terminus.222 Due to the low pH in the tumour microenvironment, pHLIP can be employed as a targeting moiety to distinguish acidic tumour and normal tissues. Yao et al. investigated tumour targeting ability of pHLIP modified gold NPs.223 They compared the binding of Au-pHLIP, Au-K-pHLIP (Asp residues replaced by Lys), and Au NPs with HeLa-GFP cells at pH 7.4 and 6.5. As shown in Fig.10, cells had higher uptake of Au-pHLIP NPs at pH 6.5 and that at pH 7.4, while the two formulas were pH insensitive. Tests in vivo in HeLa xenograft model further confirmed that pHLIP is a promising tumour acidic pH targeting peptide to enhance tumour accumulation of NPs.
Fig 10.

(A–F) Cell uptake of gold NPs and gold-pHLIP NPs at different pH values (7.4 vs. 6.5). (G–H) Biodistribution of gold NPs, gold-pHLIP NPs and gold-K-pHIP NPs with intratumoral injection (G) and intravenous injection (H). (Reprinted with permission from ref.223 with permission, Copyright 2013, National Academy of Sciences.)
4.3 Immune responses
Immune responses play important roles in our body. Immunotherapy is a broad category of anti-cancer therapies that use the body’s immune system for cancer treatment, which could enhance specific and durable anticancer responses.224 For effective therapy, immune system should be activated first, the effector cells are expanded and infiltrated to the tumour tissue. Finally, the tumour cells are destroyed.225 However, the cancerous cells can escape these immune responses, and the tumour microenvironment can remarkably dampen these processes.52 NPs are good candidates to induce antitumor immune responses for immunotherapy. Once NPs are delivered into tumour tissue, they have the ability to modulate the immunosuppressed tumour microenvironment and activate immune system. Delivery of immunostimulatory drugs to antitumor immune cells is another strategy for cancer therapy. Antigens are the molecules that can induce an immune response on the part of the host organism with high degree of specificity.226 NPs can be employed as vehicles to deliver antigens to trigger antitumor immune responses. Compared with conventional chemotherapies, immunotherapy will only need low doses of antigens to activate immune responses, and decrease the side effect of anticancer drugs.
One key regulator of immune dysregulation in the tumour microenvironment is signal transducer and activator of transcription 3 (STAT3), which has key roles in vertebrate development and mature tissue function including control of inflammation and immunity.227 STAT3 can be activated by phosphorylation of its tyrosine and serine residues via signalling from upstream regulators.227 STAT3 can mediate immune suppression by increasing the expression of MMPs, inducing suppressive cytokine secretion, and reducing the production of pro-inflammatory cytokines in tumour.228–230 Liao et al. developed ligand-targeted 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)-PEG NPs encapsulating a hydrophobic small molecule STAT3 inhibitor.231 The NPs showed an effective therapeutic inhibition of STAT3 activation in primary tumour. Alshamsan et al. prepared stearic acid modified PEI (PEI-StA) NPs to deliver siRNA for efficient STAT3 downregulation in B16 melanoma cells.232 Compared to the PEI complexes, the PEI-StA complexes showed higher potency in STAT3 silencing in B16 cells accompanied by a significant induction of IL-6 secretion and a reduction of VEGFR production. In vivo results indicated significant regression in tumour growth after siRNA/PEI-StA treatment as compared to the siRNA/PEI.
NPs can also be used as therapeutic cancer vaccines to treat existing cancer. The ability to overcome relevant tissue barriers and efficiently deliver therapeutic cancer vaccines to particular tissue destinations is the key for designing tumour vaccine delivery systems. Lymph nodes, the major sites of antigen-presenting cells (APCs), can be employed as the target site for vaccine delivery. DCs can induce antigen-specific cytotoxic T cells, which elicit an effective anti-tumour response.233 In order to realize effective vaccine delivery to lymph nodes, the vehicle should be taken up into lymphatic vessels and retained in draining lymph nodes. The size of vehicle is quite critical and should be less than 100 nm to improve the vehicle uptake into lymphatic vessels.234 Zhang’s group designed a nanovaccine that delivered 30 nm α-Ap-FNP to mature DCs directly.235 Furthermore, an adjuvant, a pharmacological or immunological agent, can enhance the efficacy of a vaccine. NPs carrying both tumour antigens and adjuvants can be designed to co-deliver vaccine components.236
Multifunctional hybrid NPs integrating different immune effectors have also been designed to overcome the immunoinhibitory nature of the tumour microenvironment and promote immunotherapy. Fahmy’s group encapsulated hydrophobic small molecule inhibitor of the immune suppressive cytokine transform growth factor-β (TGF-β) and the T cell mitogenic cytokine interleukin-2 (IL-2) into nanoscale liposomal polymeric gels (nLGs, Fig. 11A).237 The polymer can release TGF-β inhibitor (SB505124) and IL-2 to the tumour microenvironment, causing significant reduction of tumour growth. (Fig. 11B and 11C). In addition, they used dual-labelled nLGs formulated by incorporating fluorescein-labelled PEG-phosphoethanolamine into the lipid membrane of rhodamine-loaded nLGs to assess trafficking of the particles versus trafficking of the payload. Results demonstrated that both vehicle and payload accumulated in subcutaneous tumours and lung metastases (Fig. 11D). Therefore, this system can deliver both hydrophilic and hydrophobic immunomodulators to enhance anti-tumour activity against subcutaneous and metastatic melanomas. Huang’s group used lipid-calcium-phosphate (LCP) NPs to induce antigen-specific immune response and liposome-protamine-hyaluronic acid (LPH) NPs to deliver siRNA.238 The delivery of siRNA using LPH NPs resulted in efficient knockdown of TGF-β in the late stage tumour microenvironment. TGF-β down-regulation boosted the vaccine efficacy and inhibited tumour growth more than vaccine treatment alone.
Fig 11.

(A) The synthesis and structure of NPs by entrapment of the drug-loaded CD (blue) and the IL-2 (green) in the polymer matrix (red). (B) Plot of B16 melanoma tumour area vs. time after intratumoral injection of different formulas. Tumours in the nLG–SB and nLG–SB + IL-2 groups were significantly smaller when compared against all other groups from day 12 to day 22. (C) Survival rate of mice from the same study given in (B). (D) Analysis of lung tissues under bright field and fluorescent microscopy demonstrate the presence of both lipid carrier (green) and rhodamine payload (red) around individual lung tumours at early time points post injection. (Reprinted with permission from ref. 237. Copyright 2012, Nature Publishing Group.)
4.4 Tumour pathological pressure gradients
The tumour pathological pressure gradient plays an integral role to control intratumoral delivery of NPs. Pressure gradients are established in the body for the flow of interstitial fluid to exchange nutrient, oxygen and wastes between blood and cells.239 In the normal tissue, there is a balance between tissue pressure and structure to allow cell growth and realize tissue functions. By contrast, the tissue pressure is increased in the abnormal tumour region.240 Young et al. first investigated tumour tissue pressure in 1950s and found that the pressure of malignant tumours is always higher than that of normal tissues.241 The increased tumour pressure is due to the higher density of tumour cells, higher concentration of ECM, blood vessel leakiness and lymph-vessel abnormalities.240 The high tumour pressure is an obstacle to inefficient uptake of therapeutic agents for cancer treatment. Modulation of intratumoral pressure gradients is a promising strategy to enhance extravasation and therapeutic efficacy of NPs. Angiotensin II is a peptide hormone which can increase blood pressure and cause vasoconstriction. Suzuki et al. reported that infusion of Angiotensin II can result in 5.7-fold selective increase of blood flow in tumour and that in normal tissue was virtually unchanged.242 Normalization of tumour pressure gradients using VEGF, platelet-derived growth factor (PDGF) or transforming growth factor-β (TGFβ) inhibitors is another approach. VEGF inhibitor can improve blood flow by normalizing blood vessels.243 The PDGF inhibitor can decrease contraction of stromal fibroblasts with ECM.244 TGFβ inhibitor can decrease the extracellular-matrix molecules.245 Furthermore, some drugs can improve blood flow and decrease microvascular pressure, such as nicotinamide, dexamethasone and bradykinin agonists.246–248
4.5 Tumour extracellular matrix (ECM)
The extracellular matrix functions as a scaffold for tissue morphogenesis. ECM is composed of highly interconnected collagen fibres and associated large glycoproteins, proteoglycans as well as various proteins that regulate tissue homeostasis, organ development and tissue lesion.249 The components of tumour ECM are determinants for the growth and cell migration of solid tumours.250 Due to the presence of high collagen turnover, increased level of lysyl oxidase (LOX), and enhanced integrin receptors, solid tumours have the dense extracellular matrix for the transmission of extracellular signals to the cells, which increase solid stress inside tumours, and compress tumour blood vessels to reduce tumour perfusion.251–253 Furthermore, the thick tumour ECM also prevents the penetration of NPs, reducing tumour treatment efficacy. Modulation of tumour ECM provides an alternative strategy for enhancing cancer therapy.
One approach is the use of enzymes, such as hyaluronidase (HAase) and collagenase, to degrade the matrix structure.254 Liu’s group modulated the tumour microenvironment by administration of HAase, which breaks down hyaluronan, a major component of ECM in tumours (Fig. 12A).255 The C18PMH-PEG-Ce6 nanomicelles were prepared and co-injected with HAase-PEG into mice for cancer treatment. Both the tumour vascular density and effective vasculature area were increased after HAase administration, inducing enhanced perfusion inside the tumour. After treatment with HAase, the hypoxia stained signals became obviously lower within the whole tumour compared to the control group (Fig. 12B), and the tumour growth was almost completely inhibited (Fig. 12C). Cheng’s group prepared the recombinant human hyaluronidase PH20 (rHuPH20) modified PLGA-PEG NPs.256 The rHuPH20 can degrade hyaluronic acid for enhanced NPs penetration in tumour (Fig. 12E). The DOX encapsulated NPs can efficiently inhibit tumour growth and promote survival rate (Fig. 12F). Fluorescence imaging proved that HA on the diffusion path of NPs were degraded while the signal of HA maintained in the normal tissue. The HPEG-PH20-NP signal can be clearly seen around blood vessels in tumour (Fig. 12G).
Fig 12.

(A) Scheme showing the mechanism to modulate tumour microenvironment by HAase. (B) Tumour slices immunofluorescence imaging by treatment with 1500 U HAase. Red: Blood vessels; green: hypoxic regions; blue: nuclei. (C) Tumour inhibition under various treatments with saline, HAase alone, PDT alone, and PDT plus HAase at days 0 and 6. PDT effect is obviously enhanced by administration of HAase. (D) Ex vivo tumour pictures of the same study described in (C). (E) Scheme of synthesizing hyaluronidase-modified nanocarrier by conjugating thiolated rHuPH20 on the first PEG layer followed by anchoring the second PEG layer. (F) Tumour growth inhibition curves and survival rate plots for 4T1 tumor-bearing mice treated with either saline, free DOX, DOX-HPEG or DOX-HPEG-PH20 NPs at 2 mg/kg equivalent dose of DOX. (G) Staining of sectioned tumour tissues collected 24 h post-injection of saline or various NP formulas. Scale bar: 50 μm (left); 200 μm (middle); 100 μm (right). ((Fig.12 A–D was reprinted with permission from ref. 255. Copyright 2016, American Chemical Society. Fig.12 E–G was reprinted with permission from ref. 256. Copyright 2016, American Chemical Society.)
Cyclopamine, a naturally occurring steroidal jerveratrum alkaloid, can inhibit the hedgehog signalling pathway (Hh) by acting on the SMO receptor.257 Zhang et al. reported that cyclopamine can disrupt tumour extracellular fibronectins, decompress tumour blood vessels and improve tumour perfusion. The cyclopamine and paclitaxel encapsulated PEG-PLA NPs could change extracellular matrix deposition, improve pancreatic cancer tumour perfusion and achieve significant tumour growth inhibition. Oligosaccharides of hyaluronan (oHA) can also disrupt the HA matrix by replacing HA for the binding on CD44.258, 259 Gao’s group developed oHA-lipid-PTX NPs, which can efficiently disrupt the tumour HA matrix and promote the drug delivery into the cells.260
Bromelain can cleave the synthetic peptide sequence Bz-Arg-Arg-p-nitroanilide and digest extracellular matrix.261, 262 Tasciotti’s group modified MSN with bromelain and found an enhanced diffusion of MSN in tumour extracellular matrix.263 Collagenase is another enzyme that improves extracellular matrix penetration depth by breaking the peptide bonds in collagen.264, 265 Goodman et al. developed polystyrene NPs immobilized with collagenase.266 The collagenase treatment of spheroids resulted in significantly increased penetration of polystyrene NPs. Collagenase-functionalized superparamagnetic NPs synthesized by Giorgio group also demonstrated the ability to degrade the extracellular matrix for enhanced interstitial mobility.267 Recently, Vallet-Regí group designed hybrid MSN NPs attached with collagenase-polymeric nanocapsules to improve their tumour penetration.268 These polymeric nanocapsules protect the collagenase against proteolytic degradation and hydrolysis during circulation in bloodstream while allowing collagenase release at tumour pH to enhance tumour matrix degradation.
MMPs represent the most prominent family of proteinases associated with tumourigenesis.269 MMPs-mediated tumour extracellular matrix degradation leads to cancer cell invasion and metastasis.269 MMP-2 and MMP-9 are the most studied MMPs and are upregulated during progression of various tumour types, including prostate, stomach, colorectal, breast, lung and ovarian.270 Moreover, MMPs promote invasion, growth, the survival of malignant cells and metastasis to other organs.271 Therefore, direct inhibition of MMP activity is an ideal therapeutic strategy. Vyavahare group prepared MMP-inhibitor batimastat loaded poly(D,L-lactide) NPs and then conjugated with an antielastin antibody to prevent aneurysmal growth.272 Xiao and Wang developed a poly(lactic-co-glycolic acid) NPs loaded with batimastat to promote cancer chemotherapy.273 Additionally, MMPs can act a novel target for tumour therapy.274 MMP can selectively recognize and cleave MMP responsive peptides. Chen and Wang’s group coated 1,2-distearoyl-snglycero-3-phosphoethanolamine-polyethyleneglycol (DSPE-PEG) nanovesicles with a thin polymeric shell of MMP-2 degradable polymeric peptides Gly-Pro-Leu-Gly-Val-Arg-Gly-Lys (GPLGVRGK,),275 and then conjugated with a cyclic RGD peptide to promote tumour targeting. Irinotecan hydrochloride was encapsulated into the nanovesicle for cancer therapy. The GPLGVRGK peptides were degraded by MMP-2 in the tumour microenvironment. ITC can be released fast (Fig. 13A). Compared with ITC⊂N, the fluorescence intensity of ITC⊂N-G-C from the centre of the MCs is higher (Fig. 13B). This indicated the N-G-C can enhance penetration through the spheroids. The in vivo tumour suppression studies were further carried out. The ITC⊂N-G-C had better tumour suppression efficiency compared with all the other groups (Fig. 13C). The therapeutic efficacy achieved with ITC⊂N-G-C can be attributed to the synergistic contribution from improved tumour accumulation, penetration, and MMPs-responsive drug release in the tumour microenvironment. Similar designs have been applied to other nanovehicles.276–279 Li’s group developed a tumour microenvironment-adaptive hybrid micelle constructed by two amphiphilic polymers, polyethyleneimine (PEI)-block-poly[(1,4-butanediol)-diacrylate-β-5-hydroxyamyl -amine] (PDHA) (PEIPDHA) and PEG-block-PDHA (PEG-PDHA) co-loading PTX and the anti-metastasis siRNA.277 The NPs consists of a pH-sensitive core (PDHA), a cationic shell (PEI), and a matrix MMP-cleavable PEG conjugated via a peptide linker. PEG will be cut away in the tumour microenvironment and the NPs will change from neutral to positive charge, which promotes NPs internalization into tumour cells.
Fig 13.

(A) The mechanism of MMPs-responsive nanovesicles in the tumour microenvironment. Nrp-1 receptor targeting mediates tumour penetration of ITC⊂N-G-C. MMP-2 enzyme cleaves the cross-linker causes disassembly of the polymer network and the exposure of ITC⊂N-C, which enters cells through Nrp-1 mediated endocytosis. (B) Images of HT-29 multicellular spheroids (MCs). Scale bar = 50 μm. (C, D) Confocal microscopy images of HT-29 MCs incubated with Cy5-labeled ITC⊂N (C) and ITC⊂N-G-C (D) for 8 h. (E) Tumour inhibition under various treatments (N = 6). *P < 0.05. **P < 0.01. (G) Tumour volume changes of the six treatment groups (ITC⊂N-G-C, ITC⊂N-G, ITC⊂N-C, ITC⊂N, free ITC, 7.5 mg/kg equivalent of irinotecan and PBS) over the course of the treatments. (Reprinted with permission from ref. 275. Copyright 2015, John Wiley & Sons, Inc.)
Some lipases are also upregulated in the tumour microenvironment and can be employed for NPs-based activation.280, 281 Phospholipase A2 (PLA2) is a lipase overexpressed in the tumour extracellular matrix. This enzyme catalyses the hydrolysis of the ester bond of the SN-2 acyl chain of phospholipids, leading to the generation of free fatty acids and lysophospholipids.282, 283 This property can be used to create liposome-based drug delivery system that can be triggered by PLA2 at the tumour site. Gu’s group reported a tumour microenvironment responsive and transformable nanocarrier for cell membrane targeted delivery of cytokine (Fig. 14).284 In their system, PLA2 degradable liposome was employed as a shell to protect complementary DNA nanostructures (designated as nanoclews) decorated with cytokines. After PLA2 activation, the cytokine loaded DNA nanoclews were transformed into nanofibers for enhanced anticancer efficacy.
Fig 14.

(A) Schematic of phospholipase activated membrane targeted cytokine delivery system. (a) Preparation of TRAIL-NC-L. The DNA nanoclews were first prepared by rolling circle amplification (RCA) and then modified with Ni2+. After loading TRAIL protein through Ni2+-His tag affinity, the TRAIL-NC was encapsulated into a POPC liposome that could be degraded by phospholipase A2 (PLA2). (B) PLA2 in the tumour microenvironment degrades the liposome shell to release TRAIL-NC and complementary DNA NCs hybridize into microscopic fibres. TRAIL loaded spherical NPs are efficiently internalized, while hybridized DNA fibres are highly impermeable to cell membrane, facilitating the interaction of TRAIL and death receptors. (Reprinted with permission from ref. 284. Copyright 2016, Elsevier Ltd.)
5 Conclusion and perspective
This review aims to present a survey on the design of therapeutic NPs by exploiting tumour microenvironment. NPs offer many potential benefits that are now starting to be utilized in the clinic. Tumour microenvironment provides a new strategy for cancer treatment. Therefore, a deeper understanding of the realistic conditions and interactions involved in the tumour microenvironment play a significant role to develop smart NPs. Even though the tumour microenvironment has been investigated for many years, how to design NPs to make use of tumour microenvironment is still in its infancy, and there are many challenges for the design of effective therapeutic NPs for clinical cancer therapy.
First, high tumour accumulation of NPs is the key for effective cancer treatment. According to Chan’s recent review, most of the reported NPs formulas have very poor delivery efficiency to solid tumour.31 Therefore, how to reduce clearance by phagocytic blood cells during blood circulation process and increase tumour accumulation is still one of the biggest hurdles for the effective use of nanomaterials for cancer treatment. PEG and zwitterionic materials are the promising candidates to decrease the blood clearance. More recently, the cell membrane has been exploited to coat various NPs.285 This strategy provides new inspirations for enhancing blood circulation time. The development of new materials to significantly increase tumour accumulation will be of great importance. The EPR effect is another important factor for tumour accumulation due to the abnormal vascular architecture in the tumour site. The NPs with size between 20–200 nm can extravasate and accumulate in the interstitial space. However, each tumour type has its specific endothelial pore size, no single NPs size can fit all types of tumours. EPR effect also depends on the degree of tumour vascularization and angiogenesis.88 In order to further improve tumour accumulation, active targeting has been developed. The active targeting strategy also faces many challenges. Some studies showed only a modest increase in tumour accumulation over the control NPs without targeting molecules. Multi-targeting NPs system that consists of two or more targeting stages or stimuli-responsive targeting macromolecules represents a promising targeting strategy.
Second, compared with normal tissues, the solid tumour has a unique microenvironment including vascular abnormalities, hypoxia, low pH, dense tumour ECM, which has been studied to design new therapeutic NPs. Modulation and exploitation of the tumour microenvironment by NPs has been demonstrated. As cancer is a complex disease, a strategy that combines multidimensional treatment modalities together has obvious advantages over a single treatment approach. For instance, a single NPs drug delivery system can be designed to modulate the tumour pH and promote oxygen concentration at the same time. It is also possible to develop drug delivery systems with two targeting strategies and tumour pH sensitive drug release together. It is interesting that some multifunctional drug delivery systems can hold big size to enhance tumour vascular extravasation by EPR effect and discharge small NPs to improve tumour penetration by tumour microenvironment.209, 286 Thus, development of smart nanomaterials that utilize the unique tumour properties could lead to the significant progress of the NPs based cancer therapy.
Third, a deeper understanding of the tumour microenvironment and the tumour cell physiology are keys for better design of new smart and effective cancer nanotherapeutics. For example, low interstitial diffusion in solid tumours is one of the main challenges because of the dense structure of extracellular matrix. Therefore, degradation of extracellular matrix by NPs is promising to improve tumour treatment efficacy. Nowadays, radiation therapy is a critical component in cancer therapy. The mechanism is that ionizing radiation can generate oxygen-centred radicals to induce cell DNA damage.167 The hypoxic tumour environment limits the application of radiotherapy. The NPs that deliver oxygen to tumour site can improve the therapeutic outcomes of radiotherapy in cancer treatment. Based on numerous different properties of solid tumour compared with normal tissue, how to further exploit the tumour microenvironment and interaction between different kinds of cells by NPs in the tumour is still a challenge.
The ultimate goal of developing nanomedicine is clinical translation. Although numerous murine models including human xenograft models and genetically engineered mouse models have been developed to study human cancer, human cancer is different from murine models in many aspects, such as the size relative to host, stromal cells in tumour, metabolic rates and pharmacokinetic properties.287–289 Most rodent tumours grow much faster than human tumour. A mouse tumour is often grown up to 10% of the mouse body weight, which is as big as a basketball of an equivalent tumour in a 70 kg human.290 Actually, the size of human tumour is much smaller, ranging from a few millimeters to centimeters, at ime of diagnosis and treatment.290 Therefore, there are many differences between human cancer and murine models in tumour microenvironment. Athymic nude mice also lack a functional immune system and the stromal cells including cancer-associated fibroblasts, endothelial cells, and immune and inflammatory cells belong to murine cells.291 Therefore, xenograft tumour models using human cell lines to test drug responses often do not recapitulate the clinical reality in patients.289 Despite all these shortcomings, human xenograft model remains an useful tool for improving our understanding of cancer development and treatment. Further understanding of the tumour microenvironment in human cancers is critical to translate nanomedicine into the clinic.
In conclusion, this review discussed the exploitation of tumour microenvironment by NPs for cancer therapy. The use of tumour microenvironment-sensitive NPs holds promise in cancer therapy. Indeed, great progress has already been made towards this goal. Ideally, a therapeutic NPs system should be able to deliver cargos just to the tumour and be degraded without severe side effects to the body. Further work is still underway to develop new NP systems specific for tumour microenvironment.
Supplementary Material
Acknowledgments
This work is financially supported by in part by the National Key Research and Development Program of China (2016YFA0203600), National Natural Science Foundation of China (81571743, 51502251), Fundamental Research Funds for Xiamen University (20720160067), Science Foundation of Fujian Province (2014Y2004) and National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH).
References
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY. The Lancet. 2013;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen G, Roy I, Yang C, Prasad PN. Chem Rev. 2016;116:2826–2885. doi: 10.1021/acs.chemrev.5b00148. [DOI] [PubMed] [Google Scholar]
- 3.Lu Y, Aimetti AA, Langer R, Gu Z. Nat Rev Mater. 2016;1:16075. [Google Scholar]
- 4.De Crozals G, Bonnet R, Farre C, Chaix C. Nano Today. 2016;11:435–463. [Google Scholar]
- 5.Petros RA, DeSimone JM. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 6.Jain RK, Stylianopoulos T. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Misra R, Acharya S, Sahoo SK. Drug Discov Today. 2010;15:842–850. doi: 10.1016/j.drudis.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Chung C, Kim YK, Shin D, Ryoo SR, Hong BH, Min DH. Acc Chem Res. 2013;46:2211–2224. doi: 10.1021/ar300159f. [DOI] [PubMed] [Google Scholar]
- 9.Dai YL, Xiao HH, Liu JH, Yuan QH, Ma PA, Yang DM, Li CX, Cheng ZY, Hou ZY, Yang PP, Lin J. J Am Chem Soc. 2013;135:18920–18929. doi: 10.1021/ja410028q. [DOI] [PubMed] [Google Scholar]
- 10.Kumar R, Shin WS, Sunwoo K, Kim WY, Koo S, Bhuniya S, Kim JS. Chem Soc Rev. 2015;44:6670–6683. doi: 10.1039/c5cs00224a. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen KT, Zhao YL. Acc Chem Res. 2015;48:3016–3025. doi: 10.1021/acs.accounts.5b00316. [DOI] [PubMed] [Google Scholar]
- 12.Muthu MS, Leong DT, Mei L, Feng SS. Theranostics. 2014;4:660–677. doi: 10.7150/thno.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai K, He X, Song Z, Yin Q, Zhang Y, Uckun FM, Jiang C, Cheng J. J Am Chem Soc. 2015;137:3458–3461. doi: 10.1021/ja513034e. [DOI] [PubMed] [Google Scholar]
- 14.Feng Y, Panwar N, Tng DJH, Tjin SC, Wang K, Yong KT. Coord Chem Rev. 2016;319:86–109. [Google Scholar]
- 15.Janib SM, Moses AS, MacKay JA. Adv Drug Del Rev. 2010;62:1052–1063. doi: 10.1016/j.addr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bardhan R, Lal S, Joshi A, Halas NJ. Acc Chem Res. 2011;44:936–946. doi: 10.1021/ar200023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jokerst JV, Gambhir SS. Acc Chem Res. 2011;44:1050–1060. doi: 10.1021/ar200106e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma X, Zhao Y, Liang XJ. Acc Chem Res. 2011;44:1114–1122. doi: 10.1021/ar2000056. [DOI] [PubMed] [Google Scholar]
- 19.Park K, Lee S, Kang E, Kim K, Choi K, Kwon IC. Adv Funct Mater. 2009;19:1553–1566. [Google Scholar]
- 20.Yang D, Ma P, Hou Z, Cheng Z, Li C, Lin J. Chem Soc Rev. 2015;44:1416–1448. doi: 10.1039/c4cs00155a. [DOI] [PubMed] [Google Scholar]
- 21.Shin TH, Choi Y, Kim S, Cheon J. Chem Soc Rev. 2015;44:4501–4516. doi: 10.1039/c4cs00345d. [DOI] [PubMed] [Google Scholar]
- 22.Lee N, Choi SH, Hyeon T. Adv Mater. 2013;25:2641–2660. doi: 10.1002/adma.201300081. [DOI] [PubMed] [Google Scholar]
- 23.Ulbrich K, Hola K, Subr V, Bakandritsos A, Tucek J, Zboril R. Chem Rev. 2016;116:5338–5431. doi: 10.1021/acs.chemrev.5b00589. [DOI] [PubMed] [Google Scholar]
- 24.Tredan O, Galmarini CM, Patel K, Tannock IF. J Natl Cancer Inst. 2007;99:1441–1454. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 25.McKeever AE, Bloch JR, Bratic A. Clin J Oncol Nurs. 2013;17:490–495. doi: 10.1188/13.CJON.490-495. [DOI] [PubMed] [Google Scholar]
- 26.Torchilin V. Adv Drug Del Rev. 2011;63:131–135. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Greish K. In: Cancer Nanotechnology: Methods and Protocols. Grobmyer RS, Moudgil MB, editors. Humana Press; Totowa, NJ: 2010. pp. 25–37. [DOI] [Google Scholar]
- 28.Maeda H. Bioconj Chem. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 29.Maeda H, Nakamura H, Fang J. Adv Drug Del Rev. 2013;65:71–79. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Brannon-Peppas L, Blanchette JO. Adv Drug Del Rev. 2012;64(Supplement):206–212. [Google Scholar]
- 31.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Nat Rev Mater. 2016;1:16014. [Google Scholar]
- 32.Mbeunkui F, Johann DJ., Jr Cancer Chemother Pharmacol. 2009;63:571–582. doi: 10.1007/s00280-008-0881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balkwill FR, Capasso M, Hagemann T. J Cell Sci. 2012;125:5591–5596. doi: 10.1242/jcs.116392. [DOI] [PubMed] [Google Scholar]
- 34.Fukumura D, Jain RK. J Cell Biochem. 2007;101:937–949. doi: 10.1002/jcb.21187. [DOI] [PubMed] [Google Scholar]
- 35.Albini A, Sporn MB. Nat Rev Cancer. 2007;7:139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 36.Banchereau J, Palucka AK. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 37.Noy R, Pollard JW. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khawar IA, Kim JH, Kuh HJ. J Control Release. 2015;201:78–89. doi: 10.1016/j.jconrel.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 39.Junttila MR, de Sauvage FJ. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 40.Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Cancer Metastasis Rev. 2013;32:303–315. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noguera R, Nieto OA, Tadeo I, Fariñas F, Alvaro T. Histol Histopathol. 2012;27:693–705. doi: 10.14670/HH-27.693. [DOI] [PubMed] [Google Scholar]
- 42.Lu P, Weaver VM, Werb Z. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mouw JK, Ou G, Weaver VM. Nat Rev Mol Cell Biol. 2014;15:771–785. doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hida K, Ohga N, Akiyama K, Maishi N, Hida Y. Cancer Sci. 2013;104:1391–1395. doi: 10.1111/cas.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cirri P, Chiarugi P. Cancer Metastasis Rev. 2012;31:195–208. doi: 10.1007/s10555-011-9340-x. [DOI] [PubMed] [Google Scholar]
- 46.Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancers (Basel) 2015;7:2443–2458. doi: 10.3390/cancers7040902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Mol Cancer Res. 2012;10:1403–1418. doi: 10.1158/1541-7786.MCR-12-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Torsvik A, Bjerkvig R. Cancer Treat Rev. 2013;39:180–188. doi: 10.1016/j.ctrv.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 49.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 50.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pagès F. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 51.Hinrichs CS, Rosenberg SA. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker JC, Andersen MH, Schrama D, thor Straten P. Cancer Immunol, Immunother. 2013;62:1137–1148. doi: 10.1007/s00262-013-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silva JM, Videira M, Gaspar R, Préat V, Florindo HF. J Control Release. 2013;168:179–199. doi: 10.1016/j.jconrel.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 54.Gattinoni L, Powell DJ, Rosenberg SA, Restifo NP. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pei YH, Yeo Y. J Control Release. 2016;240:202–211. doi: 10.1016/j.jconrel.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 56.Cook J, Hagemann T. Curr Opin Pharm. 2013;13:595–601. doi: 10.1016/j.coph.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 57.Qian BZ, Pollard JW. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Palucka K, Banchereau J. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 60.Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, Perricaudet M, Tursz T, Maraskovsky E, Zitvogel L. Nat Med. 1999;5:405–411. doi: 10.1038/7403. [DOI] [PubMed] [Google Scholar]
- 61.Kadowaki N, Antonenko S, Ho S, Rissoan MC, Soumelis V, Porcelli SA, Lanier LL, Liu YJ. J Exp Med. 2001;193:1221–1226. doi: 10.1084/jem.193.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whiteside TL. The Link Between Inflammation and Cancer. Springer; 2006. pp. 103–124. [Google Scholar]
- 63.Kitamura T, Qian BZ, Pollard JW. Nat Rev Immunol. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsou P, Katayama H, Ostrin EJ, Hanash SM. Cancer Res. 2016;76:5597–5601. doi: 10.1158/0008-5472.CAN-16-0431. [DOI] [PubMed] [Google Scholar]
- 65.Scott DW, Gascoyne RD. Nat Rev Cancer. 2014;14:517–534. doi: 10.1038/nrc3774. [DOI] [PubMed] [Google Scholar]
- 66.Gunderson AJ, Coussens LM. Exp Cell Res. 2013;319:1644–1649. doi: 10.1016/j.yexcr.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Visser KE, Korets LV, Coussens LM. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 68.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. Trends Immunol. 37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Talmadge JE, Gabrilovich DI. Nat Rev Cancer. 2013;13:739–U779. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Katoh H, Watanabe M. Mediators Inflamm. 2015;2015 doi: 10.1155/2015/159269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hida K, Maishi N, Sakurai Y, Hida Y, Harashima H. Adv Drug Del Rev. 2016;99:140–147. doi: 10.1016/j.addr.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 72.Arap W, Pasqualini R, Ruoslahti E. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 73.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smyth MJ, Godfrey DI. Nat Immunol. 2000;1:459–460. doi: 10.1038/82698. [DOI] [PubMed] [Google Scholar]
- 76.Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. Clin Sci. 2015;128:81–93. doi: 10.1042/CS20140278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Armulik A, Genové G, Betsholtz C. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 78.Keskin D, Kim J, Cooke VG, Wu CC, Sugimoto H, Gu C, De Palma M, Kalluri R, LeBleu VS. Cell Rep. 2015;10:1066–1081. doi: 10.1016/j.celrep.2015.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu CH, Shahzad MMK, Moreno-Smith M, Lin YG, Jennings NB, Allen JK, Landen CN, Mangala LS, Armaiz-Pena GN, Schmandt R, Nick AM, Stone RL, Jaffe RB, Coleman RL, Sood AK. Cancer Biol Ther. 2010;9:176–182. doi: 10.4161/cbt.9.3.10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yonenaga Y, Mori A, Onodera H, Yasuda S, Oe H, Fujimoto A, Tachibana T, Imamura M. Oncology. 2005;69:159–166. doi: 10.1159/000087840. [DOI] [PubMed] [Google Scholar]
- 81.Dumitru CA, Lang S, Brandau S. Semin Cancer Biol. 2013;23:141–148. doi: 10.1016/j.semcancer.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 82.Dumitru CA, Gholaman H, Trellakis S, Bruderek K, Dominas N, Gu X, Bankfalvi A, Whiteside TL, Lang S, Brandau S. Int J Cancer. 2011;129:859–869. doi: 10.1002/ijc.25991. [DOI] [PubMed] [Google Scholar]
- 83.Weitzman SA, Stossel TP. Cancer Lett. 1984;22:337–341. doi: 10.1016/0304-3835(84)90172-1. [DOI] [PubMed] [Google Scholar]
- 84.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Annu Rev Immunol. 2012;30:459–489. doi: 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- 85.Baluk P, Hashizume H, McDonald DM. Curr Opin Genet Dev. 2005;15:102–111. doi: 10.1016/j.gde.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 86.Nagy JA, Dvorak HF. Clin Exp Metastasis. 2012;29:657–662. doi: 10.1007/s10585-012-9500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cerqueira BB, Lasham A, Shelling AN, Al-Kassas R. Eur J Pharm Biopharm. 2015;97:140–151. doi: 10.1016/j.ejpb.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 88.Danhier F, Feron O, Preat V. J Control Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 89.Torchilin VP. In: Drug Deliv. Schäfer-Korting M, editor. Springer Berlin Heidelberg; Berlin, Heidelberg: 2010. pp. 3–53. [DOI] [Google Scholar]
- 90.Danquah MK, Zhang XA, Mahato RI. Adv Drug Deliv Rev. 2011;63:623–639. doi: 10.1016/j.addr.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 91.Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, Bawendi MG, Fukumura D, Jain RK. Nat Nanotechnol. 2012;7:383–388. doi: 10.1038/nnano.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meng H, Xue M, Xia T, Ji Z, Tarn DY, Zink JI, Nel AE. ACS Nano. 2011;5:4131–4144. doi: 10.1021/nn200809t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Euliss LE, DuPont JA, Gratton S, DeSimone J. Chem Soc Rev. 2006;35:1095–1104. doi: 10.1039/b600913c. [DOI] [PubMed] [Google Scholar]
- 94.Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, Ferrari M. J Control Release. 2010;141:320–327. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 95.Kobayashi H, Watanabe R, Choyke PL. Theranostics. 2014;4:81–89. doi: 10.7150/thno.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fu CH, Liu TL, Li LL, Liu HY, Chen D, Tang FQ. Biomaterials. 2013;34:2565–2575. doi: 10.1016/j.biomaterials.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 97.Kim HL, Lee SB, Jeong HJ, Kim DW. RSC Adv. 2014;4:31318–31322. [Google Scholar]
- 98.Champion JA, Katare YK, Mitragotri S. J Control Release. 2007;121:3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang X, Li L, Liu T, Hao N, Liu H, Chen D, Tang F. ACS Nano. 2011;5:5390–5399. doi: 10.1021/nn200365a. [DOI] [PubMed] [Google Scholar]
- 100.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Truong NP, Whittaker MR, Mak CW, Davis TP. Expert Opin Drug Deliv. 2015;12:129–142. doi: 10.1517/17425247.2014.950564. [DOI] [PubMed] [Google Scholar]
- 102.Kumar S, Anselmo AC, Banerjee A, Zakrewsky M, Mitragotri S. J Control Release. 2015;220(Part A):141–148. doi: 10.1016/j.jconrel.2015.09.069. [DOI] [PubMed] [Google Scholar]
- 103.Kolhar P, Anselmo AC, Gupta V, Pant K, Prabhakarpandian B, Ruoslahti E, Mitragotri S. Proc Natl Acad Sci USA. 2013;110:10753–10758. doi: 10.1073/pnas.1308345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Christian DA, Cai S, Garbuzenko OB, Harada T, Zajac AL, Minko T, Discher DE. Mol Pharm. 2009;6:1343–1352. doi: 10.1021/mp900022m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hirn S, Semmler-Behnke M, Schleh C, Wenk A, Lipka J, Schäffler M, Takenaka S, Möller W, Schmid G, Simon U, Kreyling WG. Eur J Pharm Biopharm. 2011;77:407–416. doi: 10.1016/j.ejpb.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.He C, Hu Y, Yin L, Tang C, Yin C. Biomaterials. 2010;31:3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 108.Lowe S, O’Brien-Simpson NM, Connal LA. Polym Chem. 2015;6:198–212. [Google Scholar]
- 109.Garay RP, El-Gewely R, Armstrong JK, Garratty G, Richette P. Expert Opin Drug Deliv. 2012;9:1319–1323. doi: 10.1517/17425247.2012.720969. [DOI] [PubMed] [Google Scholar]
- 110.Abuchowski A, van Es T, Palczuk NC, Davis FF. J Biol Chem. 1977;252:3578–3581. [PubMed] [Google Scholar]
- 111.Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF. J Biol Chem. 1977;252:3582–3586. [PubMed] [Google Scholar]
- 112.Kingshott P, Thissen H, Griesser HJ. Biomaterials. 2002;23:2043–2056. doi: 10.1016/s0142-9612(01)00334-9. [DOI] [PubMed] [Google Scholar]
- 113.Gref R, Lück M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, Müller RH. Colloids Surf B Biointerfaces. 2000;18:301–313. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 114.Jiang S, Cao Z. Adv Mater. 2010;22:920–932. doi: 10.1002/adma.200901407. [DOI] [PubMed] [Google Scholar]
- 115.Liu S, Jiang S. Nano Today. 2016;11:285–291. [Google Scholar]
- 116.Yoshimoto K, Hirase T, Madsen J, Armes SP, Nagasaki Y. Macromol Rapid Commun. 2009;30:2136–2140. doi: 10.1002/marc.200900484. [DOI] [PubMed] [Google Scholar]
- 117.Feng W, Gao X, McClung G, Zhu S, Ishihara K, Brash JL. Acta Biomater. 2011;7:3692–3699. doi: 10.1016/j.actbio.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 118.Chang C-C, Letteri R, Hayward RC, Emrick T. Macromolecules. 2015;48:7843–7850. [Google Scholar]
- 119.Yang W, Liu S, Bai T, Keefe AJ, Zhang L, Ella-Menye J-R, Li Y, Jiang S. Nano Today. 2014;9:10–16. [Google Scholar]
- 120.Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gu L, Mooney DJ. Nat Rev Cancer. 2016;16:56–66. doi: 10.1038/nrc.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gao W, Hu C-MJ, Fang RH, Luk BT, Su J, Zhang L. Adv Mater. 2013;25:3549–3553. doi: 10.1002/adma.201300638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Enzo MV, Isenhart L, Ferrari M, Tasciotti E. Nat Nanotechnol. 2013;8:61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Toledano Furman NE, Lupu-Haber Y, Bronshtein T, Kaneti L, Letko N, Weinstein E, Baruch L, Machluf M. Nano Lett. 2013;13:3248–3255. doi: 10.1021/nl401376w. [DOI] [PubMed] [Google Scholar]
- 125.Palomba R, Parodi A, Evangelopoulos M, Acciardo S, Corbo C, de Rosa E, Yazdi IK, Scaria S, Molinaro R, Furman NET, You J, Ferrari M, Salvatore F, Tasciotti E. Sci Rep. 2016;6:34422. doi: 10.1038/srep34422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sun H, Su J, Meng Q, Yin Q, Chen L, Gu W, Zhang P, Zhang Z, Yu H, Wang S, Li Y. Adv Mater. 2016;28:9581–9588. doi: 10.1002/adma.201602173. [DOI] [PubMed] [Google Scholar]
- 127.Byrne JD, Betancourt T, Brannon-Peppas L. Adv Drug Deliv Rev. 2008;60:1615–1626. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 128.Chung AS, Lee J, Ferrara N. Nat Rev Cancer. 2010;10:505–514. doi: 10.1038/nrc2868. [DOI] [PubMed] [Google Scholar]
- 129.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Adv Drug Deliv Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lammers T, Hennink WE, Storm G. Br J Cancer. 2008;99:392–397. doi: 10.1038/sj.bjc.6604483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bae YH, Park K. J Control Release. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ferrara N. Atertio Thromb Vasc Biol. 2009;29:789–791. doi: 10.1161/ATVBAHA.108.179663. [DOI] [PubMed] [Google Scholar]
- 133.Backer MV, Gaynutdinov TI, Patel V, Bandyopadhyaya AK, Thirumamagal BTS, Tjarks W, Barth RF, Claffey K, Backer JM. Mol Cancer Ther. 2005;4:1423–1429. doi: 10.1158/1535-7163.MCT-05-0161. [DOI] [PubMed] [Google Scholar]
- 134.Xiong J-P, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 135.Nisato RE, Tille J-C, Jonczyk A, Goodman SL, Pepper MS. Angiogenesis. 2003;6:105–119. doi: 10.1023/B:AGEN.0000011801.98187.f2. [DOI] [PubMed] [Google Scholar]
- 136.Schlesinger M, Bendas G. Int J Cancer. 2015;136:2504–2514. doi: 10.1002/ijc.28927. [DOI] [PubMed] [Google Scholar]
- 137.Zhao G, Rodriguez BL. Int J Nanomed. 2013;8:61. doi: 10.2147/IJN.S37859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Senger D, Galli S, Dvorak A, Perruzzi C, Harvey V, Dvorak H. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 139.Goel S, Chen F, Hong H, Valdovinos HF, Hernandez R, Shi SX, Barnhart TE, Cai WB. ACS Appl Mater Interfaces. 2014;6:21677–21685. doi: 10.1021/am506849p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen K, Li Z-B, Wang H, Cai W, Chen X. Eur J Nucl Med Mol Imaging. 2008;35:2235–2244. doi: 10.1007/s00259-008-0860-8. [DOI] [PubMed] [Google Scholar]
- 141.Abakumov MA, Nukolova NV, Sokolsky-Papkov M, Shein SA, Sandalova TO, Vishwasrao HM, Grinenko NF, Gubsky IL, Abakumov AM, Kabanov AV, Chekhonin VP. Nanomed Nanotechnol Biol Med. 2015;11:825–833. doi: 10.1016/j.nano.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 142.Hawthorne MF. Angew Chem Int Ed Engl. 1993;32:950–984. [Google Scholar]
- 143.Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, Cheresh DA. Science. 2002;296:2404–2407. doi: 10.1126/science.1070200. [DOI] [PubMed] [Google Scholar]
- 144.Hynes RO. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 145.Cai W, Shin D-W, Chen K, Gheysens O, Cao Q, Wang SX, Gambhir SS, Chen X. Nano Lett. 2006;6:669–676. doi: 10.1021/nl052405t. [DOI] [PubMed] [Google Scholar]
- 146.Graf N, Bielenberg DR, Kolishetti N, Muus C, Banyard J, Farokhzad OC, Lippard SJ. ACS Nano. 2012;6:4530–4539. doi: 10.1021/nn301148e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Anderson JM, Shive MS. Adv Drug Deliv Rev. 2012;64(Supplement):72–82. [Google Scholar]
- 148.Zhen Z, Tang W, Chuang Y-J, Todd T, Zhang W, Lin X, Niu G, Liu G, Wang L, Pan Z, Chen X, Xie J. ACS Nano. 2014;8:6004–6013. doi: 10.1021/nn501134q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Doolittle E, Peiris PM, Doron G, Goldberg A, Tucci S, Rao S, Shah S, Sylvestre M, Govender P, Turan O, Lee Z, Schiemann WP, Karathanasis E. ACS Nano. 2015;9:8012–8021. doi: 10.1021/acsnano.5b01552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cun X, Chen J, Ruan S, Zhang L, Wan J, He Q, Gao H. ACS Appl Mater Interfaces. 2015;7:27458–27466. doi: 10.1021/acsami.5b09391. [DOI] [PubMed] [Google Scholar]
- 151.Chen F, Hong H, Goel S, Graves SA, Orbay H, Ehlerding EB, Shi S, Theuer CP, Nickles RJ, Cai W. ACS Nano. 2015;9:3926–3934. doi: 10.1021/nn507241v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yan F, Wu H, Liu H, Deng Z, Liu H, Duan W, Liu X, Zheng H. J Control Release. 2016;224:217–228. doi: 10.1016/j.jconrel.2015.12.050. [DOI] [PubMed] [Google Scholar]
- 153.Hong H, Cai W. Methods in Pharmacology and Toxicology. Humana Press; Totowa, NJ: 2016. [Google Scholar]
- 154.Liu J, Liu Q, Yang C, Sun Y, Zhang Y, Huang P, Zhou J, Liu Q, Chu L, Huang F, Deng L, Dong A, Liu J. ACS Appl Mater Interfaces. 2016;8:10726–10736. doi: 10.1021/acsami.6b01501. [DOI] [PubMed] [Google Scholar]
- 155.Makino J, Cabral H, Miura Y, Matsumoto Y, Wang M, Kinoh H, Mochida Y, Nishiyama N, Kataoka K. J Control Release. 2015;220(Part B):783–791. doi: 10.1016/j.jconrel.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 156.Patel N, Duffy BA, Badar A, Lythgoe MF, Årstad E. Bioconj Chem. 2015;26:1542–1549. doi: 10.1021/acs.bioconjchem.5b00380. [DOI] [PubMed] [Google Scholar]
- 157.Kelly KA, Allport JR, Tsourkas A, Shinde-Patil VR, Josephson L, Weissleder R. Circ Res. 2005;96:327–336. doi: 10.1161/01.RES.0000155722.17881.dd. [DOI] [PubMed] [Google Scholar]
- 158.Gosk S, Moos T, Gottstein C, Bendas G. Biochim Biophys Acta. 2008;1778:854–863. doi: 10.1016/j.bbamem.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 159.Chen Q, Zhang Xiang HF, Massagué J. Cancer Cell. 2011;20:538–549. doi: 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cao H, Zhang Z, Zhao S, He X, Yu H, Yin Q, Zhang Z, Gu W, Chen L, Li Y. J Control Release. 2015;205:162–171. doi: 10.1016/j.jconrel.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 161.He X, Yu H, Bao X, Cao H, Yin Q, Zhang Z, Li Y. Adv Healthc Mater. 2016;5:439–448. doi: 10.1002/adhm.201500626. [DOI] [PubMed] [Google Scholar]
- 162.Pouyssegur J, Dayan F, Mazure NM. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 163.Yu J, Zhang Y, Hu X, Wright G, Gu Z. Ann Biomed Eng. 2016;44:1931–1945. doi: 10.1007/s10439-016-1578-6. [DOI] [PubMed] [Google Scholar]
- 164.Gilkes DM, Semenza GL, Wirtz D. Nat Rev Cancer. 2014;14:430–439. doi: 10.1038/nrc3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Takasawa M, Moustafa RR, Baron J-C. Stroke. 2008;39:1629–1637. doi: 10.1161/STROKEAHA.107.485938. [DOI] [PubMed] [Google Scholar]
- 166.Brown JM, Wilson WR. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 167.Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Nat Rev Clin Oncol. 2012;9:674–687. doi: 10.1038/nrclinonc.2012.171. [DOI] [PubMed] [Google Scholar]
- 168.Thambi T, Deepagan VG, Yoon HY, Han HS, Kim SH, Son S, Jo DG, Ahn CH, Suh YD, Kim K, Kwon IC, Lee DS, Park JH. Biomaterials. 2014;35:1735–1743. doi: 10.1016/j.biomaterials.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 169.Denny WA. Future Oncol. 2010;6:419–428. doi: 10.2217/fon.10.1. [DOI] [PubMed] [Google Scholar]
- 170.Perche F, Biswas S, Wang T, Zhu L, Torchilin VP. Angew Chem Int Ed Engl. 2014;53:3362–3366. doi: 10.1002/anie.201308368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhao X, Li F, Li Y, Wang H, Ren H, Chen J, Nie G, Hao J. Biomaterials. 2015;46:13–25. doi: 10.1016/j.biomaterials.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 172.Zeng Y, Zhang S, Jia M, Liu Y, Shang J, Guo Y, Xu J, Wu D. Nanoscale. 2013;5:12633–12644. doi: 10.1039/c3nr04349e. [DOI] [PubMed] [Google Scholar]
- 173.Sharma R, Chen CJ. J Nanopart Res. 2009;11:671–689. [Google Scholar]
- 174.Chatterjee DK, Fong LS, Zhang Y. Adv Drug Deliv Rev. 2008;60:1627–1637. doi: 10.1016/j.addr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 175.Casas A, Di Venosa G, Hasan T, Batlle A. Curr Med Chem. 2011;18:2486–2515. doi: 10.2174/092986711795843272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhou Z, Song J, Nie L, Chen X. Chem Soc Rev. 2016;45:6597–6626. doi: 10.1039/c6cs00271d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cheng Y, Cheng H, Jiang C, Qiu X, Wang K, Huan W, Yuan A, Wu J, Hu Y. Nat Commun. 2015;6:8785. doi: 10.1038/ncomms9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Castro CI, Briceno JC. Artif Organs. 2010;34:622–634. doi: 10.1111/j.1525-1594.2009.00944.x. [DOI] [PubMed] [Google Scholar]
- 179.Que YR, Liu YJ, Tan W, Feng C, Shi P, Li YJ, Huang X. ACS Macro Lett. 2016;5:168–173. doi: 10.1021/acsmacrolett.5b00935. [DOI] [PubMed] [Google Scholar]
- 180.Song G, Liang C, Yi X, Zhao Q, Cheng L, Yang K, Liu Z. Adv Mater. 2016;28:2716–2723. doi: 10.1002/adma.201504617. [DOI] [PubMed] [Google Scholar]
- 181.Tang W, Zhen ZP, Wang MZ, Wang H, Chuang YJ, Zhang WZ, Wang GD, Todd T, Cowger T, Chen HM, Liu L, Li ZB, Xie J. Adv Funct Mater. 2016;26:1757–1768. doi: 10.1002/adfm.201504803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Luo Z, Zheng M, Zhao P, Chen Z, Siu F, Gong P, Gao G, Sheng Z, Zheng C, Ma Y, Cai L. Sci Rep. 2016;6:23393. doi: 10.1038/srep23393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhao P, Zheng M, Luo Z, Fan X, Sheng Z, Gong P, Chen Z, Zhang B, Ni D, Ma Y, Cai L. Adv Healthc Mater. 2016;5:2161–2167. doi: 10.1002/adhm.201600121. [DOI] [PubMed] [Google Scholar]
- 184.Chen H, Tian J, He W, Guo Z. J Am Chem Soc. 2015;137:1539–1547. doi: 10.1021/ja511420n. [DOI] [PubMed] [Google Scholar]
- 185.Kuang Y, Balakrishnan K, Gandhi V, Peng X. J Am Chem Soc. 2011;133:19278–19281. doi: 10.1021/ja2073824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Song G, Chen Y, Liang C, Yi X, Liu J, Sun X, Shen S, Yang K, Liu Z. Adv Mater. 2016;28:7143–7148. doi: 10.1002/adma.201602111. [DOI] [PubMed] [Google Scholar]
- 187.Chen Q, Feng L, Liu J, Zhu W, Dong Z, Wu Y, Liu Z. Adv Mater. 2016;28:7129–7136. doi: 10.1002/adma.201601902. [DOI] [PubMed] [Google Scholar]
- 188.Prasad P, Gordijo CR, Abbasi AZ, Maeda A, Ip A, Rauth AM, DaCosta RS, Wu XY. ACS Nano. 2014;8:3202–3212. doi: 10.1021/nn405773r. [DOI] [PubMed] [Google Scholar]
- 189.Fan W, Bu W, Shen B, He Q, Cui Z, Liu Y, Zheng X, Zhao K, Shi J. Adv Mater. 2015;27:4155–4161. doi: 10.1002/adma.201405141. [DOI] [PubMed] [Google Scholar]
- 190.Wilson WR, Hay MP. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 191.Qian C, Yu J, Chen Y, Hu Q, Xiao X, Sun W, Wang C, Feng P, Shen QD, Gu Z. Adv Mater. 2016;28:3313–3320. doi: 10.1002/adma.201505869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kiyose K, Hanaoka K, Oushiki D, Nakamura T, Kajimura M, Suematsu M, Nishimatsu H, Yamane T, Terai T, Hirata Y, Nagano T. J Am Chem Soc. 2010;132:15846–15848. doi: 10.1021/ja105937q. [DOI] [PubMed] [Google Scholar]
- 193.Zhang RL, Li Y, Zhang M, Tang QW, Zhang X. RSC Adv. 2016;6:30268–30276. [Google Scholar]
- 194.Liu HM, Zhang RL, Niu YW, Li Y, Qiao CM, Weng J, Li J, Zhang XN, Xiao ZB, Zhang X. RSC Adv. 2015;5:20848–20857. [Google Scholar]
- 195.Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T, Baba Y. Cancer Cell Int. 2013;13:89. doi: 10.1186/1475-2867-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Warburg O. Naturwissenschaften. 1924;12:1131–1137. [Google Scholar]
- 197.Lotz C, Kelleher DK, Gassner B, Gekle M, Vaupel P, Thews O. Oncol Rep. 2007;17:239–244. [PubMed] [Google Scholar]
- 198.Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- 199.Dong Z, Feng L, Zhu W, Sun X, Gao M, Zhao H, Chao Y, Liu Z. Biomaterials. 2016;110:60–70. doi: 10.1016/j.biomaterials.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 200.Som A, Raliya R, Tian L, Akers W, Ippolito JE, Singamaneni S, Biswas P, Achilefu S. Nanoscale. 2016;8:12639–12647. doi: 10.1039/c5nr06162h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhao Y, Luo Z, Li M, Qu Q, Ma X, Yu S-H, Zhao Y. Angew Chem Int Ed Engl. 2015;54:919–922. doi: 10.1002/anie.201408510. [DOI] [PubMed] [Google Scholar]
- 202.Pittella F, Miyata K, Maeda Y, Suma T, Watanabe S, Chen Q, Christie RJ, Osada K, Nishiyama N, Kataoka K. J Control Release. 2012;161:868–874. doi: 10.1016/j.jconrel.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 203.Li J, Chen Y-C, Tseng Y-C, Mozumdar S, Huang L. J Control Release. 2010;142:416–421. doi: 10.1016/j.jconrel.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sokolova V, Epple M. Angew Chem Int Ed Engl. 2008;47:1382–1395. doi: 10.1002/anie.200703039. [DOI] [PubMed] [Google Scholar]
- 205.Kester M, Heakal Y, Fox T, Sharma A, Robertson GP, Morgan TT, Altinoğlu Eİ, Tabaković A, Parette MR, Rouse SM, Ruiz-Velasco V, Adair JH. Nano Lett. 2008;8:4116–4121. doi: 10.1021/nl802098g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Neri D, Supuran CT. Nat Rev Drug Discov. 2011;10:767–777. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 207.Liu J, Huang Y, Kumar A, Tan A, Jin S, Mozhi A, Liang X-J. Biotechnol Adv. 2014;32:693–710. doi: 10.1016/j.biotechadv.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 208.Sun C-Y, Liu Y, Du J-Z, Cao Z-T, Xu C-F, Wang J. Angew Chem Int Ed Engl. 2016;55:1010–1014. doi: 10.1002/anie.201509507. [DOI] [PubMed] [Google Scholar]
- 209.Li HJ, Du JZ, Du XJ, Xu CF, Sun CY, Wang HX, Cao ZT, Yang XZ, Zhu YH, Nie S, Wang J. Proc Natl Acad Sci U S A. 2016;113:4164–4169. doi: 10.1073/pnas.1522080113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sun C-Y, Shen S, Xu C-F, Li H-J, Liu Y, Cao Z-T, Yang X-Z, Xia J-X, Wang J. J Am Chem Soc. 2015;137:15217–15224. doi: 10.1021/jacs.5b09602. [DOI] [PubMed] [Google Scholar]
- 211.Wang Y, Zhou K, Huang G, Hensley C, Huang X, Ma X, Zhao T, Sumer BD, DeBerardinis RJ, Gao J. Nat Mater. 2014;13:204–212. doi: 10.1038/nmat3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mizuhara T, Saha K, Moyano DF, Kim CS, Yan B, Kim Y-K, Rotello VM. Angew Chem Int Ed Engl. 2015;54:6567–6570. doi: 10.1002/anie.201411615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sundaram HS, Ella-Menye J-R, Brault ND, Shao Q, Jiang S. Chem Sci. 2014;5:200–205. [Google Scholar]
- 214.Jin Q, Xu J-P, Ji J, Shen J-C. Chem Commun. 2008:3058–3060. doi: 10.1039/b801959b. [DOI] [PubMed] [Google Scholar]
- 215.Lee ES, Shin HJ, Na K, Bae YH. J Control Release. 2003;90:363–374. doi: 10.1016/s0168-3659(03)00205-0. [DOI] [PubMed] [Google Scholar]
- 216.Lee ES, Na K, Bae YH. Nano Lett. 2005;5:325–329. doi: 10.1021/nl0479987. [DOI] [PubMed] [Google Scholar]
- 217.Hu J, Miura S, Na K, Bae YH. J Control Release. 2013;172:69–76. doi: 10.1016/j.jconrel.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 218.Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. J Control Release. 2008;129:228–236. doi: 10.1016/j.jconrel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhou K, Wang Y, Huang X, Luby-Phelps K, Sumer BD, Gao J. Angew Chem Int Ed Engl. 2011;50:6109–6114. doi: 10.1002/anie.201100884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yao L, Daniels J, Wijesinghe D, Andreev OA, Reshetnyak YK. J Control Release. 2013;167:228–237. doi: 10.1016/j.jconrel.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. Proc Natl Acad Sci USA. 2008;105:15340–15345. doi: 10.1073/pnas.0804746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Han L, Ma HJ, Guo YB, Kuang YY, He X, Jiang C. Adv Healthc Mater. 2013;2:1435–1439. doi: 10.1002/adhm.201300013. [DOI] [PubMed] [Google Scholar]
- 223.Yao L, Daniels J, Moshnikova A, Kuznetsov S, Ahmed A, Engelman DM, Reshetnyak YK, Andreev OA. Proc Natl Acad Sci USA. 2013;110:465–470. doi: 10.1073/pnas.1219665110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Couzin-Frankel J. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 225.Tang H, Qiao J, Fu Y-X. Cancer Lett. 2016;370:85–90. doi: 10.1016/j.canlet.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. ACS Nano. 2014;9:16–30. doi: 10.1021/nn5062029. [DOI] [PubMed] [Google Scholar]
- 227.Banerjee K, Resat H. Int J Cancer. 2016;138:2570–2578. doi: 10.1002/ijc.29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dewitte H, Verbeke R, Breckpot K, De Smedt SC, Lentacker I. Nano Today. 2014;9:743–758. [Google Scholar]
- 229.Cheung AS, Mooney DJ. Nano Today. 2015;10:511–531. doi: 10.1016/j.nantod.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, Drake C, Pardoll D, Yu H. Cancer Cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liao D, Liu Z, Wrasidlo WJ, Luo Y, Nguyen G, Chen T, Xiang R, Reisfeld RA. Cancer Res. 2011;71:5688–5696. doi: 10.1158/0008-5472.CAN-11-1264. [DOI] [PubMed] [Google Scholar]
- 232.Alshamsan A, Hamdy S, Samuel J, El-Kadi AOS, Lavasanifar A, Uludağ H. Biomaterials. 2010;31:1420–1428. doi: 10.1016/j.biomaterials.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 233.Bachmann MF, Jennings GT. Nat Rev Immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 234.Hawley AE, Davis SS, Illum L. Adv Drug Deliv Rev. 1995;17:129–148. [Google Scholar]
- 235.Qian Y, Jin H, Qiao S, Dai Y, Huang C, Lu L, Luo Q, Zhang Z. Biomaterials. 2016;98:171–183. doi: 10.1016/j.biomaterials.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 236.Silva JM, Videira M, Gaspar R, Preat V, Florindo HF. J Control Release. 2013;168:179–199. doi: 10.1016/j.jconrel.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 237.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Licona Limon P, Ferrandino AF, Gonzalez D, Habermann A, Flavell RA, Fahmy TM. Nat Mater. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Xu Z, Wang Y, Zhang L, Huang L. ACS Nano. 2014;8:3636–3645. doi: 10.1021/nn500216y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Stapleton S, Milosevic MF. Cancer targeted drug delivery. Springer; 2013. pp. 241–272. [Google Scholar]
- 240.Heldin C-H, Rubin K, Pietras K, Ostman A. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 241.Young JS, Llumsden CE, Stalker AL. J Pathol Bacteriol. 1950;62:313–333. doi: 10.1002/path.1700620303. [DOI] [PubMed] [Google Scholar]
- 242.Suzuki M, Hori K, Abe I, Saito S, Sato H. J Natl Cancer Inst. 1981;67:663–669. [PubMed] [Google Scholar]
- 243.Lee C-G, Heijn M, di Tomaso E, Griffon-Etienne G, Ancukiewicz M, Koike C, Park K, Ferrara N, Jain RK, Suit HD. Cancer Res. 2000;60:5565–5570. [PubMed] [Google Scholar]
- 244.Pietras K, Rubin K, Sjöblom T, Buchdunger E, Sjöquist M, Heldin C-H, Östman A. Cancer Res. 2002;62:5476–5484. [PubMed] [Google Scholar]
- 245.Lammerts E, Roswall P, Sundberg C, Gotwals PJ, Koteliansky VE, Reed RK, Heldin NE, Rubin K. Int J Cancer. 2002;102:453–462. doi: 10.1002/ijc.10722. [DOI] [PubMed] [Google Scholar]
- 246.Lee I, Boucher Y, Jain RK. Cancer Res. 1992;52:3237–3240. [PubMed] [Google Scholar]
- 247.Emerich DF, Dean RL, Snodgrass P, Lafreniere D, Agostino M, Wiens T, Xiong H, Hasler B, Marsh J, Pink M, Kim BS, Perdomo B, Bartus RT. J Pharmacol Exp Ther. 2001;296:632–641. [PubMed] [Google Scholar]
- 248.Kristjansen PEG, Boucher Y, Jain RK. Cancer Res. 1993;53:4764–4766. [PubMed] [Google Scholar]
- 249.Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. Mol Cell Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ricciardelli C, Rodgers RJ. Semin Reprod Med. 2006;24:270–282. doi: 10.1055/s-2006-948556. [DOI] [PubMed] [Google Scholar]
- 251.Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR, Hornicek FJ, Boucher Y, Munn LL, Jain RK. Proc Natl Acad Sci USA. 2012;109:15101–15108. doi: 10.1073/pnas.1213353109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kolacna L, Bakesova J, Varga F, Kostakova E, Plánka L, Necas A, Lukas D, Amler E, Pelouch V. Physiol Res. 2007;56:S51. doi: 10.33549/physiolres.931302. [DOI] [PubMed] [Google Scholar]
- 253.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popović Z, Huang P, Bawendi MG, Boucher Y, Jain RK. Nat Commun. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Barua S, Mitragotri S. Nano Today. 2014;9:223–243. doi: 10.1016/j.nantod.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Gong H, Chao Y, Xiang J, Han X, Song GS, Feng LZ, Liu JJ, Yang GB, Chen Q, Liu Z. Nano Lett. 2016;16:2512–2521. doi: 10.1021/acs.nanolett.6b00068. [DOI] [PubMed] [Google Scholar]
- 256.Zhou H, Fan ZY, Deng JJ, Lemons PK, Arhontoulis DC, Bowne WB, Cheng H. Nano Lett. 2016;16:3268–3277. doi: 10.1021/acs.nanolett.6b00820. [DOI] [PubMed] [Google Scholar]
- 257.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, DeNicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Rückert F, Grützmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Slomiany MG, Dai L, Bomar PA, Knackstedt TJ, Kranc DA, Tolliver L, Maria BL, Toole BP. Cancer Res. 2009;69:4992–4998. doi: 10.1158/0008-5472.CAN-09-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lesley J, Hascall VC, Tammi M, Hyman R. J Biol Chem. 2000;275:26967–26975. doi: 10.1074/jbc.M002527200. [DOI] [PubMed] [Google Scholar]
- 260.Yang CX, Liu YW, He YQ, Du Y, Wang WJ, Shi XX, Gao F. Biomaterials. 2013;34:6829–6838. doi: 10.1016/j.biomaterials.2013.05.036. [DOI] [PubMed] [Google Scholar]
- 261.Lee KL, Albee KL, Bernasconi RJ, Edmunds T. Biochem J. 1997;327:199–202. doi: 10.1042/bj3270199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Istrati D, Vizireanu C, Dima F, Dinică R. St Cerc St CICBIA. 2012;13:81–89. [Google Scholar]
- 263.Parodi A, Haddix SG, Taghipour N, Scaria S, Taraballi F, Cevenini A, Yazdi IK, Corbo C, Palomba R, Khaled SZ, Martinez JO, Brown BS, Isenhart L, Tasciotti E. ACS Nano. 2014;8:9874–9883. doi: 10.1021/nn502807n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, Bawendi MG, Boucher Y, Breakefield XO, Jain RK. Cancer Res. 2006;66:2509–2513. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- 265.Kuriyama N, Kuriyama H, Julin CM, Lamborn KR, Israel MA. Cancer Res. 2001;61:1805–1809. [PubMed] [Google Scholar]
- 266.Goodman TT, Olive PL, Pun SH. Int J Nanomed. 2007;2:265. [PMC free article] [PubMed] [Google Scholar]
- 267.Kuhn SJ, Finch SK, Hallahan DE, Giorgio TD. Nano Lett. 2006;6:306–312. doi: 10.1021/nl052241g. [DOI] [PubMed] [Google Scholar]
- 268.Villegas MR, Baeza A, Vallet-Regí M. ACS Appl Mater Interfaces. 2015;7:24075–24081. doi: 10.1021/acsami.5b07116. [DOI] [PubMed] [Google Scholar]
- 269.Kessenbrock K, Plaks V, Werb Z. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Himelstein B, Canete-Soler R, Bernhard E, Dilks D, Muschel R. Invasion Metastasis. 1994;14:246–258. [PubMed] [Google Scholar]
- 271.Guo F, Tian J, Cui M, Fang M, Yang L. Mol Med Report. 2015;12:753–759. doi: 10.3892/mmr.2015.3425. [DOI] [PubMed] [Google Scholar]
- 272.Nosoudi N, Nahar-Gohad P, Sinha A, Chowdhury A, Gerard P, Carsten CG, Gray BH, Vyavahare NR. Circ Res. 2015;117:E80–E89. doi: 10.1161/CIRCRESAHA.115.307207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Xiao L, Wang M. Cell Biochem Biophys. 2014;70:269–272. doi: 10.1007/s12013-014-9893-8. [DOI] [PubMed] [Google Scholar]
- 274.Vandooren J, Opdenakker G, Loadman PM, Edwards DR. Adv Drug Deliv Rev. 2016;97:144–155. doi: 10.1016/j.addr.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 275.Liu Y, Zhang D, Qiao ZY, Qi GB, Liang XJ, Chen XG, Wang H. Adv Mater. 2015;27:5034–5042. doi: 10.1002/adma.201501502. [DOI] [PubMed] [Google Scholar]
- 276.van Rijt SH, Bolukbas DA, Argyo C, Datz S, Lindner M, Eickelberg O, Konigshoff M, Bein T, Meiners S. ACS Nano. 2015;9:2377–2389. doi: 10.1021/nn5070343. [DOI] [PubMed] [Google Scholar]
- 277.Tang S, Meng QS, Sun HP, Su JH, Yin Q, Zhang ZW, Yu HJ, Chen LL, Chen Y, Gu WW, Li YP. Adv Funct Mater. 2016;26:6033–6046. [Google Scholar]
- 278.Ansari C, Tikhomirov GA, Hong SH, Falconer RA, Loadman PM, Gill JH, Castaneda R, Hazard FK, Tong L, Lenkov OD, Felsher DW, Rao JH, Daldrup-Link HE. Small. 2014;10:566–575. doi: 10.1002/smll.201301456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Grünwald B, Vandooren J, Locatelli E, Fiten P, Opdenakker G, Proost P, Krüger A, Lellouche JP, Israel LL, Shenkman L, Comes Franchini M. J Control Release. 2016;239:39–48. doi: 10.1016/j.jconrel.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 280.de la Rica R, Aili D, Stevens MM. Adv Drug Deliv Rev. 2012;64:967–978. doi: 10.1016/j.addr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 281.Blenke EO, Mastrobattista E, Schiffelers RM. Expert Opin Drug Deliv. 2013;10:1399–1410. doi: 10.1517/17425247.2013.805742. [DOI] [PubMed] [Google Scholar]
- 282.Touqui L, Alaoui-El-Azher M. Curr Mol Med. 2001;1:739–754. doi: 10.2174/1566524013363258. [DOI] [PubMed] [Google Scholar]
- 283.Six DA, Dennis EA. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 284.Sun WJ, Ji WY, Hu QY, Yu JC, Wang C, Qian CG, Hochu G, Gu Z. Biomaterials. 2016;96:1–10. doi: 10.1016/j.biomaterials.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kroll AV, Fang RH, Zhang L. Bioconjug Chem. 2017;28:23–32. doi: 10.1021/acs.bioconjchem.6b00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Rao J, Khan A. J Am Chem Soc. 2013;135:14056–14059. doi: 10.1021/ja407514z. [DOI] [PubMed] [Google Scholar]
- 287.Danhier F. J Control Release. 2016;244(Part A):108–121. doi: 10.1016/j.jconrel.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 288.Peterson J, Houghton P. Eur J Cancer. 2004;40:837–844. doi: 10.1016/j.ejca.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 289.Richmond A, Su Y. Dis Model Mech. 2008;1:78–82. doi: 10.1242/dmm.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Nichols JW, Bae YH. Nano Today. 2012;7:606–618. doi: 10.1016/j.nantod.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Martinez-Garcia R, Juan D, Rausell A, Muñoz M, Baños N, Menéndez C, Lopez-Casas PP, Rico D, Valencia A, Hidalgo M. Genome Med. 2014;6:27. doi: 10.1186/gm544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.