

Abstract
Rationale:
Adult-onset Still disease (AOSD) is a rare systemic inflammatory disease of unknown etiology characterized by evanescent salmon-pink rash, fever spikes, arthralgia, and lymphadenopathy. AOSD usually has a good prognosis, but it can sometimes be fatal, especially when it is complicated by systemic inflammatory response syndrome (SIRS) and multiple organ failure.
Patient concerns:
A previously healthy 26-year-old woman was referred to our hospital for persistent high fever and mild systemic edema. Five days later, the patient presented with dyspnea, hypotension, and anuria. Anasarca developed with massive pleural effusion, ascites, and systemic edema, resulting in an increase of 47 kg in body weight.
Diagnoses:
The patient was diagnosed as AOSD after infection, malignancy, hematologic disorders, and other autoimmune diseases were excluded.
Interventions:
We administered tocilizumab, an IL-6 receptor inhibitor, intravenously in addition to cyclosporine, prednisolone, plasma exchange, and continuous hemodiafiltration.
Outcomes:
The patient's systemic condition improved. After stabilization by all medications, the patient was managed and responded to tocilizumab alone. To the best of our knowledge, this was the first case of severe SIRS complicating AOSD that was successfully treated with an anti- IL-6 receptor antibody.
Lessons:
SIRS should not be overlooked in a patient with steroid-resistant AOSD and edema. Inhibitors of the IL-6 receptor can be used safely and effectively to control AOSD complicated with severe SIRS.
Keywords: adult-onset Still disease, interleukin 6, systemic inflammatory response syndrome, tocilizumab
1. Introduction
Adult-onset Still disease (AOSD), which is classified as a subgroup of autoimmune disease, is characterized by fever spikes, salmon-pink rash, and arthralgia.[1,2] It is a relatively rare disease, but also a well-known differential diagnosis of fever of unknown origin. AOSD generally has a mild clinical course and is rarely fatal. Herein, we report a severe case of AOSD presenting with systemic inflammatory response syndrome (SIRS), uncontrollable extravascular fluid volume expansion, and third space accumulation leading to anasarca, 47-kg weight gain, and multiple organ failure. Treatment with tocilizumab in addition to cyclosporine, plasma exchange, and continuous hemodiafiltration (CHDF) improved the patient's general condition and vascular leakage. The patient was salvaged without complications. These results indicated that tocilizumab is a promising treatment for uncontrollable extravascular fluid volume expansion caused by severe SIRS complicating AOSD.
2. Methods
This is a case report of a patient at our medical practice. No Institutional Review Board approval was obtained. Our IRB designates a single-patient case report as not subject to IRB review because it does not meet the definition of human subject research. The patient has signed a consent form allowing disclosure of medical records.
3. Case presentation
A 26-year-old Japanese woman, without any pertinent past medical history, was referred to our hospital because of fever, abdominal pain in the lower quadrant, nausea, and edema. She had initially consulted a gynecologist and admitted that she was diagnosed as pelvic inflammatory disease.
On admission, she had a fever of 38°C, but the other vital signs were within normal limits. Physical examination showed mild tenderness on the left lower quadrant of the abdomen. Laboratory tests at the time of admission revealed severe inflammation [white blood cell (WBC) 13,900/mm3, neutrophil 76%; C-reactive protein (CRP) 27.83 mg/dL; and ferritin 371.4 ng/mL] and thrombocytopenia (platelet 82,000 /mm3) with normal renal function (creatinine, 0.59 mg/dL). The possibility of sepsis was ruled out by negative results of blood, sputum, and urine cultures; endotoxin; β-d-glucan; procalcitonin; and a broad range bacterial polymerase chain reaction (PCR) from blood. Rheumatoid factor and autoantibody tests, including anti-ADAMTS13 autoantibody, which is a marker of thrombotic thrombocytopenic purpura, were negative. Immunoglobulin (Ig) G, IgM, IgA, and IgE were not elevated. Computed tomography (CT) scan showed bilateral pleural effusion, ascites, and lymphadenopathy in the neck and paraaortic station.
Initially, she was treated with ceftriaxone and azithromycin. However, a few days later, she developed hypotension and progressive dyspnea and she was transferred to the intensive care unit. Chest x-ray revealed massive bilateral pleural effusion. Laboratory tests showed acute reduction of total protein (4.6 g/dL) and albumin (2.1 g/dL) without elevation in transaminase and creatinine levels. Additional test results were as follows: serum interleukin (IL)-6, 57.5 pg/mL (normal ≤ 4.0 pg/mL); tumor necrosis factor alpha (TNFα), 13.2 pg/mL (0.6–2.8 pg/mL); and vascular endothelial growth factor (VEGF) 534 pg/mL (≤115 pg/mL). These results suggested a progressing acute inflammatory response. On the 5th day of admission, contrast-enhanced CT showed increased pleural effusion and ascites, hepatosplenomegaly, and cervical lymph node enlargement (Fig. 1 A–C). A diagnosis of SIRS was made and infusion of noradrenaline and methylprednisolone (mini-boluses of 125 mg/ day for 3 days) was started. However, on the 11th day, anasarca progressed and the patient was transferred to our Department of Internal Medicine for further evaluation and management.
Figure 1.
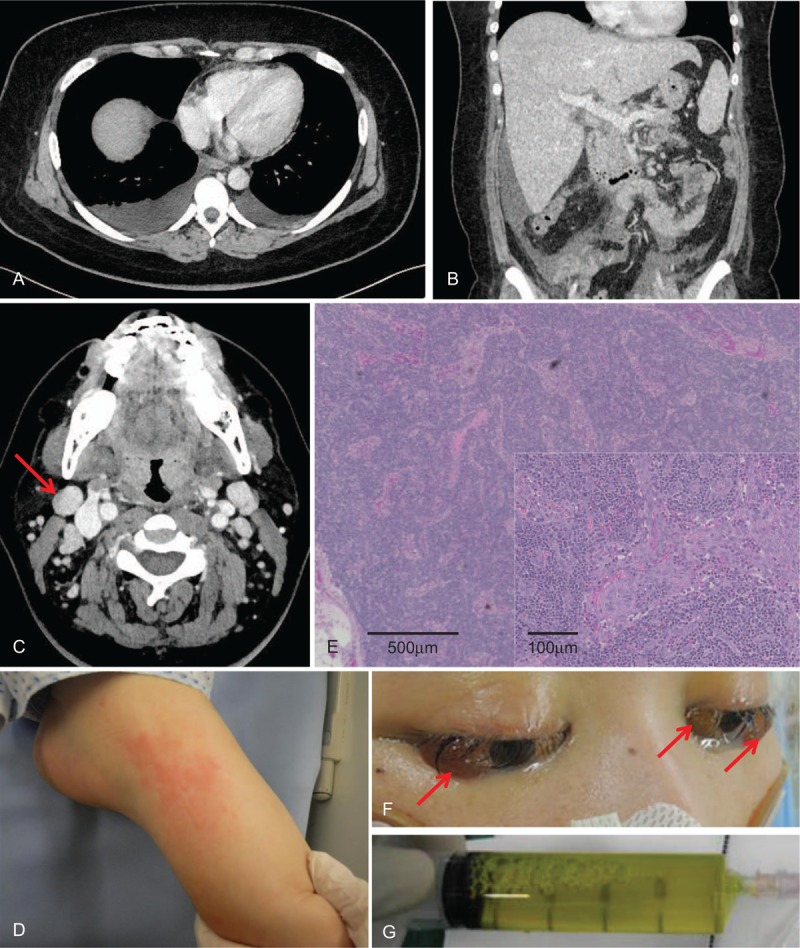
Severe anasarca caused by the uncontrollable systemic inflammatory response syndrome complicating adult-onset Still disease. A, Chest and (B) abdominal computed tomography show effusion in the third spaces and hepatomegaly. C, Both internal jugular lymph nodes are enlarged. D, Salmon-pink purpura on the left foot is seen. E, Pathologic examination of the biopsy sample from the right lymph node (arrow) shows no evidence of proliferative diseases, such as malignancy or Castleman disease. F, Cataract edema (arrows) during the peak of anasarca, with 47-kg weight gain. G, Paracentesis drainage shows a transudate.
On examination, we noticed the presence of pink purpura on both feet (Fig. 1 D), which disappeared 10 hours later. Laboratory tests revealed marked inflammation (WBC 33,900/ mm3, CRP 27.6 mg/dL, and ferritin 3,119 ng/mL); disseminated intravascular coagulation (DIC) (platelet 36,000/mm3 and prothrombin time international normalized ratio 2.0); acute kidney injury (creatinine 2.95 mg/dL, blood urea nitrogen 89 mg/dL, and estimated glomerular filtration rate 17.2 mL/min/1.73 m2); and liver failure [total bilirubin 8.1 mg/dL, aspartate transaminase (AST) 226 U/L (normal, 13–30 U/L), and alanine transaminase (ALT) 15 U/L (normal, 7–23 U/L)].
After consultation with hematologists, bone marrow biopsy was performed and showed trilineage hyperplasia without dysplasia and myeloblasts. These results ruled out hematologic disorders, such as leukemia and hemophagocytic syndrome. Biopsy under endotracheal intubation and systemic anesthesia was performed on the right internal jugular lymph node, which was the largest of the lymphadenopathies. Pathologic examination showed atrophic, ill-defined follicle formation with plasma cell and neutrophil infiltration and no proliferative malignant cells or vasculature (Fig. 1 E).
We continued to manage the patient with tracheal intubation and mechanical ventilation because of progressing respiratory failure secondary to accumulating pleural effusion and ascites. Noradrenaline drip, plasma exchange, and CHDF were needed to keep blood pressure within the normal range when there was vascular leakage because of SIRS. Indeed, stroke volume variation was as high as 29% (normal range <13%), suggesting endovascular dehydration.[3] Her physical presentation and laboratory tests were re-evaluated. Based on Yamaguchi's classification criteria,[4] the patient was diagnosed as AOSD complicated with severe SIRS. Pulsed methylprednisolone therapy (1 g/day for 3 days) and plasma exchange were administered, with no remarkable results. The patient remained severely hypotensive, requiring large amounts of crystalloids and high doses of noradrenaline because of profuse plasma leakage from increased vascular permeability. Fluid continued to accumulate in the interstitial space, leading to generalized edema, large amount of pleural effusion, ascites, and even cataract edema (Fig. 1 F). On day 26, weight gain reached its maximum at 47 Kg. Frequent drainage of the bilateral pleural effusion and paracentesis (3–4 times, 1–2 L per drainage) were required to prevent respiratory failure and abdominal compartment syndrome. All fluid aspirates were characterized as clear, yellow transudates (Fig. 1 G).
At this time, vascular permeability caused by severe SIRS was considered to be the most critical mechanism for the poor systemic condition of the patient. Anti-IL6 receptor antibody (tocilizumab) treatment has been used in Japan for both juvenile idiopathic arthritis (juvenile-onset Still disease) and Castleman disease, especially when standard steroid therapy is ineffective. Therefore, we decided to administer tocilizumab intravenously to ameliorate the patient's condition. Careful consideration was made because the patient was still in an unknown state of shock and administration of these drugs could be fatal, if complicated with bacterial or fungal infection. Within a week after the addition of tocilizumab to conventional treatments, vascular permeability improved, and enabled us to introduce extracorporeal ultrafiltration method. Subsequently, her hemodynamic and respiratory status improved, followed by resolution of DIC and recovery from multiple organ failure.
After infusion of tocilizumab, serum IL-6 levels escalated to 2040 pg/mL, which was expected based on the manufacturer's information. Initially, we gave 8 mg/Kg of tocilizumab weekly for 3 times, then every 2 weeks for 3 times, and every 4 weeks thereafter (Fig. 2). Cyclosporine was also used at an initial dose of 3 mg/kg daily, and then was tapered to a minimum dose (0.2 mg/kg) 3 weeks later before discontinuation. With tocilizumab alone, we observed that there was no remission of SIRS and AOSD. Although she could not move her extremities, except for the finger tips, just after extubation on day 43 and needed rehabilitation program for 3 months, she was eventually discharged from our hospital with her previous body weight and without residual complications.
Figure 2.
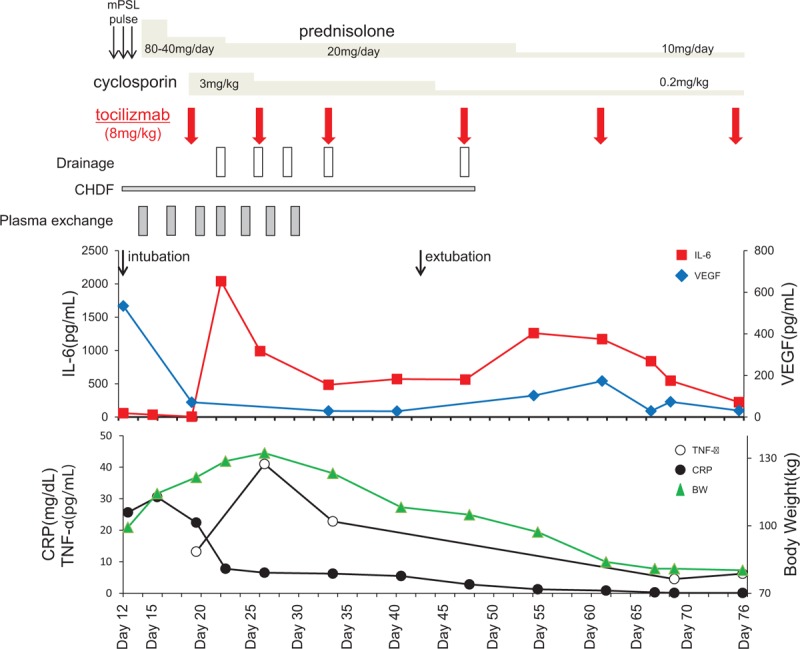
Temporal course of a 26-year-old woman with systemic inflammatory response syndrome complicating adult-onset Still disease. BW = body weight, CHDF = continuous hemodiafiltration, CRP = C-reactive protein, IL-6 = interleukin-6, mPSL = methylprednisolone, TNF-α = tumor necrosis factor alpha, VEGF = vascular endothelial growth factor.
4. Discussion
We reported a severe case of SIRS complicating AOSD that presented with generalized edema, multiple organ failure, and hypovolemic shock because of extravascular fluid volume expansion and third space accumulation. Pulsed methylprednisolone, plasma exchange, and CHDF were unable to control the anasarca and hypovolemic shock in this patient. On the other hand, combination therapy of tocilizumab and cyclosporine led to dramatic recovery after a week with no complications. To the best our knowledge, this was the first case that survived SIRS complicating AOSD after treatment with tocilizumab.
AOSD is a rare systemic inflammatory disease that is characterized by evanescent salmon-pink rash, fever spikes, arthralgia, and lymphadenopathy.[1,2] Its etiology remains unknown and there are no specific diagnostic tests. Therefore, the diagnosis of AOSD is usually based on clinical findings after exclusion of other diseases. AOSD usually has a good prognosis, but can be life-threatening,[5] especially when it is complicated with multiple organ failure and DIC caused by SIRS,[6–10] hemophagocytic syndrome,[11–13] thrombotic thrombocytopenic purpura,[5] acute respiratory distress syndrome,[14] diffuse alveolar hemorrhage,[15] pulmonary arterial hypertension,[16] and liver failure.[17] Therefore, these severe complications should not be overlooked when we see patients with AOSD.
Our patient presented with shock because of profuse plasma leakage caused by increased vascular permeability caused by SIRS. To our knowledge, available literature showed only a few reports of cases with severe plasma leakage leading to shock. AOSD can also induce vascular leakage in the peripheral arteries and lead to complications of angioedema and urticarial, which can be successfully treated with pulsed methylprednisolone.[15] In our patient, there was no angioedema; therefore, it remains unknown whether the mechanism of angioedema in AOSD is similar to that of SIRS.
Accumulating evidence suggests that serum and tissue levels of IL-1, IL-6, and TNFα are increased in patients with AOSD and that the level of IL-6 is correlated with disease activity.[1] IL-6 also plays an important role in SIRS [18] and induces severe vascular inflammation and leakage by activation of VEGF.[19] Therefore, it is considered reasonable to use anti-IL 6 antibody for patients with SIRS complicating AOSD.[19] Indeed, in the present case, there was marked elevation of IL-6, TNFα, and VEGF; after recovery from SIRS induced by anti-IL-6 receptor treatment, VEGF decreased (Fig. 2).
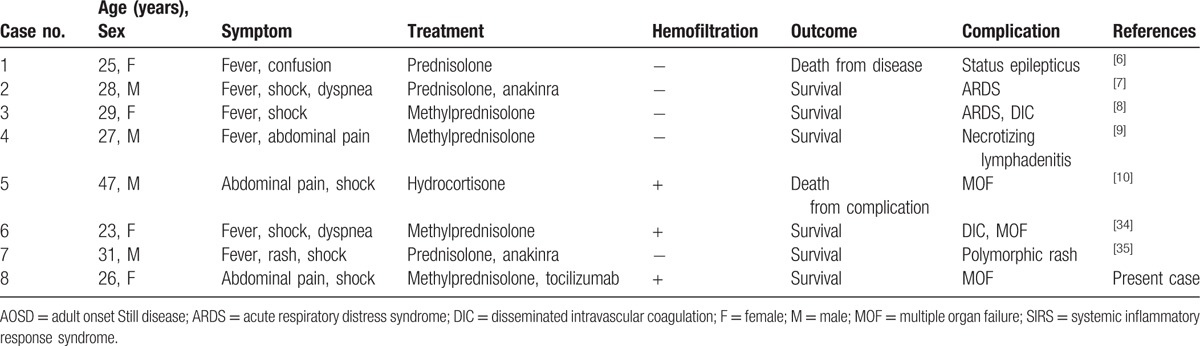
The causative roles of IL-1 and IL-6 in AOSD have been demonstrated through the responses to direct inhibition of anti-IL-1 receptor antibody (anakinra) and anti-IL-6 receptor antibody (tocilizumab).[20–25] On the other hand, anti-TNFα agents were not effective in patients with refractory AOSD.[26] Therefore, anakinra and tocilizumab have been recently recommended for refractory systemic AOSD.[1,27] Indeed, tocilizumab has been shown to be effective for refractory AOSD around the world.[25,28–33] However, it remains unknown whether tocilizumab would be effective for SIRS complicating AOSD. Although SIRS complicating AOSD is extremely rare and mild cases are responsive to high-dose steroids,[8,34] it can also be fatal.[6] We searched for MEDLINE from January 1, 1991 to December 1, 2016 using a search strategy with the terms “Adult onset Still disease” and “Systemic inflammatory response syndrome,” and/or “edema” and/or “shock”. Eight published cases of SIRS complicating AOSD, including the present case, were found. The mean age of these eight cases was 29.5 years (range, 23–47 years) (Table 1).[6–10,34,35] To the best of our knowledge, this patient represents the first reported case of severe SIRS complicating steroid-resistant AOSD that was successfully treated with anti-IL-6 antibody (Table 1).
Table 1.
It has been reported and reviewed that tocilizumab has been effective for AOSD complicated with macrophage activation syndrome (MAS), which is another form of acute systemic inflammatory syndrome characterized by hemophagocytic syndrome leading to pancytopenia and organ dysfunction.[36] MAS is also an important complication in AOSD, although bone marrow biopsy showed no hemophagocytic syndrome in the present case. Furthermore MAS and even SIRS should be recognized as a complication of tocilizumab treatment.[19,37]
Although the pathologic examination did not show hyperplastic lymph nodes containing hyalinized follicles and/or marked interfollicular vascular proliferation, the possibility of Castleman disease could not be ruled out completely because steroid therapy has already been started before the patient was transferred to our department. A new entity of Castleman disease has been proposed as TAFRO syndrome,[38] which comprise a combination of thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, organomegaly, and hypergammaglobulinemia. The present case showed thrombocytopenia, anasarca, and renal dysfunction, but did not show myelofibrosis and hypergammaglobulinemia. In Japan, tocilizumab is generally used for the treatment of Castleman disease and polyarticular juvenile idiopathic arthritis. These similar clinical manifestations to TAFRO syndrome accompanying SIRS with AOSD in this patient made it seem reasonable to administer tocilizumab in addition to cyclosporine, steroids, and plasma exchange.
SIRS complicating AOSD is rare, but can lead to multiple organ failure and be life-threatening. Therefore, SIRS should not be overlooked as a potential complication of AOSD, especially when a patient presents with systemic edema. Anti-IL-6 receptor antibody can be used safely and effectively for severe AOSD refractory to usual immunosuppressive agents and plasma exchange.
Footnotes
Abbreviations: AOSD = adult-onset Still disease, CHDF = continuous hemodiafiltration, IL-6 = interleukin-6, SIRS = systemic inflammatory response syndrome, TNFα = tumor necrosis factor alpha, VEGF = vascular endothelial growth factor.
The authors declare no conflicts of interest.
References
- [1].Gerfaud-Valentin M, Jamilloux Y, Iwaz J, et al. Adult-onset Still's disease. Autoimmun Rev 2014;13:708–22. [DOI] [PubMed] [Google Scholar]
- [2].Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int 2010;30:855–62. [DOI] [PubMed] [Google Scholar]
- [3].Slagt C, Malagon I, Groeneveld AB. Systematic review of uncalibrated arterial pressure waveform analysis to determine cardiac output and stroke volume variation. Br J Anaesth 2014;112:626–37. [DOI] [PubMed] [Google Scholar]
- [4].Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol 1992;19:424–30. [PubMed] [Google Scholar]
- [5].Efthimiou P, Kadavath S, Mehta B. Life-threatening complications of adult-onset Still's disease. Clin Rheumatol 2014;33:305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hong YH, Lee CK. A case of adult onset Still's disease with systemic inflammatory response syndrome complicated by fatal status epilepticus. Rheumatol Int 2008;28:931–3. [DOI] [PubMed] [Google Scholar]
- [7].Guignard S, Dien G, Dougados M. Severe systemic inflammatory response syndrome in a patient with adult onset Still's disease treated with the anti-IL1 drug anakinra: a case report. Clin Exp Rheumatol 2007;25:758–9. [PubMed] [Google Scholar]
- [8].Iglesias J, Sathiraju S, Marik PE. Severe systemic inflammatory response syndrome with shock and ARDS resulting from Still's disease: clinical response with high-dose pulse methylprednisolone therapy. Chest 1999;115:1738–40. [DOI] [PubMed] [Google Scholar]
- [9].Assimakopoulos SF, Karamouzos V, Papakonstantinou C, et al. Suppurative necrotizing granulomatous lymphadenitis in adult-onset Still's disease: a case report. J Med Case Rep 2012;6:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mejjad O, Vittecoq O, Tamion F, et al. A shock associated with adult-onset Still's disease. Joint Bone Spine 2001;68:76–8. [DOI] [PubMed] [Google Scholar]
- [11].Namas R, Nannapaneni N, Venkatram M, et al. An unusual case of adult-onset Still's disease with hemophagocytic syndrome. Necrotic leukoencephalopathy and disseminated intravascular coagulation. Case Rep Rheumatol 2014;2014:128623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mimura T, Shimodaira M, Kibata M, et al. Adult-onset Still's disease with disseminated intravascular coagulation and hemophagocytic syndrome: a case report. BMC Res Notes 2014;7:940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Park JH, Bae JH, Choi YS, et al. Adult-onset Still's disease with disseminated intravascular coagulation and multiple organ dysfunctions dramatically treated with cyclosporine A. J Korean Med Sci 2004;19:137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dua AB, Manadan AM, Case JP. adult onset Still's disease presenting with acute respiratory distress syndrome: case report and review of the literature. Open Rheumatol J 2013;7:125–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mora Alfonso SA, Rodriguez DM, Londono JD, et al. Acute adult-onset still's disease presenting as pulmonary hemorrhage, urticaria, angioedema and leukemoid reaction: a case report and literature review. Springer Plus 2015;4:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mubashir E, Ahmed MM, Hayat S, et al. Pulmonary hypertension in a patient with adult-onset Stills disease. Clin Rheumatol 2007;26:1359–61. [DOI] [PubMed] [Google Scholar]
- [17].Naniwa T, Tamechika S, Iwagaitsu S, et al. Successful use of higher-dose etanercept for multirefractory systemic flare of adult-onset Still's disease with liver failure with no response to tocilizumab therapy. Case Rep Rheumatol 2013;2013:923497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Oda S, Hirasawa H, Shiga H, et al. Sequential measurement of IL-6 blood levels in patients with systemic inflammatory response syndrome (SIRS)/sepsis. Cytokine 2005;29:169–75. [DOI] [PubMed] [Google Scholar]
- [19].Kang S, Tanaka T, Kishimoto T. Therapeutic uses of anti-interleukin-6 receptor antibody. Int Immunol 2015;27:21–9. [DOI] [PubMed] [Google Scholar]
- [20].Naniwa T, Ito R, Watanabe M, et al. Case report: successful use of short-term add-on tocilizumab for multirefractory systemic flare of adult-onset Still's disease. Clin Rheumatol 2013;32(suppl 1):S103–6. [DOI] [PubMed] [Google Scholar]
- [21].Nakahara H, Mima T, Yoshio-Hoshino N, et al. A case report of a patient with refractory adult-onset Still's disease who was successfully treated with tocilizumab over 6 years. Mod Rheumatol 2009;19:69–72. [DOI] [PubMed] [Google Scholar]
- [22].Matsumoto K, Nagashima T, Takatori S, et al. Glucocorticoid and cyclosporine refractory adult onset Still's disease successfully treated with tocilizumab. Clin Rheumatol 2009;28:485–7. [DOI] [PubMed] [Google Scholar]
- [23].De Bandt M, Saint-Marcoux B. Tocilizumab for multirefractory adult-onset Still's disease. Ann Rheum Dis 2009;68:153–4. [DOI] [PubMed] [Google Scholar]
- [24].Iwamoto M, Nara H, Hirata D, et al. Humanized monoclonal anti-interleukin-6 receptor antibody for treatment of intractable adult-onset Still's disease. Arthritis Rheum 2002;46:3388–9. [DOI] [PubMed] [Google Scholar]
- [25].de Boysson H, Fevrier J, Nicolle A, et al. Tocilizumab in the treatment of the adult-onset Still's disease: current clinical evidence. Clin Rheumatol 2013;32:141–7. [DOI] [PubMed] [Google Scholar]
- [26].Fautrel B, Sibilia J, Mariette X, et al. Tumour necrosis factor alpha blocking agents in refractory adult Still's disease: an observational study of 20 cases. Ann Rheum Dis 2005;64:262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Al-Homood IA. Biologic treatments for adult-onset Still's disease. Rheumatology (Oxford) 2014;53:32–8. [DOI] [PubMed] [Google Scholar]
- [28].Puechal X, DeBandt M, Berthelot JM, et al. Tocilizumab in refractory adult Still's disease. Arthritis Care Res (Hoboken) 2011;63:155–9. [DOI] [PubMed] [Google Scholar]
- [29].Elkayam O, Jiries N, Dranitzki Z, et al. Tocilizumab in adult-onset Still's disease: the Israeli experience. J Rheumatol 2014;41:244–7. [DOI] [PubMed] [Google Scholar]
- [30].Song ST, Kim JJ, Lee S, et al. Efficacy of tocilizumab therapy in Korean patients with adult-onset Still's disease: a multicentre retrospective study of 22 cases. Clin Exp Rheumatol 2016;34:S64–71. [PubMed] [Google Scholar]
- [31].Nishina N, Kaneko Y, Kameda H, et al. The effect of tocilizumab on preventing relapses in adult-onset Still's disease: a retrospective, single-center study. Mod Rheumatol 2015;25:401–4. [DOI] [PubMed] [Google Scholar]
- [32].Ortiz-Sanjuan F, Blanco R, Calvo-Rio V, et al. Efficacy of tocilizumab in conventional treatment-refractory adult-onset Still's disease: multicenter retrospective open-label study of thirty-four patients. Arthritis Rheumatol 2014;66:1659–65. [DOI] [PubMed] [Google Scholar]
- [33].Kobayashi D, Ito S, Murasawa A, et al. Two cases of adult-onset Still's disease treated with tocilizumab that achieved tocilizumab-free remission. Intern Med 2015;54:2675–9. [DOI] [PubMed] [Google Scholar]
- [34].Ames PR, Walker E, Aw D, et al. Multi-organ failure in adult onset Still's disease: a septic disguise. Clin Rheumatol 2009;28(suppl 1):S3–6. [DOI] [PubMed] [Google Scholar]
- [35].Michailidou D, Shin J, Forde I, et al. Typical evanescent and atypical persistent polymorphic cutaneous rash in an adult Brazilian with Still's disease: a case report and review of the literature. Auto Immun Highlights 2015;6:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Watanabe E, Sugawara H, Yamashita T, et al. Successful tocilizumab therapy for macrophage activation syndrome associated with adult-onset Still's disease: a case-based review. Case Rep Med 2016;2016: 5656320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tsuchida Y, Sumitomo S, Shoda H, et al. Macrophage activation syndrome associated with tocilizumab treatment in adult-onset Still's disease. Mod Rheumatol 2017;27:556–7. [DOI] [PubMed] [Google Scholar]
- [38].Kawabata H, Takai K, Kojima M, et al. Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop 2013;53:57–61. [DOI] [PubMed] [Google Scholar]