

Abstract
Cohort studies of large series of patients with sarcoidosis over a long period of time are scarce. The aim of this study is to report a 40-year clinical experience of a large series of patients at Bellvitge University Hospital, a tertiary university hospital in Barcelona, Spain. Diagnosis of sarcoidosis required histological confirmation except in certain specific situations. All patients underwent a prospective study protocol. Clinical assessment and follow-up of patients were performed by a multidisciplinary team.
From 1976 to 2015, 640 patients were diagnosed with sarcoidosis, 438 of them (68.4%) were female (sex ratio F/M 2:1). The mean age at diagnosis was 43.3 ± 13.8 years (range, 14–86 years), and 613 patients (95.8%) were Caucasian. At diagnosis, 584 patients (91.2%) showed intrathoracic involvement at chest radiograph, and most of the patients had normal pulmonary function. Erythema nodosum (39.8%) and specific cutaneous lesions (20.8%) were the most frequent extrapulmonary manifestations, but there was a wide range of organ involvement. A total of 492 patients (76.8%) had positive histology. Follow-up was carried out in 587 patients (91.7%), over a mean of 112.4 ± 98.3 months (range, 6.4–475 months). Corticosteroid treatment was administered in 255 patients (43.4%), and steroid-sparing agents in 49 patients (7.7%). Outcomes were as follows: 111 patients (18.9%) showed active disease at the time of closing this study, 250 (42.6%) presented spontaneous remission, 61 (10.4%) had remission under treatment, and 165 (28.1%) evolved to chronic sarcoidosis; among them, 115 (19.6%) with mild disease and 50 (8.5%) with moderate to severe organ damage. A multivariate analysis showed that at diagnosis, age more than 40 years, the presence of pulmonary involvement on chest radiograph, splenic involvement, and the need of treatment, was associated with chronic sarcoidosis, whereas Löfgren syndrome and mediastinal lymphadenopathy on chest radiograph were indicators of good outcome.
Sarcoidosis is a multisystem disease with protean clinical-radiographic manifestations. Although almost half of patients follow a spontaneous resolution or under treatment, a significant number of them may have several degrees of organ damage. This study emphasizes the value of a multidisciplinary approach and long-term follow-up by specialized teams in sarcoidosis.
Keywords: Löfgren's syndrome, observational, prognosis, sarcoidosis
1. Introduction
Sarcoidosis is a multisystem disease of unknown etiology, characterized by the presence of non-caseating granulomatous inflammation in 1 or more organs.[1] Although the diagnosis warrants a tissue biopsy, in special situations a presumptive diagnosis may be made based on clinical-radiographic findings alone. The exclusion of other granulomatous diseases is equally essential.[2] From the first descriptions of the disease in the 19th century, knowledge has expanded across multiple fields such as epidemiology, etiology, genetics, pathogenesis, and also with regard to clinical aspects, including treatment, evolution, and prognosis.[2–4] The clinical expression of sarcoidosis and the long-term natural history of the disease may vary according to different ethnicities and countries.[5–7] During the second half of the last century, a number of reports, including several historical series of patients describing the general clinical characteristics and prognosis of the disease, contributed to the clinical knowledge of sarcoidosis.[8–26] However, observational studies of large series of patients over a long period of time are scarce. One of the most outstanding studies was the revision by Johns and Michele[27] at the Johns Hopkins University School of Medicine in Baltimore, MD in which the authors reported a 50-year experience at the institution, mainly focusing on clinical presentation, case series, issues in clinical management and extrathoracic manifestations of sarcoidosis. However, the data relating to the series of patients was reported in a fractioned way, and an overall view of the series as a whole was not provided.[31]
In our institution, the Bellvitge University Hospital in Barcelona, Spain, the care of patients with sarcoidosis started during the mid-1970s in a special unit devoted to this disease. Since then, the everyday care of these patients has been carried out in close collaboration with a multidisciplinary team comprising different specialists. The senior author of this paper has become involved in the clinical assessment and management of patients with sarcoidosis from the beginning of the unit, and since 1992, he has been the director of the group. The aim of this study is to present an observational and prognostic investigation of a large cohort of patients with sarcoidosis, studied prospectively and followed up at our institution over the last 4 decades.
2. Methods
2.1. Diagnosis of sarcoidosis
During the period 1976 to 2015, 640 patients were diagnosed with sarcoidosis at the Bellvitge University Hospital, an 800-bed tertiary university hospital in Barcelona, Spain. The criteria for acceptance of a diagnosis of sarcoidosis were: a compatible clinical and radiological picture; histological demonstration of non-caseating granulomas in 1 or more tissues with negative stains and cultures for mycobacteria and fungus, or, a positive Kveim–Siltzbach skin test; and exclusion of other granulomatous diseases. One positive biopsy was considered enough if the thoracic radiology was consistent with sarcoidosis, while 2 or more were required, if available, when the radiology was normal or not typical for sarcoidosis. The diagnosis of sarcoidosis was accepted without histological confirmation in the following circumstances: patients with Löfgren syndrome; asymptomatic finding of typical bilateral hilar lymphadenopathy (BHL), both in a chest radiograph with a lambda pattern in 67-gallium scan and increased serum angiotensin converting enzyme (SACE) level, or in a thoracic computed tomography (CT); a CT pulmonary pattern typical for sarcoidosis with or without a bronchoalveolar lavage with a lymphocytic alveolitis and a CD4/CD8 >3.5. During the period 1977 to 1981, the Kveim–Siltzbach suspensions were provided by the Royal Brompton Hospital, London, UK, and from 1996 to 2000 by the Mount Sinai Medical Center, New York, NY.
2.2. Definitions and classifications
The chest radiograph stages were classified according to the Scadding criteria.[15] Doubtful or atypical hilar lymph node enlargement and/or pulmonary involvement on chest radiograph were confirmed by 67-gallium scan and/or thoracic CT. Löfgren syndrome was defined as the association of erythema nodosum and/or periarticular ankle inflammation with BHL with or without pulmonary involvement. Extrapulmonary organ involvement was defined according to previous reported criteria.[28,29] Since 2011, Fluorodeoxyglucose F 18 combined positron emission tomography and computer tomography (FDG PET/CT) substituted the practice of 67-gallium scan. FDG PET/CT was performed only in selected cases according to the indications, such as initial assessment of difficult cases, looking for hidden activity sites on which to perform a biopsy, and assessment of activity in patients with chronic fibrotic pulmonary sarcoidosis for assessing treatment.[30]
2.3. Study protocol
Since 1976, the patients diagnosed with sarcoidosis were submitted to a prospective study protocol at diagnosis. During 2015, the protocols were transferred to a recently created ACCES (Microsoft Office Access Database 2003) database. The study protocol included a clinical history and physical examination (including ocular examination), chest radiograph, general hematologic, and biochemical exams, including (SACE) level, tuberculin purified protein derivate skin test (PPD), and pulmonary function tests (PFT), including spirometry, pulmonary volumes, and diffusing capacity for carbon monoxide. Other ancillary tests to assess both intrathoracic and extrapulmonary sarcoidosis and the types of biopsies were performed according to clinical indications and involved organs. In order to assess changes in clinical aspects of sarcoidosis over time, and investigate whether the advent of more advanced imaging techniques had some impact in the detection of organ involvement, a comparison was made between the first 2 and last 2 decades of the study. Data regarding treatment and outcome were collected from patients who were followed up. No specific therapeutic schedule was followed, and corticosteroids and other immunosuppressive agents were administered according to indications recommended in medical literature.[31] Patients were followed up every 3 to 6 months until the disease became inactive, and thereafter, once a year.
The prospective study protocol, diagnosis of sarcoidosis, adjudication of outcomes, and treatment decisions, were always made ultimately by the leading physician of the group, F. Badrinas (see acknowledgements) from 1976 to 1992 and J. Mañá from 1992 to 2015. They personally evaluated all the patients so the criteria were completely homogeneous. Any patient with a suspicion of sarcoidosis initially seen by other specialists was referred to the outpatient sarcoidosis clinic. According to the type of organ involvement and treatment requirements, the patients were periodically discussed in clinical sessions with the participation of the members of the multidisciplinary team constituted by specialists in Internal Medicine, Pulmonary, Dermatology, Ophthalmology, Pathology, Neurology, Radiology, Nuclear Medicine, and others. The present study was approved by the Ethics Committee at Bellvitge University Hospital.
2.4. Outcomes assessment
The outcomes of sarcoidosis were classified according to the presence or absence of ongoing disease after 2 years, between 2 and 5 years, and more than 5 years (chronic disease) from diagnosis. Chronic sarcoidosis was classified as having mild or moderate to severe organ damage. Organ damage was defined as disease sequel of sarcoidosis that is not reversible, and therefore unlikely to respond to immunosuppressant agents or resolve on its own. In the absence of validated criteria, moderate to severe organ damage was defined as the presence of 1 of the following: radiological signs of pulmonary fibrosis and forced vital capacity (FVC) <60% of predicted and/or diffusing capacity for carbon monoxide (DLco) <50% of predicted, presence of moderate to severe pulmonary hypertension suggested by echocardiogram (systolic pulmonary arterial pressure >40 mm Hg), neurosarcoidosis with sequel (other than facial nerve palsy), cardiac sarcoidosis, chronic cutaneous involvement (plaques and lupus pernio), chronic uveitis, chronic renal failure, portal hypertension, or chronic symptomatic bone involvement. Relapse was defined as the reappearance of the disease during the decreasing phase or until 2 years after the suppression of treatment. Recurrence was defined as the reappearance of the disease after at least 1 year of spontaneous remission.[32] To examine factors predicting chronic sarcoidosis, a series of variables at diagnosis including age, sex, organ involvement, PFT, SACE, and need of treatment were analyzed.
2.5. Statistical analysis
A descriptive analysis was performed, by expressing the results as means and standard deviations for continuous variables, and absolute values and percentages for categorical variables. A t test (and Mann–Whitney U test in the absence of parametric distribution) was performed for the comparison between continuous variables, and the chi-square test or Fisher exact test, when appropriate, for the comparison of categorical variables. A univariate and multivariate analyses by means of binary logistic regression were performed in order to assess the prognostic factors at diagnosis related to chronic sarcoidosis. Those variables that reached statistical significance in the univariate analysis were introduced in the multivariate analysis. SPSS (SPSS 15.0. 2009, Chicago, IL) was used for statistical analyses (P < .05).
3. Results
3.1. Demographic data and clinical presentation
Six hundred and forty patients were diagnosed with sarcoidosis over 40 years at Bellvitge University Hospital in Barcelona, Spain. Four hundred and thirty-eight patients (68.4%) were women and 202 (31.6%) were men (sex ratio F/M 2:1). The mean age at diagnosis was 43.3 ± 13.8 years (age range, 14–86). Six hundred and thirteen patients (95.8%) were Caucasian, 14 (2.2%) North African, 5 (0.8%) Hispanic, 5 (0.8%) Black, and 3 (0.4%) Asian. At diagnosis, 498 patients (79.4%) were non-smokers, 67 (10.7%) smokers, 62 (9.9%) former smokers, and 13 had no data regarding smoking behavior. Fifteen patients (2.3%) in 9 families had familial sarcoidosis. The time between the onset of symptoms and diagnosis was less than 6 months in 418 patients (71.3%), and more than 6 months in 168 (28.7%). Table 1 shows the clinical presentation of sarcoidosis.
Table 1.
Mode of onset of sarcoidosis in 640 patients.
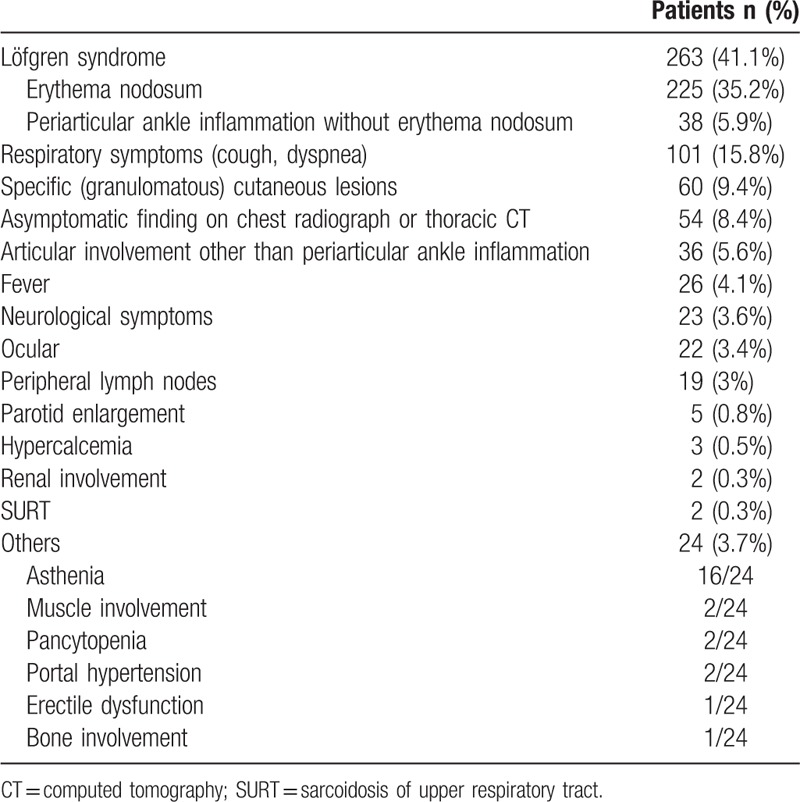
3.2. Pulmonary and extrapulmonary manifestations
Two hundred patients (31.3%) complained of respiratory symptoms (dyspnea and/or cough). Table 2 shows the chest radiograph stages and Table 3 the relationship with thoracic CT, both at diagnosis. In most patients, PFT were within the normal range at diagnosis (Table 4). Table 5 shows the relationship between functional respiratory patterns and the chest radiograph stages at diagnosis. The presence of pulmonary involvement correlated with decline in pulmonary function. Pulmonary hypertension was detected at some stage of the follow-up in 8 patients (1.3%).
Table 2.
Radiological stages at diagnosis in 640 patients.

Table 3.
Relationship among chest radiograph stages and thoracic CT at diagnosis in 313 patients.
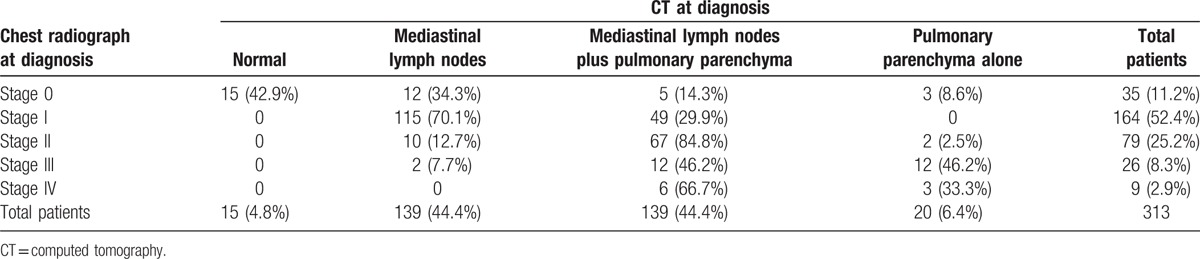
Table 4.
Pulmonary function tests at diagnosis.

Table 5.
Relationship among pulmonary function test patterns and chest radiograph stage at diagnosis. Percentages do not add up since not all patients underwent complete functional tests. Chi-square test.
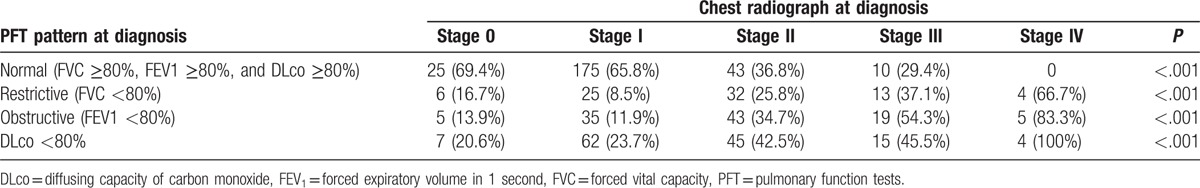
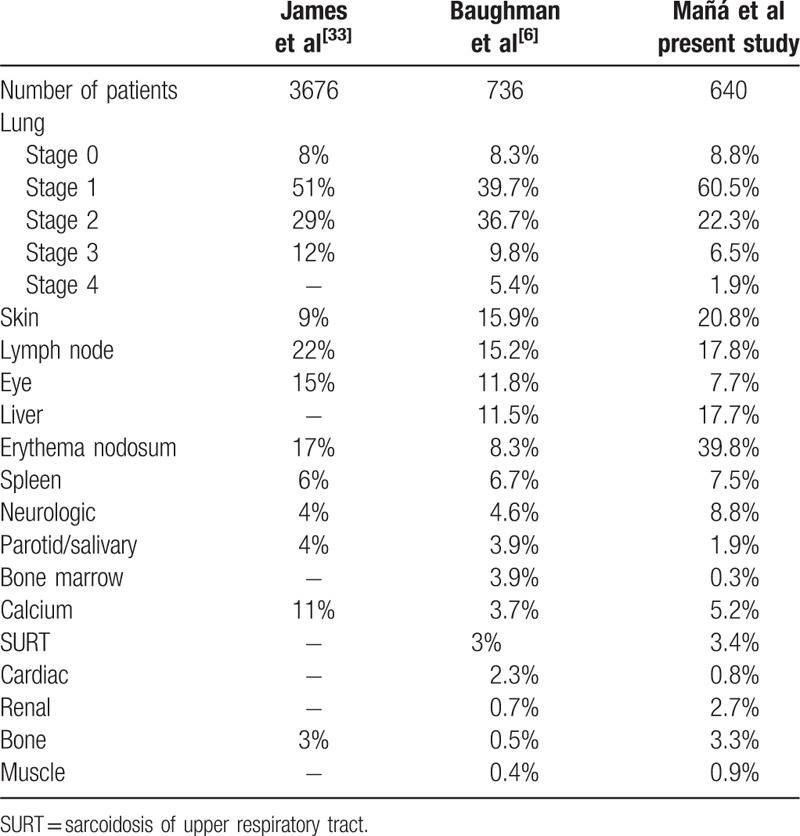
Table 6 illustrates the findings of extrapulmonary manifestations, by organ involvement occurring during the first 6 months after diagnosis (first column), and in a detailed clinical expression at some time during the follow-up (second column). The skin was the second organ involved after the lung either as erythema nodosum or as specific (granulomatous) cutaneous lesions. It was not infrequent to find more than 1 type of skin lesion coexisting in the same patient. Table 7 shows a comparison of specific organ involvement in the present study and 2 other historical large series of sarcoidosis.[6,33]
Table 6.
Extrapulmonary sarcoidosis at diagnosis or at any time during the follow-up in 640 patients.

Table 7.
Comparison of organ involvement with other historical large series of sarcoidosis.
3.3. Ancillary tests and histological confirmation
PPD was negative in 447 out of 513 patients (87.1%). One hundred and sixty-five of 436 patients (37.8%) showed hypergammaglobulinemia at diagnosis. SACE levels at diagnosis were increased in 278 out of 543 patients (51.2%) in whom SACE had been performed. 67-gallium scan was performed in 406 patients at diagnosis. Increased uptake was present in mediastinal lymph nodes in 339 patients (83.5%), in pulmonary parenchyma in 96 (23.5%), and in parotid and salivary glands in 74 (18.1%). Twenty-nine patients (7.1%) did not show increased 67-gallium uptake. FDG PET-CT was performed in 85 patients either at diagnosis or at any time during the follow-up. Mediastinal involvement was present in 62 patients (42.5%), pulmonary parenchyma in 36 (24.7%), and extrapulmonary sites in 34 (23.3%). Table 8 displays the different types of positive biopsy procedures.
Table 8.
Histological diagnosis of sarcoidosis. Percentage was calculated in basis of the total of the series, including patients without histological confirmation.

3.4. Treatment, follow-up, and outcome
A total of 587 patients (91.7%) were followed-up and 53 (8.3%) were not, since they had been referred to our sarcoidosis unit from other hospitals only for diagnosis assessment. The mean follow-up was 112.4 ± 98.3 months (range, 6.4–475 months). Of this group, 266 (45.3%) received treatment for at least 3 months at some time during the follow-up. Corticosteroid treatment was administered in 255 patients (43.4%). Steroid-sparing agents, usually with a low-dose of corticosteroids, or a combination of them, were administered as follows: methotrexate in 25 patients (4.3%), antimalarials in 23 (4%), and azathioprine in 22 (3.7%). Other agents were used in a few cases: mycophenolate mofetil in 8 patients, cyclosporine in 3, infliximab in 3, minocycline in 2, cyclophosphamide in 2, rituximab in 2, and pentoxifylline in 1.
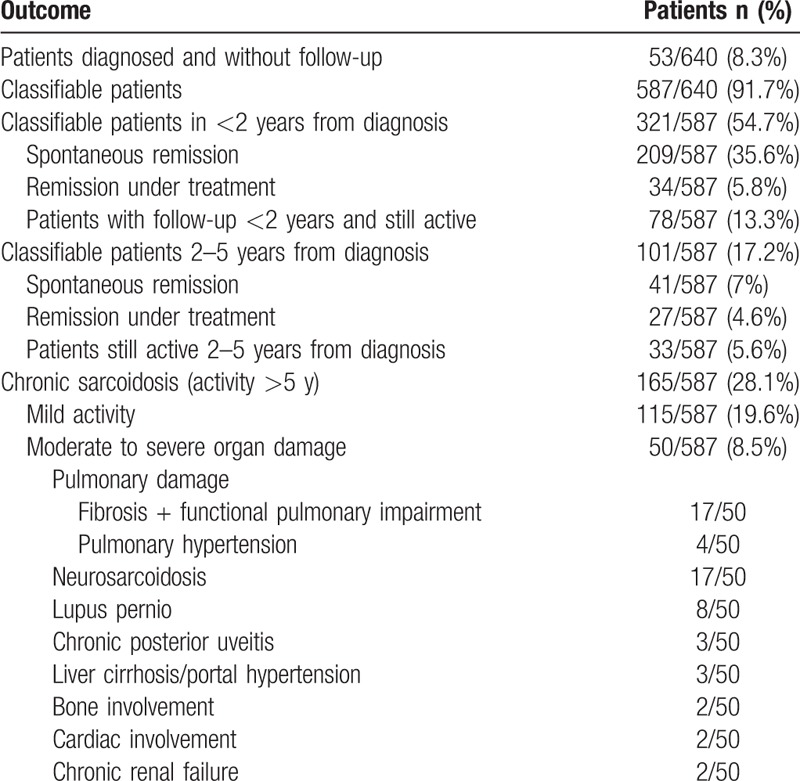
Table 9 summarizes the overall outcome of the 587 patients that were followed-up. One hundred and eleven patients (18.9%) showed active disease at the time of closing the present study. Two hundred and fifty patients (42.6%) showed spontaneous remission without treatment. Sixty-one patients (10.4%) had remission under treatment. One hundred and sixty-five patients (28.1%) showed active disease more than 5 years after diagnosis and were classified as chronic sarcoidosis. Among these, 115 patients (19.6%) showed mild activity and 50 patients (8.5%) were classified as having moderate to severe organ damage. The most frequently damaged organ was the lung (particularly pulmonary fibrosis), the nervous system, and the skin. Four patients are currently under assessment for pulmonary transplantation, and 1 is under hemodialysis and being assessed for a kidney transplant. One patient had a complication of mycetoma. Thirty out of 266 treated patients (11.3%) presented relapse after decreasing the dose of corticosteroids or the suppression of therapy. Twenty-two out of 587 patients (3.7%), 21 Löfgren, and 1 non-Löfgren, had recurrence of the disease years after spontaneous remission without treatment. Nine out of 587 patients (1.5%) died due to causes related to sarcoidosis (6 pulmonary fibrosis, 2 pulmonary hypertension, and 1 portal hypertension).
Table 9.
Patients’ classification throughout a 40-year follow-up. Part of the cohort cannot be classified since they are patients yet on activity and others due to lack of follow-up, so that numbers do not add up. Different severe organ damage can be present in the same patient, so numbers do not add up as well.
3.5. Predictor factors at diagnosis of chronic sarcoidosis
Table 10 shows the results of univariate and multivariate analysis of factors at diagnosis predicting active disease at 5 years (chronic sarcoidosis, including both mild and moderate to severe organ damage). Age more than 40 years, the presence of pulmonary involvement on chest radiograph, splenic involvement, and the need of treatment (both corticosteroids and steroid-sparing agents) were associated with a poor prognosis. Löfgren syndrome and the presence of mediastinal lymphadenopathy on chest radiograph were associated with a good outcome.
Table 10.
Predictor factors at diagnosis of chronic sarcoidosis in 165 patients.
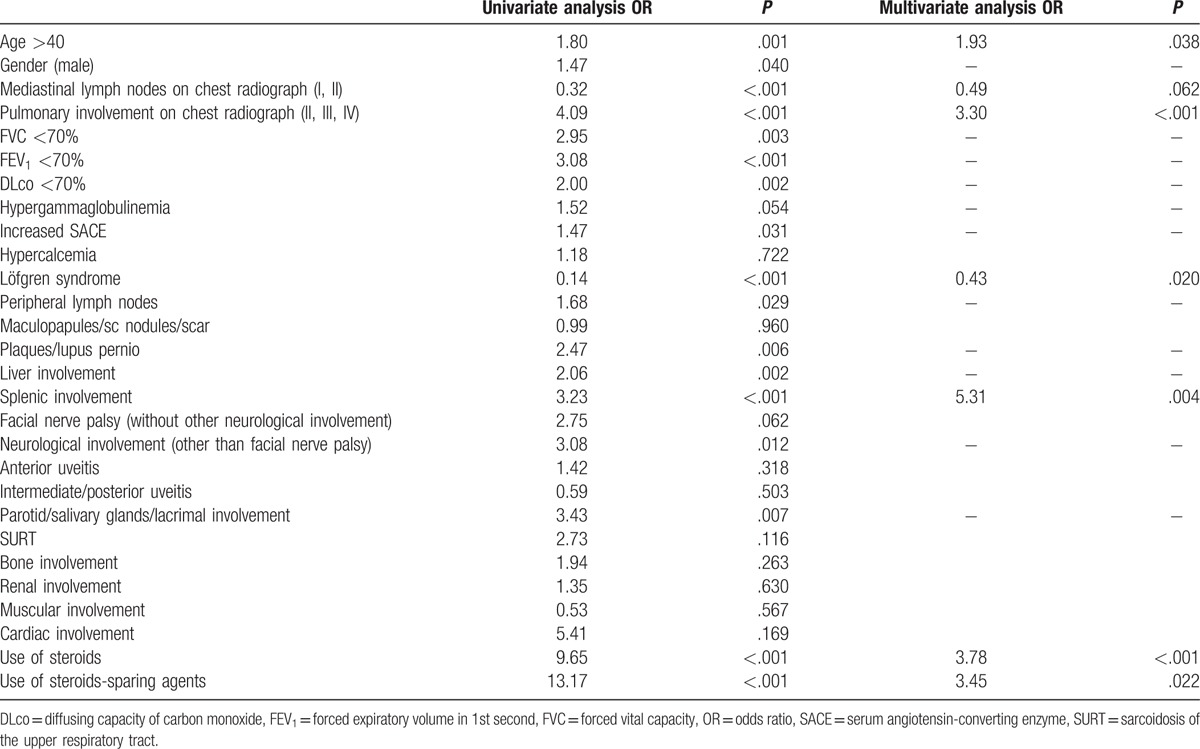
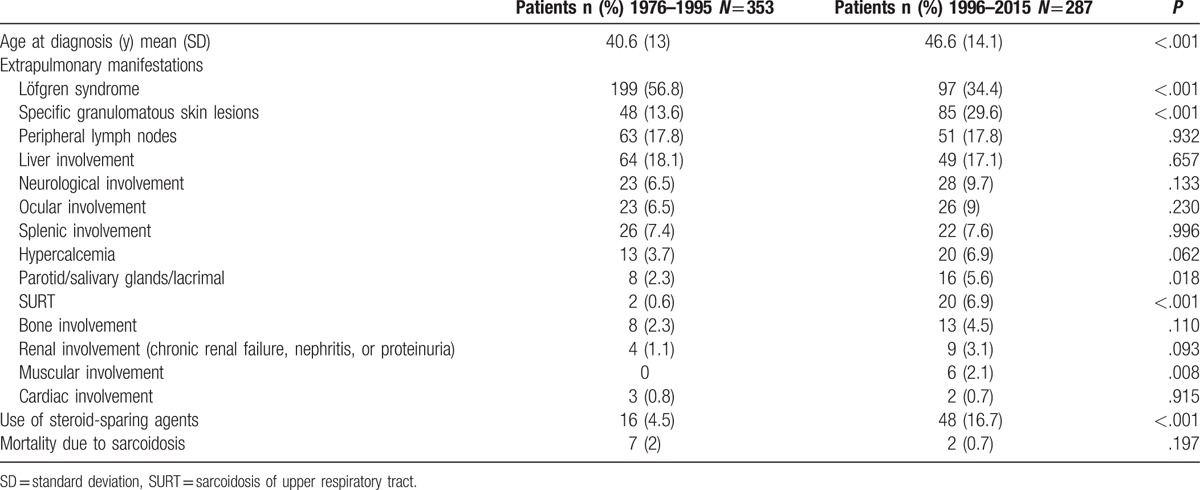
Table 11 shows the comparison between the first 2 and last 2 decades of the study as regards age of diagnosis, extrapulmonary organ involvement, treatment with steroid-sparing agents, and mortality.
Table 11.
Mean age, extrapulmonary sarcoidosis, use of steroid-sparing agents and mortality over time. Chi-square or Fisher exact test for categorical variables and t test for quantitative variables.
4. Discussion
The present study describes our 40-year clinical experience of a large cohort of 640 consecutive patients with sarcoidosis at Bellvitge University Hospital, Barcelona, Spain. Most of our patients were Caucasians, because immigration in Spain has been minimal until recently. Since there is no reliable official epidemiological data on sarcoidosis in our country, this large series of patients and its prolonged period of follow-up provide a representation of the situation of sarcoidosis in Spain.
4.1. Mode of presentation of sarcoidosis
Knowledge of the wide variety of presentations of sarcoidosis is crucial in initial diagnostic suspicion.[34] The most outstanding feature of this series was the high frequency of erythema nodosum as a way of onset of the disease. The high frequency of Löfgren syndrome in Spain had been already reported in a cooperative study among 3 hospitals in Barcelona, performed more than 20 years ago.[24] Several studies have also described a high prevalence of Löfgren syndrome in northern European countries.[35,36] It has been hypothesized that Löfgren syndrome is a consequence of a combination of environmental factors, with a genetic predisposition.[37] Earlier studies showed that symptoms of erythema nodosum were, significantly, first noticed during springtime.[36,38,39] In addition, a genetic study involving Swedish and Spanish patients confirmed previous reports that the CCR2 haplotype 2 and HLA-DRB1∗0301 were independent risk factors predisposing to Löfgren syndrome.[36,37,40] A small but significant number of patients (6.4%) presented only with ankle swelling without erythema nodosum, which is considered a variant of Löfgren syndrome.[35,39,41]
Three other forms of presentations in our series deserve some comments. In spite of 91% of our patients having intrathoracic radiographic involvement, respiratory symptoms were the way of presentation in only 16% of cases, which corroborates the frequent clinical-radiographic dissociation seen in sarcoidosis. A systemic work-up for sarcoidosis is indicated in any patient presenting with granulomatous cutaneous lesions (9.4% in our series).[42] In previous reports we have stressed the presence of papules in the knees as a frequent form of onset of systemic sarcoidosis.[43,44] Not infrequently, sarcoidosis is discovered by chance in a chest radiograph or a thoracic CT performed for other reasons (8.4% in our series).[45] Currently, the detection of sarcoidosis as an asymptomatic finding has increased with the use of FDG PET/CT scan performed during the follow-up of patients with cancer.[46] The wide clinical variety of the other forms of presentations shown in Table 1 highlights the multisystem nature of sarcoidosis.
4.2. Pulmonary sarcoidosis
The chest radiograph stages distribution based on Scadding classification in our series is in concordance with most series of sarcoidosis.[2] Thoracic CT was performed in 49% of our patients. Significantly, it demonstrated the presence of pulmonary involvement in 23% of patients with stage 0, and in 30% of those with stage I on chest radiograph. However, this finding by itself is not an indication for treatment, unless there is the concomitant presence of respiratory symptoms and/or decline in respiratory function.[47] In sarcoidosis, thoracic CT is considered more important as a diagnostic modality than for treatment indications.[4] However, it should not be performed systematically in patients with a typical chest radiograph.[48] Most of our patients showed normal PFT at diagnosis. This finding reaffirms that clinical-radiographic dissociation is also extensive to pulmonary function in sarcoidosis. In our series the most frequent impairments were restrictive and/or obstructive patterns and/or decrease in DLco. Concomitant irreversible airflow obstruction is frequently present in stage IV. Although there is a significant overlap between radiological stages and impairment of PFT (as shown in Table 5), in general, the more advanced the radiological stage, the greater the decline in functional status. FVC is considered the simplest and most accurate parameter to reflect the impact of pulmonary involvement.[49,50]
4.3. Extrapulmonary sarcoidosis
The frequency of extrapulmonary manifestations in our cohort of patients agrees with other large international series.[6,7,33] The skin, specific (granulomatous) cutaneous lesions, was the second organ involved after the lung. The more frequent granulomatous cutaneous lesions were maculopapules and plaques. Lupus pernio, that has been reported to be frequent in African American and West Indian patients, with a woman predominance, and a hallmark of chronicity,[51–53] is not frequent in Spain. Mild cholestasis caused by granulomatous hepatitis is common, while severe cholestasis and portal hypertension are rare. It is important to recognize the presence of low-attenuation nodules on CT on the liver and spleen as a not infrequent finding of sarcoidosis.[3] Eye involvement was not frequent in our series, in spite of systematically performing an ophthalmologic examination in the initial assessment.
Neurosarcoidosis was present in 8% of patients in our series. The most frequent manifestation was cranial neuropathy, particular facial palsy, sometimes bilateral. Leptomeningeal involvement, either as aseptic meningitis or as focal or diffuse gadolinium meningeal enhancement on MRI, on isolation or associated with cranial neuropathies, is a common abnormality in neurosarcoidosis and occasionally may result in hydrocephalus. The hypothalamus and the pituitary gland are the most frequent parenchymal brain involvement and may result in endocrinopathies. However, as shown in Table 6, any region of the brain and spinal cord can be involved, resulting in a variety of neurological symptomatology. Periventricular white matter lesions on MRI are not infrequently seen, but they are considered non-specific. Peripheral neuropathy is uncommon and small fiber neuropathy causing disabling pain is being increasingly recognized.[54]
Table 6 also shows that other forms of extrapulmonary involvement, such as hypercalcemia and salivary and lacrimal glands, were less frequent. Recognition of sarcoidosis of the upper respiratory tract (SURT) is important since subtle symptoms of nasal obstruction may be overlooked by physicians.[55] Bone sarcoidosis is being detected with increasing frequency in asymptomatic patients following the introduction of FDG PET/CT. Renal involvement is usually a consequence of granulomatous or lymphocytic interstitial nephritis.[56] Symptomatic muscle involvement is rare. Cardiac sarcoidosis is very uncommon and difficult to recognize, although is frequent in Japan.[57] Apart from the different organs involved, a considerable burden of disease may be caused by nonspecific but troublesome symptoms, termed by Lazar and Culver as “sarcoidosis penumbra.” [31] The most significant is fatigue, which accounted for almost 10% of patients in our series. Fatigue may be present even when the disease is already in remission, and may frequently be accompanied by depression and impair quality of life.[58,59]
Interestingly, although most extrapulmonary organ involvement took place in the first 6 months after diagnosis, some organs were affected during follow-up. Therefore, longitudinal screening should be maintained over time.[60] Only symptoms related to Löfgren syndrome always occurred at onset.
4.4. Histological diagnosis
One positive biopsy may be sufficient when clinical-radiographic findings are typical; otherwise, 2 or more may be necessary. However, in some classic presentations, such as Löfgren syndrome, asymptomatic BHL, and Heerfordt syndrome, the diagnosis can be accepted without histological support.[4] Given the multisystem nature of the disease, a wide variety of biopsies have been used in sarcoidosis. Currently, endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA) is becoming the diagnostic procedure of choice in many centers because of high sensitivity and low complications rate, and it is replacing the practice of mediastinoscopy and transbronchial lung biopsy.[61,62] Conjunctival and nasal mucosa biopsies also have a high yield, and their practice has been recommended as silent sites of granulomatous inflammation in patients with diagnostic difficulties, for instance, in isolated neurosarcoidosis. However, the demonstration of granulomatous inflammation by itself is not specific to sarcoidosis, and a differential diagnosis with other granulomatous diseases, according to the involved organ, is mandatory.[2]
4.5. Treatment, follow-up and outcome
In sarcoidosis there is often a struggle to distinguish between treatable active disease and irreversible untreatable disease-related damage. In most patients, the disease resolves spontaneously without treatment (42.6% of patients in our series) or with treatment (10.4% of our patients). Some authors have suggested classifying sarcoidosis as chronic when the disease remains active for more than 2 years.[21] However, chronicity is currently accepted when the disease remains active for more than 5 years, since a significant number of patients become inactive between 2 and 5 years spontaneously or under treatment.[53] In our series, 28.1% of patients showed active disease for more than 5 years and were classified as chronic sarcoidosis. However, patients with chronic sarcoidosis may show a mild degree of activity without clinically significant organ dysfunction, in what has been called smoldering sarcoidosis (19.6% in our series), or with moderate to severe organ damage (8.5% of our patients). Signs and symptoms caused by organ damage may be a consequence of still active disease, which is theoretically susceptible to some response to therapy, or irreversible fibrosis, both often coexisting in the same organ. Signs and symptoms caused by active granulomatous inflammation and irreversible damage may be initially similar and a major challenge is to distinguish between these states in order to indicate appropriate therapy.
Treatment indications in sarcoidosis have been controversial for many years, as clinicians have to bear in mind a range of possibilities in evolution of the disease: spontaneous remission, mild persistent disease for years with almost no clinical repercussion, chronic disabling active disease, irreversible damage, and, equally important, a balance between the benefits and side effects of treatment. In our view, irreversible side effects caused by treatment should be included in the concept of damage in sarcoidosis. The main indications for treatment are symptomatic pulmonary progressive disease with functional derangement, and major extrapulmonary organ involvement.[31,47,63–68] In our series, 45.3% of patients received treatment, most of them with corticosteroids. A small but significant number of patients received other immunosuppressive agents, usually in combination with low-doses of corticosteroids. Anti-TNF therapies were used in very few cases. However, only about 10% of patients did complete remission take place during treatment. In addition, in most patients with chronic sarcoidosis, treatment just helped to keep the disease under relative control. These findings may put in question the true efficacy of current treatments in sarcoidosis. The most frequent and clinically significant forms of end-stage organ damage were pulmonary fibrosis, neurosarcoidosis, and cutaneous disfiguring lesions such as lupus pernio. Pulmonary fibrosis from sarcoidosis is usually slowly progressive, and may range from asymptomatic to be life-threatening because of the development of respiratory failure, bronchiectasis, or pulmonary hypertension.[69–71]
4.6. Predictor factors at diagnosis of chronic sarcoidosis
This study corroborates that Löfgren syndrome and the presence of mediastinal lymphadenopathy are clinical factors associated with good prognosis, whereas advanced age, pulmonary, and splenic involvement are associated with poor outcomes. These results are similar to those reported by our previous and other prognostic studies.[21,25,26,72] However, variables representing some of the extrathoracic manifestations that have been previously associated with poor prognosis in the literature, such as cardiac sarcoidosis and others, did not reach statistical significance because of the few patients with these manifestations in our series (Table 6). In several studies the use of corticosteroids has been associated with chronic sarcoidosis. An intense immune response, as noted by BAL studies, is usually indicative of a favorable outcome, and it has been hypothesized that its suppression with corticosteroids may be harmful.[73] However, it seems more plausible that the indications of therapy, according to current guidelines, simply reflexes the more severe cases at the beginning of the disease.[74] The important differences between Löfgren syndrome and other forms of sarcoidosis in clinical presentation and outcome, and the existence of at least 2 clear genetic markers of Löfgren syndrome, such as HLA class II haplotype DRB1∗0301 and CCR2-haplotype 2, strongly suggest that they may represent distinct entities.[40]
4.7. Comparison between the first 2 and last 2 decades
Our study allows a number of interesting comparisons to be made. Firstly, the age of diagnosis increased slightly in the last 2 decades. This may be due to the frequency of patients with Löfgren syndrome being much higher during the first 2 decades. We believe that the main reason for the decrease of patients with Löfgren syndrome in the last 2 decades is a more accurate selection of difficult cases for referral from other centers. Secondly, the detection of other organ involvement may have increased with the introduction of more advanced imaging techniques, despite data not always being statistically significant. Finally, a clear increase in the use of steroid-sparing agents can be observed in the last 2 decades.
Our study has some limitations. The high frequency of Löfgren syndrome in our country limits comparisons with other series of sarcoidosis based on different ethnicities. Besides, as we only analyzed patients assessed at a tertiary reference center, the sample selection could be somehow slightly biased. The study is also limited by the observational nature inherent to studies of clinical series of sarcoidosis. The follow-up of the patients was not uniform, the analysis of cases was made at different moments of their disease course, and some data were missed. The absence of validated criteria to define the presence of activity of sarcoidosis and in particular to evaluate the degree of organ damage is an additional important limitation of the study as well.
In summary, the present observational and prognostic investigation reports a 40-year overview of the clinical spectrum and long-term follow-up of the disease in a large series of 640 patients with sarcoidosis at a single reference center in Barcelona, Spain. This extended and consistent experience in managing a large number of patients with sarcoidosis firmly emphasizes the value of a multidisciplinary approach by specialized teams in the clinical assessment and long-term follow-up of the disease.
Acknowledgments
The authors would like to acknowledge the fundamental contribution to this study made by Dr Francesc Badrinas, who set up the Sarcoidosis Unit at the Bellvitge University Hospital in the 1970s, working as director until his death in 1992.
Footnotes
Abbreviations: BHL = bilateral hilar lymphadenopathy, CNS = central nervous system, CT = computer tomography, DLco = diffusing capacity for carbon monoxide, EBUS-TBNA = endobronchial ultrasound-guided transbronchial needle aspiration, FDG PET/CT = Fluorodeoxyglucose F 18 combined positron emission tomography and computer tomography, FEV1 = forced expiratory volume in 1st second, FVC = forced vital capacity, PFT = pulmonary function tests, PPD = tuberculin purified protein derivate skin test, SACE = serum angiotensin converting enzyme, SURT = sarcoidosis of the upper respiratory tract.
The authors have no conflicts of interest to disclose.
References
- [1].Mitchell DN, Scadding JG. Sarcoidosis. Am Rev Respir Dis 1974;110:774–802. [DOI] [PubMed] [Google Scholar]
- [2].Hunninghake GW, Costabel U, Ando M, et al. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:149–73. [PubMed] [Google Scholar]
- [3].Iannuzzi MC, Ribicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007;357:2153–65. [DOI] [PubMed] [Google Scholar]
- [4].Valeyre D, Prasse A, Nunes H, et al. Sarcoidosis. Lancet 2014;383:1155–67. [DOI] [PubMed] [Google Scholar]
- [5].Siltzbach LE, James DG, Turiaf J, et al. Course and prognosis of sarcoidosis around the world. Am J Med 1974;57:847–52. [DOI] [PubMed] [Google Scholar]
- [6].Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit care Med 2001;164:1885–9. [DOI] [PubMed] [Google Scholar]
- [7].Judson MA, Boan AD, Lackland DT. The clinical course of sarcoidosis: presentation, diagnosis, and treatment in a large white and black cohort in the United States. Sarcoidosis Vasc Diffuse Lung Dis 2012;29:119–27. [PubMed] [Google Scholar]
- [8].Longcope WT, Freiman DG. A study of sarcoidosis. Based on a combined investigation of 160 cases including 30 autopsies from the Johns Hopkins hospital and Massachusetts General hospital. Medicine 1952;31:1–32. [PubMed] [Google Scholar]
- [9].Israel HL, Sones M. Sarcoidosis. Clinical observation on 160 cases. Arch Inter Med 1958;102:766–76. [DOI] [PubMed] [Google Scholar]
- [10].Sones M, Israel HL. Course and prognosis of sarcoidosis. Am J Med 1960;29:84–93. [DOI] [PubMed] [Google Scholar]
- [11].Bacharach T. Sarcoidosis. A clinical review of 111 cases. Am Rev Respir Dis 1961;84:12–6. [DOI] [PubMed] [Google Scholar]
- [12].Sones M, Israel HL. Course and prognosis of sarcoidosis: Philadelphia. Am Rev Respir Dis 1961;84:60–5. [DOI] [PubMed] [Google Scholar]
- [13].James DG. Course and prognosis of sarcoidosis: London. Am Rev Respir Dis 1961;84:66–70. [DOI] [PubMed] [Google Scholar]
- [14].Löfgren S, Stavenow S. Course and prognosis of sarcoidosis: Stockolm. Am Rev Respir Dis 1961;84:71–3. [DOI] [PubMed] [Google Scholar]
- [15].Scadding JG. Prognosis of intrathoracic sarcoidosis in England. A review of 136 cases after five years’ observation. Br J Med 1961;2:1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mayock RL, Bertrand P, Morrison CE, et al. Manifestations of sarcoidosis. Analysis of 145 patients, with a review of nine series selected from the literature. Am J Med 1963;35:67–89. [DOI] [PubMed] [Google Scholar]
- [17].Kitamura K, Shigematsu I, Hosoda Y. Sarcoidosis in Japan. Observations on 700 cases. Am Rev Respir Dis 1967;96:952–6. [DOI] [PubMed] [Google Scholar]
- [18].Hannuksela M, Salo OP, Mustakallio K. The prognosis of acute untreated sarcoidosis. Ann Clin Res 1970;2:57–61. [PubMed] [Google Scholar]
- [19].Thygesen K, Viskum K. Manifestations and course of the disease in intrathoracic sarcoidosis. Scand J Respir Dis 1972;53:174–80. [PubMed] [Google Scholar]
- [20].Romer FK. Presentattion of sarcoidosis and outcome of pulmonary changes. A review of 243 patients followed up for up to 10 years. Dan Med Bull 1982;29:27–32. [PubMed] [Google Scholar]
- [21].Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. QJM 1983;208:525–33. [PubMed] [Google Scholar]
- [22].Hillerdal G, Nöu E, Osterman K, et al. Sarcoidosis: epidemiology and prognosis. A 15-year European study. Am Rev Respir Dis 1984;130:29–32. [DOI] [PubMed] [Google Scholar]
- [23].Reich JM, Johnson RE. Course and prognosis of sarcoidosis in a nonreferral setting. Analysis of 86 patients observed for a 10 years. Am J Med 1985;78:61–7. [DOI] [PubMed] [Google Scholar]
- [24].Mañá J, Badrinas F, Morera J, et al. Sarcoidosis in Spain. Sarcoidosis 1992;9:118–22. [PubMed] [Google Scholar]
- [25].Mañá J, Salazar A, Manresa F. Clinical factors predicting persistence of activity in sarcoidosis: a multivariate analysis of 193 cases. Respiration 1994;61:219–25. [DOI] [PubMed] [Google Scholar]
- [26].Judson MA, Baughman RP, Thompson BW, et al. Two year prognosis of sarcoidosis: the ACCESS experience. Sarcoidosis Vasc Diffuse Lung Dis 2003;20:204–11. [PubMed] [Google Scholar]
- [27].Johns CJ, Michel TM. The clinical management of sarcoidosis. A 50-year experience at the Johns Hopkins hospital. Medicine 1999;78:65–111. [DOI] [PubMed] [Google Scholar]
- [28].Judson MA, Baughman RP, Teirstein AS, et al. and the ACCESS Research Group. Defining organ involvement in sarcoidosis: the ACCESS proposed instrument. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:75–86. [PubMed] [Google Scholar]
- [29].Judson MA, Costabel U, Drent M, et al. The WASOG sarcoidosis organ assessment instrument: an update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:19–27. [PubMed] [Google Scholar]
- [30].Teirsten AS, Machac J, Almeida O, et al. Results of whole-body fluorodeoxyglucose positron emission tomography scans in 137 patients with sarcoidosis. Chest 2007;132:1949–53. [DOI] [PubMed] [Google Scholar]
- [31].Lazar CA, Culver DA. Treatment of sarcoidosis. Semin Respir Crit Care Med 2010;31:501–18. [DOI] [PubMed] [Google Scholar]
- [32].Mañá J, Montero A, Vidal M, et al. Recurrent sarcoidosis: a study of 17 patients with 24 episodes of recurrence. Sarcoidosis Vasc Diffuse Lung Dis 2003;20:212–21. [PubMed] [Google Scholar]
- [33].James DG, Neville E, Siltzbach LE, et al. A worldwide review of sarcoidosis. Ann NY Acad Sci 1976;278:321–34. [DOI] [PubMed] [Google Scholar]
- [34].Valeyre D, Bernaudin JF, Uzunhan Y, et al. Clinical presentation of sarcoidosis and diagnostic work-up. Semin Respir Crit Care Med 2014;35:336–51. [DOI] [PubMed] [Google Scholar]
- [35].Grunewald J, Eklund A. Sex-specific manifestations of Löfgren's syndrome. Am J Respir Crit Care Med 2007;175:40–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Grunewald J, Eklund A. Löfgren's syndrome. Human leukocyte antigen strongly influences the disease course. Am J Respir Crit Care Med 2009;179:307–12. [DOI] [PubMed] [Google Scholar]
- [37].Grunewald J. HLA associations and Löfgren's syndrome. Expert Rev Clin Immunol 2012;8:55–62. [DOI] [PubMed] [Google Scholar]
- [38].Badrinas F, Morera J, Fité E, et al. Seasonal clustering of sarcoidosis. Lancet 1989;2:455–6. [DOI] [PubMed] [Google Scholar]
- [39].Mañá J, Gómez-Vaquero C, Montero A, et al. Löfgren's syndrome revisited: A study of 186 patients. Am J Med 1999;107:240–5. [DOI] [PubMed] [Google Scholar]
- [40].Spagnolo P, Sato H, Grunewald J, et al. A common haplotype of the C-C chemokine receptor 2 gene and HLA-DRB1∗0301 are independent genetic risk factors for Löfgren's syndrome. J Intern Med 2008;264:433–41. [DOI] [PubMed] [Google Scholar]
- [41].Mañá J, Gómez-Vaquero C, Salazar A, et al. Periarticular ankle sarcoidosis: a variant of Löfgren's syndrome. J Rheumatol 1996;23:874–7. [PubMed] [Google Scholar]
- [42].Mañá J, Marcoval J, Rubio M, et al. Granulomatous cutaneous sarcoidosis: diagnosis, relationship to systemic disease, prognosis and treatment. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:268–81. [PubMed] [Google Scholar]
- [43].Marcoval J, Moreno A, Mañá J. Papular sarcoidosis of the knees. A clue for the diagnosis of the erythema nodosum-associated sarcoidosis. J Am Acad Dermatol 2003;49:75–8. [DOI] [PubMed] [Google Scholar]
- [44].Marcoval J, Mañá J. Papular sarcoidosis of the knees. A frequent form of presentation of systemic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2016;33:59–65. [PubMed] [Google Scholar]
- [45].Judson MA. The clinical features of sarcoidosis: a comprehensive review. Clinic Rev Allerg Immunol 2015;49:63–78. [DOI] [PubMed] [Google Scholar]
- [46].Chowdhury FU, Sheerin F, Bradley KM, et al. Sarcoid-like reaction to malignancy on whole-body integrated 18F-FDG PET/TC: prevalence and disease pattern. Clin Radiol 2009;64:675–81. [DOI] [PubMed] [Google Scholar]
- [47].Judson MA. The treatment of pulmonary sarcoidosis. Respir Med 2012;106:1351–61. [DOI] [PubMed] [Google Scholar]
- [48].Mañá J, Teirstein AS, Mendelson DS, et al. Excessive thoracic computed tomographic scanning in sarcoidosis. Thorax 1995;50:1264–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Valeyre D, Bernaudin JF, Jeny F, et al. Pulmonary sarcoidosis. Clin Chest Med 2015;36:631–41. [DOI] [PubMed] [Google Scholar]
- [50].Baughman RP, Drent M, Culver DA, et al. Endpoints for clinical trials of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2012;29:90–8. [PubMed] [Google Scholar]
- [51].Spiteri MA, Matthey F, Gordon T, et al. Lupus pernio: a clinico-radiological study of thirty-five cases. Br J Dermatol 1985;112:315–22. [DOI] [PubMed] [Google Scholar]
- [52].Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis. Relationship to severity and chronicity of disease. Clin Exp Dermatol 2011;6:739–44. [DOI] [PubMed] [Google Scholar]
- [53].Baughman RP, Lower EE. Features of sarcoidosis associated with chronic disease. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:275–81. [PubMed] [Google Scholar]
- [54].Tavee JO, Stern BJ. Neurosarcoidosis. Clin Chest Med 2015;36:643–56. [DOI] [PubMed] [Google Scholar]
- [55].Cohen Aubart F, Ouayoun M, Brauner M, et al. Sinonasal involvement in sarcoidosis. A case-control study of 20 patients. Medicine 2006;85:365–71. [DOI] [PubMed] [Google Scholar]
- [56].Mahévas M, Lescure FX, Boffa JJ, et al. Renal sarcoidosis. Clinical, laboratory, and histological presentation and outcome in 47 patients. Medicine 2009;88:98–106. [DOI] [PubMed] [Google Scholar]
- [57].Birnie D, Ha A, Gula LJ, et al. Cardiac sarcoidosis. Clin Chest Med 2015;36:657–68. [DOI] [PubMed] [Google Scholar]
- [58].Drent M, Strookappe B, Hoitsma E, et al. Consequences of sarcoidosis. Clin Chest Med 2015;36:727–37. [DOI] [PubMed] [Google Scholar]
- [59].Judson MA. Quality of life assessment in sarcoidosis. Clin Chest Med 2015;36:739–50. [DOI] [PubMed] [Google Scholar]
- [60].Inoue Y, Inui N, Hashimoto D, et al. Cumulative incidence and predictors of progression in corticosteroid-naïve patients with sarcoidosis. PloS ONE 2015;10:e0143371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Navarri N, Lawrence DR, Kolvekar S, et al. Endobronchial ultrasound-guided transbronchial needle aspiration prevents mediastinoscopies in the diagnosis of isolated mediastinal lymphadenopathy: a prospective trial. Am J Respir Crit Care Med 2012;186:255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Culver DA. Diagnosing sarcoidosis. Curr Opin Pulm Med 2015;21:499–509. [DOI] [PubMed] [Google Scholar]
- [63].Hunninghake GW, Gilbert S, Pueringer R, et al. Outcome of the treatment for sarcoidosis. Am J Respir Crit Care Med 1994;149:893–8. [DOI] [PubMed] [Google Scholar]
- [64].Gibson GJ, Prescott RJ, Muers MF, et al. British Thoracic Society sarcoidosis study: effects of long term corticosteroid treatment. Thorax 1996;51:238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gottlieb JE, Israel HL, Steiner RM, et al. Outcome in sarcoidosis. The relationship of relapse to corticosteroid therapy. Chest 1997;111:623–31. [DOI] [PubMed] [Google Scholar]
- [66].Baughman RP, Nunes H, Sweiss NJ, et al. Established and experimental medical therapy of pulmonary sarcoidosis. Eur Respir J 2013;41:1424–38. [DOI] [PubMed] [Google Scholar]
- [67].Baughman RP, Lower EE. Treatment of sarcoidosis. Clinic Rev Allerg Immunol 2015;49:79–92. [DOI] [PubMed] [Google Scholar]
- [68].Wijsenbeek MS, Culver DA. Treatment of sarcoidosis. Clin Chest Med 2015;36:751–67. [DOI] [PubMed] [Google Scholar]
- [69].Valeyre D, Nunes H, Bernaudin JF. Advanced pulmonary sarcoidosis. Curr Opin Pulm Med 2014;20:488–95. [DOI] [PubMed] [Google Scholar]
- [70].Baughman RP, Engel PJ, Nathan S. Pulmonary hypertension in sarcoidosis. Clin Chest Med 2015;36:703–14. [DOI] [PubMed] [Google Scholar]
- [71].Kouranos V, Jacob J, Wells A. Severe sarcoidosis. Clin Chest Med 2015;36:715–26. [DOI] [PubMed] [Google Scholar]
- [72].Salazar A, Mañá J, Albareda JM, et al. Splenomegaly in sarcoidosis. Sarcoidosis 1995;12:131–4. [PubMed] [Google Scholar]
- [73].Reich JM. Con: the treatment of the granulomatous response is beneficial in acute sarcoidosis. Respir Med 2010;104:1778–81. [DOI] [PubMed] [Google Scholar]
- [74].Baughman RP, Judson MA, Teirstein AS, et al. Presenting characteristics as predictors of duration of treatment in sarcoidosis. Q J Med 2006;99:307–15. [DOI] [PubMed] [Google Scholar]