

SUMMARY
Mutations in cancer reprogram amino acid metabolism to drive tumor growth, but the molecular mechanisms are not well understood. Using an unbiased proteomic screen, we identified mTORC2 as a critical regulator of amino acid metabolism in cancer via phosphorylation of the cystine-glutamate antiporter xCT. mTORC2 phosphorylates serine 26 at the cytosolic N-terminus of xCT, inhibiting its activity. Genetic inhibition of mTORC2, or pharmacologic mTOR kinase inhibition, promotes glutamate secretion, cystine uptake and incorporation into glutathione linking growth factor receptor signaling with amino acid uptake and utilization. These results identify an unanticipated mechanism regulating amino acid metabolism in cancer, enabling tumor cells to adapt to changing environmental conditions.
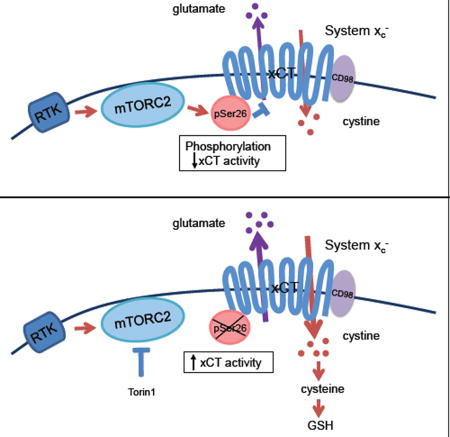
Graphical Abstract
INTRODUCTION
Dysregulated amino acid metabolism is an emerging hallmark of cancer (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). Tumor cells take up amino acids from the extracellular environment as a carbon and nitrogen source for protein and nucleotide synthesis (DeBerardinis et al., 2008). Uptake of amino acids from the tumor microenvironment also contributes to one-carbon metabolism and redox maintenance (Altman et al., 2016; Yang and Vousden, 2016). Macropinocytosis, a recently described opportunistic pathway of amino acid uptake (Commisso et al., 2013; Pavlova and Thompson, 2016), provides one mechanism for coupling cancer cell proliferation with amino acid availability. However, tumor cells may also regulate amino acid uptake by modulating the level or activity of specific amino acid transporters (Bhutia et al., 2015). Currently, the underlying molecular mechanisms of amino acid transporter regulation in cancer are not well understood.
The cystine-glutamate antiporter xCT encoded by the SLC7A11 gene, is highly expressed in multiple human cancer types, including triple negative breast cancer and glioblastoma (GBM) (Chung et al., 2005; Takeuchi et al., 2012; Timmerman et al., 2013). xCT is a 12-pass transmembrane protein, which together with its binding partner CD98 (SLC3A2) forms the amino acid transporter system xc−. The primary function of system xC− is to take up cystine, the oxidized dimeric form of cysteine, in exchange for glutamate, contributing to tumor growth (Bassi et al., 2001; Lewerenz et al., 2012). In nutrient depleted conditions, cystine uptake is critical for glutathione synthesis to buffer reactive oxygen species (ROS), whereas in nutrient replete conditions, glutamate can contribute to many anabolic reactions (Commisso et al., 2013; Conrad and Sato, 2012; DeBerardinis et al., 2008; Kim et al., 2001). Thus, post-translational mechanisms of xCT regulation may be important for enabling tumor cells to rapidly respond to changing environmental conditions. In triple negative breast cancer, extracellular glutamate inhibits xCT through a paracrine mechanism, inducing HIF to drive tumor growth (Briggs et al., 2016), suggesting that xCT may be highly responsive to extracellular amino acids. Suppression of xCT activity results in intracellular cysteine depletion, which directly inhibits HIF prolyl hydroxylases, thereby inducing HIF to promote tumor growth (Briggs et al., 2016). We hypothesized that cell autonomous signaling mechanisms could provide an additional route of xCT regulation.
To identify complementary pathways of xCT regulation, we performed an unbiased mass spectrometry proteomics screen to identify xCT binding partners. Here we discovered an unanticipated mechanism of cross talk between altered growth factor receptor signaling and glutamate-cystine metabolism in tumor cells, linking growth factor receptor signaling with amino acid metabolism in cancer.
RESULTS
Unbiased screen identifies mTORC2 as a binding partner of xCT
We stably expressed a FLAG-tagged xCT or vector control in GBM cells and used Stable Isotope Labeling in Cell culture (SILAC) (Ong and Mann, 2006) coupled to mass spectrometry to identify xCT binding partners. xCT-bound complexes were immunoprecipitated and subjected to quantitative LC/MS-MS (Figure 1A), revealing 125 potential xCT binding proteins with a median fold enrichment of xCT/vector > 10, Log10 (xCT/vector) > 1 (Figure 1B and Table S1), that enriched in pathways involved in cellular and protein metabolism by DAVID (Huang da et al., 2009) Gene Ontology (GO) analysis (Figure 1C and Table S2). Established xCT binding partners including CD98 (SLC3A2), part of system xC−, and CD44, an obligate binding partner (Ishimoto et al., 2011), were identified as well as the recently described binding partner epidermal growth factor receptor (EGFR) (Tsuchihashi et al., 2016). Surprisingly, Rictor and mTOR, which are core components of mTOR complex 2 (mTORC2), were also identified as potential xCT binding partners (Figure 1B). No additional AGC kinases were detected.
Figure 1. xCT physically interacts with mTOR Complex 2 in GBM cells.

(A) A brief schematic of the SILAC labeling and Mass Spectrometry experiment performed to identify xCT specific binding proteins in U87EGFRvIII cells.
(B) The median fold enrichment of the identified proteins was plotted on a Log10 scale as xCT versus vector. A cutoff of Log10 (xCT/vector) <1 was applied and indicated by the shaded area below the dash line. Known xCT binding proteins as well as mTOR and Rictor were labeled in red.
(C) DAVID gene ontology (GO) analysis was performed using the list of 125 potential xCT binding proteins identified in (B). Top 10 enriched biological pathways were plotted using the – (Log10 FDR). The enriched pathway that contain both mTOR and Rictor were indicated in red and the full gene list of each pathway can be found in Table S2.
(D) Co-immunoprecipitation (Co-IP) was performed to validate mTOR and Rictor as xCT binding proteins in GBM (T98G), breast cancer (MDA-MB-231, Hs578T), and lung cancer (A549) cell lines stably expressing the FLAG-tagged xCT or vector control.
In GBM, lung cancer and triple-negative breast cancer cell lines, Co-Immunoprecipitation (Co-IP) experiments confirmed the physical association between xCT and endogenous mTORC2 components mTOR and Rictor. Importantly, Raptor, which is specific to mTORC1 (Figures 1D and S1A), was not identified in the SILAC screen and was not detected by Co-IP analysis, thus confirming that the physical association with xCT was specific to mTORC2. Reverse Co-IP confirmed the binding of xCT to both a FLAG-tagged mTOR and a myc-tagged Rictor, in GBM cells (Figure S1B). These data demonstrate that xCT specifically interacts with mTORC2, but not mTORC1, in GBM cells.
mTORC2 phosphorylates xCT downstream of growth factor signaling in GBM
mTORC2 is a serine/threonine kinase that is a core-component of altered growth factor receptor signaling in some cancer types, including GBM (Masui et al., 2015a; Masui et al., 2013; Tanaka et al., 2011). EGFRvIII mutation in GBM cells, or ligand stimulation of EGFR and/or PTEN loss, activates mTORC2 to promote tumor growth (Tanaka et al., 2011), potentially by phosphorylating AGC kinases including Akt, SGK1 as well as several members in the PKC family such as PKCα, PKCδ and PKCξ (Jacinto and Lorberg, 2008; Kennedy and Lamming, 2016; Pearce et al., 2010). mTORC2 has also recently been identified as being responsive to nutrient levels (Masui et al., 2013; Moloughney et al., 2016), and is involved in regulating a number of essential metabolic pathways in cancer, including glycolysis, glutaminolysis, de novo lipid synthesis and nucleotide and ROS metabolism (Aramburu et al., 2014; Dang, 2012; Lamming and Sabatini, 2013). Rictor overexpression did not affect the levels of xCT mRNA, excluding effects of mTORC2 on xCT transcription, at least in the time course studied (Figure S2A). Therefore, we hypothesized that mTORC2 could possibly regulate xCT activity through phosphorylation. A number of serine and threonine residues on xCT that have been previously reported to be potential phosphorylation sites including S26, S51 and S481 (Hornbeck et al., 2015; Lundby et al., 2012; Schweppe et al., 2013; Yu et al., 2011; Zhou et al., 2013), could potentially serve as targets of mTORC2. Importantly, S26, S51 and S481 are all preceded by an arginine at the −3 position (RXXS/T) (Figure 2A), which suggest that they could belong to the broad category of AGC kinase family substrates (Alessi et al., 1996; Pearce et al., 2010).
Figure 2. mTORC2 phosphorylates xCT downstream of growth factor signaling.

(A) RXXS/T motifs on xCT were listed by analyzing xCT protein sequence. S26 (in red) phosphorylation was detected in our study and has been reported by others. S51 and S481 (in blue) phosphorylation were reported on PhosphoSitePlus but were not detected in our experiments. Phosphorylation of the remaining RXXS/T sites (in black) on xCT has not been reported in any other studies or observed in our experiments (Hornbeck et al., 2015). (http://www.phosphosite.org/uniprotAccAction?id=Q9UPY5.)
(B) Immunoprecipitation (IP) - western blot was performed in U87 cells stably expressing wtEGFR and myc-tagged xCT. Cells were serum starved for 24 h post 24 h of transfection with siRNA before stimulation with 25 ng/ml EGF. Cell lysates were collected at indicated time points and subjected to pRXXS/T IP and western blotting analysis.
(C) U87EGFRvIII cells stably overexpressing xCT or vector control were treated with 250 nM Torin1. Protein lysates were collected over a time course of 24 h for IP-western blot to determine xCT phosphorylation on RXXS/T motifs.
See also Figure S2.
To test the hypothesis that mTORC2 regulates xCT phosphorylation in response to growth factor signaling, we knocked down Rictor or Raptor with siRNAs in GBM cells stably expressing wild-type EGFR, and examined xCT phosphorylation after stimulation with EGF. To broadly monitor the state of xCT serine/threonine phosphorylation, we performed immunoprecipitation of cellular lysates using phospho-RXXS/T antibody conjugated beads, followed by immunoblotting for myc-tagged xCT. As shown in Figure 2B, EGF stimulation increased xCT phosphorylation, which was abrogated by Rictor knockdown, demonstrating that EGF signaling promotes xCT serine/threonine phosphorylation in an mTORC2-dependent manner. Inhibition of mTORC1 has been shown to hyperactivate mTORC2 signaling through relief of IRS-1 dependent feedback inhibition (Dibble et al., 2009; Harrington et al., 2004; Manning, 2004). Consistent with this model, Raptor knockdown increased mTORC2 signaling in an IRS-dependent fashion (Figure S2B), leading to elevated xCT phosphorylation (Figure 2B). In addition, the mTOR kinase inhibitor Torin1, which blocks both mTORC1 and mTORC2 activity (Liu et al., 2010), but not the mTORC1-specific inhibitor Rapamycin (Figure S2F), significantly inhibited xCT phosphorylation on RXXS/T motifs in GBM cells (Figures 2C and S2F).
mTORC2 phosphorylates and activates downstream AGC kinases including PKCa, Akt and SGK1, amplifying the signaling cascade by phosphorylating a much broader range of downstream substrates involved in various cellular processes (Laplante and Sabatini, 2009, 2012). Therefore, we tested the possibility of whether xCT phosphorylation was regulated by any of the AGC kinases downstream of mTORC2. Surprisingly, we did not detect physical interaction between any AGC kinases including PKCα, Akt or SGK1 and xCT in either the SILAC or Co-IP experiments across different cell lines (Table S1 and Figures S2C and S2D). Furthermore, neither siRNA-mediated genetic knockdown, nor pharmacological inhibition of PKCα, Akt and SGK1 (Halland et al., 2015) suppressed xCT phosphorylation upon EGF stimulation (Figures S2E and S2F), suggesting that downstream effector AGC kinases are not required for mTORC2-mediated xCT phosphorylation.
xCT is phosphorylated at serine 26 in the cytosolic N-terminus by mTORC2
xCT is a twelve-transmembrane protein (Gasol et al., 2004). We hypothesized that mTORC2-dependent phosphorylation of xCT would be more likely to occur on cytosolic domains, which are more accessible to kinases including mTORC2 (Figure 3A). Consistent with this hypothesis, deletion of xCT’s cytosolic N-terminus completely abrogated the phosphorylation of xCT on RXXS/T motifs. In contrast, deletion of xCT’s cytosolic C-terminus had no effect on xCT phosphorylation (Figures 3B and 3C). Several previous large-scale quantitative LC/MS-MS phosphoproteomic studies identified phosphorylation of xCT on serine 26 at the cytosolic N-terminus (Schweppe et al., 2013; Zhou et al., 2013), including the demonstration that xCT serine 26 phosphorylation was decreased by an mTOR kinase inhibitor Ku-0063794 but not rapamycin in mouse embryonic fibroblasts (Yu et al., 2011). In addition, xCT serine 26 resides within an mTOR substrate motif defined in part by a proline or glycine at −1 position and a phenylalanine, proline or leucine at the +1 position previously identified by Hsu et al (Hsu et al., 2011). These data raised the possibility that mTORC2 might regulate xCT by phosphorylating serine 26 of xCT’s N-terminus cytosolic domain. Serine 26 of xCT is largely conserved across species (Figure 3D) (Hornbeck et al., 2015), suggesting that it may be a biologically important phosphorylation site.
Figure 3. mTORC2 phosphorylates xCT on serine26.

(A) A simplified schematic diagram of xCT 2D structure constructed based on sequence and predicted domains of xCT obtained from UniProt-KB. Transmembrane domains were shown as cylinders. Potential phosphorylation sites within RXXS/T motifs were labeled with the same color code as in Fig.2A.
(B) A simplified schematic diagram depicting xCT mutants generated and used in the following experiments.
(C) Phosphorylation on RXXS/T motifs in wildtype and mutant xCT were analyzed by pRXXS/T IP and western blot.
(D) Phosphorylation on xCT serine 26 is conserved across species. (Hornbeck et al., 2015). (http://www.phosphosite.org/uniprotAccAction?id=Q9UPY5.)
(E) Schematic of LC-MS/MS to identify potential phosphorylation sites on xCT in GBM cells.
(F) Tandem mass spectrometry (MS/MS) spectra showing phosphorylation of xCT on serine 26 in U87EGFRvIII cells.
(G) In vitro kinase assay was carried out by incubating mTORC2 IP-purified from HEK293T cells, peptide substrates and [γ-32P]-ATP in kinase reaction buffer at room temperature for 1 h. Scintillation counts from three independent replicates were presented as mean counts per minute (cpm) ± SEM. Statistical analysis was performed using one-way ANOVA. *** refers to p value < 0.001. n.s. refers to not statistically significant.
(H) IP-western blot detecting wild-type or S26A mutant xCT phosphorylation on RXXS/T motifs upon EGF stimulation.
See also Figure S3.
To test the hypothesis that mTORC2 regulates xCT by phosphorylating it on serine 26, we immunoprecipitated the FLAG-tagged xCT protein, and subjected samples to LC/MS-MS analysis after peptide fractionation by hydrophilic interaction chromatography (HILIC) (Figure 3E). As shown in Figure 3F, xCT is phosphorylated on serine 26 in GBM cells. To determine whether serine 26 of xCT is indeed an mTORC2 substrate, we performed an in vitro kinase assay using purified mTORC2 and peptides containing the xCT serine 26 sequence (Figure S3A). xCT S26 (serine 26) phosphorylation at even higher levels compared to Akt S473 (serine 473), an established mTORC2 substrate (Sarbassov et al., 2005), was detected. Further, the phosphorylation resistant mutant xCT S26A (serine 26 to alanine mutation) was not phosphorylated by mTORC2. (Figure 3G). In contrast, xCT S26 could not be phosphorylated by SGK1, and phosphorylation of xCT S26 by Akt1 was markedly less than that of GSK3β, a known Akt1 substrate (Figure S3B). Importantly, xCT S26A mutant could no longer be phosphorylated upon growth factor stimulation in GBM cells (Figures 3H and S3C) and phosphorylation of the S26A xCT did not change in response to Torin1 treatment (Figure 2C). Taken together, these data suggest that mTORC2 phosphorylates xCT on serine 26 in response to EGFR signaling.
Phosphorylation resistant mutant S26A increases xCT activity
To examine the effect of mTORC2-dependent phosphorylation of serine 26 on xCT function, we measured glutamate secretion using a series of strategic mutants. First, we deleted either the cytosolic N or C terminus of xCT, revealing that both domains were important for xCT function, as measured by glutamate secretion (Figure S4A), although the mechanisms by which each domain regulates xCT activity appeared to differ. Deletion of the C-terminus of xCT prevented it from binding to CD98 (Figures S4B and S4C), which is required for xCT recruitment onto the plasma membrane (Bassi et al., 2001). In contrast, the interaction between CD98 and xCT remained intact in the N-terminus deletion mutant, indicating that the cytosolic N-terminus regulates xCT function through alternative mechanisms (Figures S4B and S4C). This prompted us to test whether serine 26 phosphorylation could be the point of regulation at the N-terminus of xCT.
In the previous Co-IP experiment, we observed that the endogenous xCT could bind to the exogenously overexpressed xCT protein (Figure S2C). Thus, to exclude potential effects of endogenous xCT binding, we obtained xCT knockout MEFs and generated stable cell lines overexpressing the wild-type xCT, or the phosphorylation resistant mutant S26A (Figure 4A). Since the glutamate transport function of xCT is Na+ independent and requires the presence of extracellular cystine, we measured glutamate secretion in xCT KO MEF cell lines in a Na+ free PBS buffer system as reported previously (Kobayashi et al., 2015), and compared glutamate secretion in the absence or presence of cystine as well as the xCT inhibitor sulfasalazine (SAS) (Gout et al., 2001) to exclude possible glutamate efflux through other transporters. The phosphorylation resistant mutant S26A significantly increased glutamate secretion (Figure 4B). Together, these data suggest that xCT activity is increased when mTORC2 mediated phosphorylation on serine 26 is ablated.
Figure 4. Inhibition of xCT phosphorylation on serine 26 increases glutamate/cystine transport and supports glutathione synthesis.

(A) Cell surface and total protein levels of xCT were analyzed by western blotting in xCT KO MEFs stably overexpressing vector control, wt xCT and xCT S26A. Band intensities were quantified by densitometry and relative surface xCT levels was calculated, and normalized to Na, K-ATPase as loading control of cell surface proteins.
(B) xCT activity assay was performed in xCT KO MEFs stably overexpressing vector control, wt xCT and xCT S26A. Glutamate secretion was measured using the AmplexRed glutamate assay kit, calculated and normalized to cell counts and cell surface xCT protein levels. Results were obtained from three replicates and data are presented as mean ± SEM. Statistical analysis was performed using one-way ANOVA. *** refers to p value < 0.001.
(C) Glutamate secretion was measured using the AmplexRed glutamate assay kit in U87EGFRvIII cells with stable Rictor knockdown using two different shRNAs. Results were obtained from three independent replicates and data are presented as mean ± SEM. *** refers to p value < 0.001. Statistical analysis was performed using two-way ANOVA comparing the mean of shRictor1 and shRictor2 to shscramble control.
(D) Glutamate secretion was measured in U87EGFRvIII cells treated with Torin1 for 24 h after transfected with 50 nM siRNA targeting xCT for 48 h by NOVA Bioprofile 400 analyzer. Results were obtained from three independent replicates and data are presented as mean ± SEM. **refers to p value < 0.01. n.s. refers to not statistically significant. Statistical analysis was performed using two-way ANOVA comparing the mean of Torin1 to DMSO control in each conditions.
(E) Cystine uptake was measured in U87EGFRvIII cells after treatment with 250 nM Torin1 for 24 h using a sodium cyanide and sodium nitroprusside based assay (Egea et al., 2015; Nakagawa and Coe, 1999). Results were obtained from seven replicates and data are presented as mean ± SEM. *** refers to p value < 0.001.
(F) A brief schematic showing the labeling process of GSH and GSSG with [3,3’-13C2] L-Cystine.
(G) Exogenous cystine incorporation into newly synthesized glutathione was determined by labeling cells with [3,3’-13C2] L-Cystine together with 24 h of DMSO or Torin1 treatment in DMEM supplemented with 5% dialyzed FBS. The labeling percentage of both GSH and GSSG by [3,3’-13C2] L-Cystine were calculated. Each column represents three replicate samples collected at each time point. Data are presented as mean ± SEM. *** refers to p value < 0.001. n.s. refers to not statistically significant.
(H) Total cellular GSH levels were measured using the GSH/GSSG-Glo™ Glutathione Assay Kit (Promega). U87EGFRvIII cells were treated with DMSO or 250 nM Torin1 for 24 h after being transfected with xCT siRNA for 48 h. Total cellular GSH levels were normalized to blank control and cell counts. Results were obtained from three replicate samples and data are presented as mean ± SEM. Statistical analysis was performed using one-way ANOVA. *** refers to p value < 0.001.
(I) U87EGFRvIII cells were treated with DMSO or 250 nM Torin1 with or without µM erastin which has been reported to induce ferroptosis in several cancer cell lines. Cells were collected and stained with FITC-Annexin V and PI after 24 h of treatment and cell death was analyzed by flow cytometry. %Ferroptotic cells shown in the bar graph on the right was calculated by adding up the percentage of cells in the upper and lower right quadrant in each graph on the left.
See also Figure S4.
mTORC2 inhibition increases xCT activity, cystine uptake, and incorporation into glutathione
Having shown that inhibition of the mTORC2-mediated phosphorylation on serine 26 increases xCT activity, we hypothesized that inhibition of mTORC2 should also have the same effect. Genetic inhibition of mTORC2 by two different Rictor shRNAs significantly increased glutamate secretion through xCT (Figures 4C, S4D and S4E). Treatment with Torin1 phenocopied Rictor knockdown, significantly increased xCT-specific glutamate secretion (Figures 4D, S4D and S4E) and cystine uptake (Figure 4E) in multiple GBM cell lines, as well as in triple negative breast cancer and lung cancer cell lines - which have high levels of xCT and mTORC2 activity (Figure S4E) (Briggs et al., 2016; Masui et al., 2013), while xCT is not affected by Torin1 in normal human astrocytes (NHA) (Figure S4F). These results demonstrate that genetic inhibition of mTORC2 or pharmacological inhibition of mTOR kinase increases xCT activity.
GBM cells as well as many other cancer cells are well known for relying on uptake of extracellular cystine for glutathione synthesis (Chung et al., 2005). Therefore, we performed metabolic tracer analysis using 13C-labeled cystine, to examine whether cystine incorporation into glutathione is consistent with increased cystine uptake by mTOR kinase inhibition. As predicted by our model, Torin1 treatment enhanced cystine uptake (Figure 4E) and incorporation of the labeled cystine into glutathione (Figure 4G). Labeled cystine incorporation into both GSH and GSSG was increased over time in Torin1 treated cells before steady state was reached with 100% labeling at 24h (Figures 4F and 4G). In addition, xCT knockdown further significantly decreased glutathione levels in addition to Torin1 (Figure 4H), and combination of an xCT inhibitor erastin with Torin1 resulted in significant GBM cell death while cell survival was not affected by either drug alone (Figure 4I), indicating that increased xCT activity has a major contribution to glutathione synthesis and GBM cell survival upon pharmacological mTOR kinase inhibition. More importantly, consistent with our previous findings that mTORC2 activity is responsive to glucose availability in GBM cells, glucose deprivation also inhibited mTORC2-mediated xCT phosphorylation upon EGF stimulation (Figure S4G). Taken together, these data demonstrate a critical role for mTORC2 in linking growth factor receptor signaling with glucose, amino acid and glutathione metabolism in cancer.
DISCUSSION
By using an unbiased proteomic screen for xCT binding partners, followed by functional validation, we have made the surprising discovery that mTORC2 regulates amino acid metabolism in tumor cells by phosphorylating serine 26 of the cystine-glutamate antiporter xCT on its cytosolic N-terminus to suppress glutamate secretion. Aberrant growth factor receptor signaling and or c-MYC activation increase glutamine uptake, converting it to glutamate to provide tumor cells with a carbon source for TCA anaplerosis as well as a nitrogen source for protein and nucleotide synthesis (Altman et al., 2016; DeBerardinis et al., 2008; Masui et al., 2015a). Thus, when microenvironmental nutrient levels are sufficient to support tumor cell proliferation, it would be disadvantageous for cancer cells to secrete glutamate. The mechanism identified here ensures that glutamine-derived glutamate can be used primarily for tumor growth when extracellular nutrient levels can support it. However, when nutrients become scarce, it would be advantageous for tumor cells to increase xCT-dependent cystine uptake at the expense of glutamate efflux, enabling tumor cells to buffer cellular redox stress by synthesizing glutathione from xCT-derived cystine. Therefore, the mechanism described here enables tumor cells to adapt to changing nutrient levels, linking proliferative signals to environmental conditions. It is interesting to note that mTORC2 has recently been shown to require either glucose or acetate in order to phosphorylate its downstream substrates (Masui et al., 2015b), raising the possibility that under nutrient poor conditions, lower mTORC2 signaling could tilt the balance from proliferation to survival, at least in part by favoring glutamate efflux, cystine uptake and glutathione synthesis to protect tumor cells from cellular stress.
xCT is a 12-pass transmembrane protein that has two serine residues preceded by an arginine at the −3 position (RXXS/T) , S26, S51 on its N-terminus that may serve as consensus phosphorylation sites for mTORC2. Unlike S51, which lies in the transmembrane domain, S26 is predicted to reside on the cytoplasmic face of the membrane, where it could be engaged by mTORC2 (Gasol et al., 2004; UniProt, 2015). Interestingly, in a SILAC-based mass spectrometric screen of TSC null MEFs to identify mTOR regulated proteins which identified Grb10 as an mTORC1 substrate, Yu and colleagues identified serine 26 of xCT as a site whose phosphorylation is inhibited by the mTOR kinase inhibitor Ku-0062794, but not by rapamycin (Yu et al., 2011), consistent with our finding that xCT serine 26 is an mTORC2 substrate. mTORC2 is thought to promote its biological activity by phosphorylating AGC kinases such as AKT, PKC and SGK1 which in turn phosphorylate their downstream substrates (Laplante and Sabatini, 2009, 2012). It is interesting to note that we found no evidence of xCT binding to these AGC kinases either by SILAC-Mass Spectrometry, or in the Co-IP studies, suggesting that mTORC2 may directly regulate xCT serine 26 phosphorylation.
High xCT levels are associated with poor outcome in a number of cancer types, including glioblastoma (Robert et al., 2015) and triple negative breast cancer (Timmerman et al., 2013). The mTORC2-dependent mechanism reported here, in addition to a recently described paracrine mechanism of xCT reported by Briggs and colleagues (Briggs et al., 2016), suggests that regulation of xCT function by post-translational modification may be critical for its tumor promoting effects. In triple negative breast cancer cells, high extracellular glutamate levels were demonstrated to suppress xCT function, depleting tumor cells of intracellular cysteine. Intracellular cysteine depletion was shown to cause oxidation of specific cysteine residues of the prolyl hydroxylase EglN1, thereby suppressing EglN1-dependent HIF1 α degradation, thus elevating intra-tumoral HIF1 α levels to drive tumor growth (Briggs et al., 2016). In addition, a recent study suggests that xCT plays an important in regulating nutrient flexibility (Shin et al., 2017). Our results identified an important molecular mechanism linking growth factor signaling with anapleurotic flux through phosphorylation of xCT on serine 26. Future studies will be needed to determine whether there is any cooperation between these complementary post-translational regulatory mechanisms.
The diversity of metabolic adaptations employed by cancer cells in response to rapidly changing conditions, contributes to their biological aggressiveness and therapeutic resistance by enabling them to proliferate when nutrients are plentiful and to shift their resources to survival when nutrients are scarce (Palm et al., 2015). The results presented here demonstrate that mTORC2 controls cystine uptake and glutathione metabolism by directly phosphorylating xCT, thus linking altered growth factor receptor signaling with amino acid metabolism and ROS buffering in cancer.
Star Methods Text
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Paul Mischel (pmischel@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and cell culture
All cell lines used in this study are listed in the Key Resource Table. Human cell lines (including U87, U87wtEGFR, U87EGFRvIII, U251, T98G, U373, Hs578T, MDA-MB-231, A549, HEK293T) were cultured in DMEM supplemented with 10% FBS. U87wtEGFR and U87EGFRvIII isogenic cell lines were established as described previously (Wang et al., 2006) Cell lines were not authenticated as they were obtained from ATCC. xCT KO MEFs were a kind gift from Dr. Hideyo Sato, Yamagata University, Japan (Kobayashi et al., 2015) and were cultured in DMEM supplemented with 10% FBS and additionally supplemented with 50uM β-mercaptoethanol. xCT KO MEFs were authenticated using PCR to confirm knockout of the xCT gene. GBM39, GBM6 and GSC11 patient-derived neurosphere lines were cultured in NeuroCult medium supplemented with epidermal growth factor, fibroblast growth factor, and heparin. Normal Human Astrocytes (NHA) were obtained from LONZA and were not authenticated. NHAs were cultured according to the manufacturer’s protocol using the AGM BulletKit. All cells were cultured in a humidified incubator with 5% CO2 at 37 °C.
METHOD DETAILS
Glutamate Secretion Assay
Glutamate secretion from cells were measured using a Nova BioProfile Basic Analyzer (Nova Biomedical), or with the Amplex® Red Glutamic Acid/Glutamate Oxidase Kit. Briefly, cells were seeded in triplicates in 6-well plates at optimal density, and 24 h before measurement cells were washed three times with 1 × PBS and changed to 1 ml fresh DMEM media supplemented with 5% dialyzed FBS, including three wells without cells as blank control. After incubation, media were collected from each well and analyzed by the BioProfile Basic Analyzer or using the Amplex® Red Glutamic Acid/Glutamate Oxidase Kit according to manufacturer’s instructions. Cell numbers were determined using the TC20TM Automated Cell Counter (Bio-Rad). Glutamate secretion was calculated by subtracting the levels of glutamate in the blank control and normalized to cell counts for each sample.
SILAC Labeling and Mass Spectrometry
U87EGFRvIII cells stably expressing the vector control or FLAG-xCT were cultured in DMEM SILAC media that lack lysine and arginine and supplemented with 10% dialyzed FBS. 12C6-L-arginine and 12C6-L-lysine were supplemented to the vector control cells and 13C6, 15N4-L-arginine and 13C6, 15N2-L-lysine were supplemented to the FLAG-xCT cells. Cells were passaged at least five times to ensure complete labeling (Ong and Mann, 2006). SILAC labelled cells were lysed in Pierce IP lysis buffer supplemented with Halt Protease and Phosphatase Inhibitor Cocktail. Protein lysates were cleared by centrifugation and incubated with anti-FLAG M2 affinity gel overnight at 4 °C. FLAG-xCT and its binding proteins were eluted with 0.1 M glycine, pH 2.5 at room temperature with rotation for 2 min and then neutralized with 1 M Tris-HCl, pH 7.8. The eluted proteins were then reduced, alkylated and digested with 1 µg of trypsin. Digested peptides were desalted using a 50 mg Sep-Pak C18 cartridge and fractionated using HILIC (Hydrophilic Interaction Liquid Chromatography) with a linear gradient from 19%-32% H2O with 0.01% TFA over 24 minutes on an TSKgel Amide-80 1 mm inner diameter column (TOSOH BioSci). The HILIC fractions were analyzed by LC-MS/MS on a LTQ XL-Orbitrap Discovery mass spectrometer with one full scan followed by 10 MS2 dependent scans. A Dionex UltiMate 3000 RSLC nano pump was used with a 70min gradient from 12–33% acetonitrile with 0.1% formic acid and a flow rate of 300 nl/min.
Western Blotting
Cultured cells were lysed with RIPA lysis buffer, and supplemented Halt Protease and Phosphatase Inhibitor Cocktail. Protein concentration of each sample was determined with Bradford Assay using the Protein Assay Dye Reagent Concentrate. Equal amounts of protein extracts were mixed with 4 × Laemmli sample buffer and separated by electrophoresis on 4–12% NuPAGE Bis-Tris Mini Gel, and then transferred using the Trans-Blot® TurboTM Transfer System (BioRad) onto nitrocellulose membranes. Membranes were blocked with 5% bovine serum albumin (BSA) and incubated with corresponding primary antibodies and horseradish peroxidase-conjugated secondary antibodies. The immunoreactivity was detected with SuperSignal™ West Pico Chemiluminescent Substrate or SuperSignal™ West Femto maximum Sensitivity Substrate. Signals were captured and analyzed using the Bio-Rad ChemiDoc™ MP Imaging system and the Image Lab™ Software (Bio-Rad).
Co-immunoprecipitation (Co-IP) and Immunoprecipitation (IP)
Cells were lysed with Pierce IP lysis buffer and supplemented with Halt Protease and Phosphatase Inhibitor Cocktail. Protein concentrations were determined by Bradford Assay using the Protein Assay Dye Reagent Concentrate for each sample and equal amounts of protein lysates were incubated with antibody-conjugated beads as indicated at 4 °C overnight with end-to-end rotation. Protein-bound beads were then washed 3–4 times with wash buffer according to manufacturer’s instructions provided for different beads used in the experiment. Proteins were then eluted with 3 × FLAG peptide for Co-IP with the anti-FLAG®M2 affinity gel; or with 0.1 M glycine, pH 2.5 at room temperature with rotation for 2 min and then neutralized with 1 M Tris-HCl, pH 7.8 for all the other Co-IP and IP experiments. Both input and eluate samples were analyzed by SDS-PAGE and immunoblotting.
Generation of transient and stable protein overexpression cell lines
Transient overexpression of mTOR, Rictor and Raptor in U87 cells was performed by transfecting one 10mm plate of 80–90% confluent U87 cells with 10 µg of FLAG-mTOR (Vilella-Bach et al., 1999), myc-Rictor-corrected (Sarbassov et al., 2004), or HA-Raptor (Kim et al., 2002) DNA plasmids using X-tremeGENE™ HP DNA Transfection Reagent at 1:3 plasmid / reagent ratio in DMEM media supplemented with 10% FBS. Media were changed after 24 h of incubation and cells were harvested at 48 h post-transfection. The FLAG-Rictor plasmid was generated by cloning the Rictor gene from the myc-Rictor plasmid into the p3xFLAG-CMV-10 Expression Vector. Stable overexpression of wild type and mutant xCT cell lines were established using the lentiviral expression system. Briefly, Myc-DDK-tagged SLC7A11 cDNA was cloned into the lentiviral expression vector pLVX-Puro using a pair of pLVX-puro-xCT primers. The xCT point mutants S26A was generated using the QuikChange Multi Site-Directed Mutagenesis Kit. The xCT N-terminus deletion mutant was generated by PCR, and the C-terminus deletion mutants was generated by fusion PCR using two sets of primers. Cloning primers are listed in Table S3. Lentivirus were packaged in HEK293T cells by transfecting cells with pLVX-Puro-xCT plasmids together with lentiviral packaging plasmids TAT, Gag/Pol, VSVG and Rev using the X-tremeGENE™ HP DNA Transfection Reagent. Media was changed after 16 h of transfection and virus were collected after 48 h. U87EGFRvIII cells were infected with virus in the presence of 12.5 µg/ml Polybrene for 24 h and selected with 1 µg/ml puromycin for at least one week to establish stable overexpression cell lines before used for experiments.
Transient and stable knockdown of proteins using siRNA and shRNA
Transient knockdown of proteins was achieved by transfection of siRNAs using Lipofectamine® RNAiMAX Transfection Reagent in DMEM media supplemented with 10% FBS. Media were changed after 24 h of transfection and cells were harvested 48 h post-transfection or with an additional 24 h of drug treatment. All siRNAs were obtained from GE Dharmacon except for the siRNA targeting xCT was custom synthesized by GE Dharmacon. Lentiviral shRNA plasmids scramble shRNA, Rictor_1 shRNA and Rictor_2 shRNA (Sarbassov et al., 2005) were used to generate stable mTORC2 knockdown cell lines shscramble, shRictor1 and shRictor2. Generation of stable knockdown cell lines was also performed using the lentiviral delivering system similar to the procedure described above for stable overexpression cell lines. Cells were all selected for at least one week and kept in 1 µg/ml puromycin before used for further experiments.
RNA Extraction and Real-Time PCR (RT-PCR)
Total RNA was extracted using the RNeasy Mini Kit. RNA concentrations were measured and 1 µg of RNA was used from each sample for cDNA synthesis using the SuperScript® VILOTM cDNA Synthesis Kit. RT-PCR was performed using the 2 × SYBR Green qPCR Master Mix (Bimake, B21202) on the CFX96 Real-Time PCR Detection System (Bio-Rad) following the manufacturer’s instructions. Results were analyzed using the delta delta Ct method and TPB was used as the reference gene. Primer sequences were listed in Table S3.
Protein Sequence Analysis
Human xCT protein sequence was downloaded from UniProtKB with the accession number Q9UPY5. The complete sequence was scanned through for serine/threonine residues proceeded with arginine at the −3 position, hence the RXXS/T motif. xCT 2D structure was constructed based on the sequence analysis and predicted topology information available on UniProtKB (UniProt, 2015).
In Vitro Kinase Assay
mTORC2 was purified from HEK293T cells transiently overexpressing FLAG-Rictor. Cells were lysed as described above using IP lysis buffer and lysates were subjected to IP using anti-FLAG®M2 affinity gel at 4 °C for 2 h, and mTORC2 was eluted using 3xFLAG peptide at 4 °C for an additional 30 min. Biotinylated Peptide substrates [GYXXXX(S/A)XXXXGRRRRR] were custom synthesized by EZBiolab, and peptide sequences were listed in Figure S3A. In vitro kinase assay was carried out by incubating 0.1 mM peptide with IP-purified mTORC2, recombinant Akt1 or SGK1 kinase in kinase reaction buffer (25 mM HEPES pH 7.4, 50 mM KCl, 10 mM MgCl2) together with 50 µM cold ATP and 5 µCi [γ-32P]ATP for 1 h at room temperature and terminated with 0.5 volume of M guanidine hydrochloride. Each reaction was performed in triplicates and µl of reaction mix was spotted onto SAM2® Biotin Capture Membrane. Membranes were washed and dried according to manufacturer’s instructions. Radioactivity was determined by autoradiography and quantified by scintillation counting. Data was analyzed using GraphPad Prism 7.
xCT activity assay
Glutamate secretion in xCT KO MEF cell lines were measured using a Na+ free PBS buffer system to exclude other glutamate transporter activity as reported (Kobayashi et al., 2015). Briefly, cells were seeded in 6-well plates at optimal density, washed three times with prewarmed Na+ free PBS buffer (137 mM choline chloride, 3 mM KCl, 0.01% CaCl2, 0.01% MgCl2 and 0.1% glucose, pH 7.4) and incubated in 1 ml Na+ free PBS buffer without cystine, with 500 µM cystine, or with 500 µM cystine and 500 µM SAS at 37 °C for 1 h. After incubation, supernatants were collected from each well and analyzed using the Amplex® Red Glutamic Acid/Glutamate Oxidase Kit. Glutamate secretion was calculated by subtracting the blank control, normalized to cell counts as well as cell surface xCT levels for each sample.
Cell Surface Protein Purification
Cell Surface proteins were purified using the PierceTM Cell Surface Protein Isolation Kit according to manufacturer’s instructions. Briefly, cells were washed with cold PBS and incubated with Sulfo-NHS-SS-Biotin at 4°C for 30min to label cell surface proteins. After labeling the reaction was quenched and cells were collected and lysed. Protein concentrations of lysates were determined using Bradford assay and equal amount of proteins from each sample were incubated with NeutrAvidin Agarose gels at 4 °C overnight to purify labeled cell surface proteins. After incubation proteins were eluted and subject to SDS-PAGE and immunoblotting analysis.
Cystine Uptake Assay
Sodium-independent cystine uptake through xCT was measure using a sodium nitroprusside based assay as described previously (Nakagawa and Coe, 1999). Briefly, cells were seeded at optimal confluency in 6-well plates and treated with drugs as indicated in the paper. After drug treatment, cells were first washed three times with 1 × PBS at room temperature, and pre-incubated in 1 ml cystine uptake buffer (122 mM choline chloride, 1.8 mM KCl, 1.3 mM CaCl2, 1.2 mM potassium phosphate, 25 mM Triethylammonium bicarbonate, 10 mM glucose, 0.4 mM MgSO4, pH7.4) for 15 min before 1 µM L-cystine was added and further incubated for 1 h at 37 °C. 500 µl uptake buffer was collected from each well and centrifuged at 14,000 × rpm for 2 min. 400 µl of the supernatant was added to cuvettes containing 300 µl 10% NaCN, 100 µl ddH2O and 1 ml 150 mM choline chloride, pipetted to mix and incubated at room temperature for 20 min. Then 100 µl of 20% sodium nitroprusside solution was added to the cuvette, mixed and absorbance was read at 521 nm using a NanoDrop 2000c Spectrometer immediately within 1 min. Cystine concentrations was calculated using a standard curve and cystine uptake was calculated by subtracting from blank controls without cells and normalized to cell counts from each well.
Metabolite Extraction and LC-MS/MS Metabolomics Analysis
Cells were washed three times with 1 × PBS and incubated in DMEM supplemented with 5% dialyzed FBS for 24 h before sample extraction. Cells were rinsed quickly on ice with ice cold 150 mM ammonium acetate (NH4AcO) and scraped off in 1 ml ice cold 80% methanol and collected into Eppendorf tubes. 5 nmol of norvaline was added to the cell suspension as internal control and the tubes were vortexed and spun down at 15,000 rpm for 5 min at 4 °C. Cell pellets were re-extracted with additional 200 µl of cold 80% methanol and supernatants were combined and transferred into glass vials and dried under vacuum. Metabolites were resuspended in 50 µl 70% acetonitrile (ACN) and 5 µl was used for analysis on a Q Exactive Orbitrap Mass Spectrometer (Thermo Scientific) in polarity-switching mode with positive voltage 3.0 kV and negative voltage 2.25 kV. The mass spectrometer was coupled to an UltiMate 3000RSLC (Thermo Scientific) UHPLC system. Mobile phase A was 5 mM NH4AcO, pH 9.9, B was ACN, and the separation achieved on a Luna 3 mm NH2 100 A (150 × 2.0 mm) (Phenomenex) column. The flow was 300 µl/min, and the gradient ran from 15% A to 95% A in 18 min, followed by an isocratic step for 9 minutes and reequilibration for 7 minutes.
Total Glutathione Measurement
Total cellular glutathione was measured using the GSH/GSSG-Glo™ Assay Kit. Briefly, cells were seeded at 1000 cells/well in 96 well plates after 48 h of siRNA transfection and treated with DMAO or Torin1 for an additional 24 h. After treatment media was removed and cells were lysed on a plate shaker for 5 min with Total Glutathione Reagent provided by the kit and transferred to a white 96 well plate. Subsequent reagents were added following manufacturer’s protocol and luminescence was measured using a Tecan Infinite M1000 microplate reader (Tecan) and normalized to cell counts from parallel wells for each treatment condition.
FITC-Annexin V/PI Apoptosis Assay
Cells were seeded in 6-well plates at 80,000–100,000 cells/well. Next day media was changed to regular DMEM supplemented with 5% FBS together with indicated drug treatment for 24 h. Cells were then trypsinized and resuspended in fresh media and combined with media supernatant to ensure collection of the dead cells. Cell suspension were spanned down at 400xg for 4 min and resuspended in 100ul 1x Annexin V binding buffer. Annexin V / PI staining was performed using the FITC Annexin V Apoptosis Detection Kit I according to the manufacturer’s protocol. After 15min of incubation additional 1 × binding buffer was added before samples were analyzed by flow cytometry using the BD LSRII flow cytometer (BD Biosciences). Data analysis was performed using the Kaluza Analysis Software and GraphPad Prism 7.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Data were all presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using two-tail unpaired Student’s t test for experiments where two means are compared unless specified otherwise. One-way analysis of variance (ANOVA) and two-way ANOVA was used when needed to compare means of three or more experimental groups and were indicated in the figure legend. Details of calculation and how statistical significance was determined, number of replicates, as well as p values were specified in corresponding figure legend.
LC/MS Proteomics Data Analysis
MS data were searched on Sorcerer2-SEQUEST using the reviewed Swiss-Prot human database with the following static and variable modifications for the two IPs. The modifications for the SILAC IP were K 8.0142 (variable, heavy lysine), R 10.00827(variable, heavy arginine), M 15.994920 (static, oxidation), and C 57.021465 (static, carbamidomethyl). The modifications for the phosphorylation identification IP were STY 79.963311(variable, phosphorylation), M 15.994920 (static, oxidation), and C 57.021465 (static, carbamidomethyl). The Trans-Proteomic Pipeline V4.3 JETSTREAM rev1 was used to analyze the search result. A PeptideProphet of 0.8 was applied and the common contaminants were removed; the identified peptides were quantified using XPRESS and a minimal ion intensity of 1.0E3 was used to calculate the abundance ratio. At least three unique peptides were required for a protein or a protein complex to be identified as an xCT binding protein, and the median abundance ratio for each identified protein was calculated and plotted.
Gene Ontology (GO) Analysis
The enrichment of GO terms (http://www.geneontology.org/) of xCT physically interacted proteins were calculated by Fisher’s exact test using the DAVID bioinformatics resources (Huang da et al., 2009). A Benjamini-Hochberg-corrected false discovery rate (FDR) ≤ 0.05 was used to determine the enriched functions.
LC/MS Metabolomics Data Analysis
Metabolites were detected based on a delta ppm of 3 or less and retention time accuracy relative to purchased standards of 30 sec or less and quantified as area under the curve (AUC) using the TraceFinder 3.3 (Thermo Scientific) software. Relative amounts of metabolites as well as percentage of labeling were calculated and normalized to control samples (DMSO treatment or si scramble knockdown) as well as total cell numbers.
DATA AND SOFTWARE AVAILABILITY
Original imaging data have been deposited to Mendeley Data and are available at: http://dx.doi.org/10.17632/46v4njmrs3.1. DAVID bioinformatics resources are available at: https://david.ncifcrf.gov/home.jsp. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaino et al., 2013) with the dataset identifier PXD006461.
Supplementary Material
The full list of proteins identified from the SILAC experiment was shown.
The top 10 enriched pathways and corresponding gene list obtained from the DAVID gene ontology analysis performed using the list of 125 candidate xCT binding proteins was shown.
Acknowledgments
We thank Dr. Hideyo Sato for kindly providing us with the xCT KO MEFs and helpful suggestions. We thank past and present members of the Paul S. Mischel Lab for helpful discussions, and Minh Thai, Carolina Espindola-Camacho and Abby Krall from the Heather R. Christofk Lab for help with the metabolomics experiments. We also want to thank Zhipeng Meng, Haixin Yuan and Jenna Jewell from the Kun-Liang Guan Lab for help with the in vitro kinase assay. This work was supported by grants from National Institute for Neurological Diseases and Stroke (NS73831) and the National Cancer Institute F31CA186668 (G.R.V), the Defeat GBM Program of the National Brain Tumor Society, the Ben and Catherine Ivy Foundation, and generous donations from the Ziering Family Foundation in memory of Sigi Ziering (P.S.M). C.P.A and H.Z. were supported by NIH Grant R01-GM116897. HRC was supported by a Research Scholar Grant, RSG-16-111-01-MPC, from the American Cancer Society. K-L.G. was supported by NIH Grant R35ca196878. K-L.G. is a co-founder of Vivace Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Y.G. and P.S.M. designed the experiments and wrote the manuscript. Y.G. conducted most of the experiments and data analysis. Y.G. and C.P.A. performed SILAC labeling, LC/MS proteomic analysis, and in vitro kinase assay. H.Z. helped design the LC/MS proteomic experiment, in vitro kinase assay and data analysis. D.B. performed the metabolomics measurements and analyzed data. W.Z. conducted the Gene Ontology Analysis. G.R.V., J.B., S.I., B.G., K.M., H.Y. helped with conducting the experiments and data analysis. T.C.G., A.K.S., T.F.C., H.R.C., K.G. helped design the experiments and data interpretation. All authors discussed the results and helped revise the manuscript.
References
- Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS letters. 1996;399:333338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nature reviews Cancer. 2016;16:619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu J, Ortells MC, Tejedor S, Buxade M, Lopez-Rodriguez C. Transcriptional regulation of the stress response by mTOR. Science signaling. 2014;7:re2. doi: 10.1126/scisignal.2005326. [DOI] [PubMed] [Google Scholar]
- Bassi MT, Gasol E, Manzoni M, Pineda M, Riboni M, Martin R, Zorzano A, Borsani G, Palacin M. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc. Pflugers Archiv : European journal of physiology. 2001;442:286296. doi: 10.1007/s004240100537. [DOI] [PubMed] [Google Scholar]
- Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer research. 2015;75:1782–1788. doi: 10.1158/0008-5472.CAN-14-3745. [DOI] [PubMed] [Google Scholar]
- Briggs KJ, Koivunen P, Cao S, Backus KM, Olenchock BA, Patel H, Zhang Q, Signoretti S, Gerfen GJ, Richardson AL, et al. Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell. 2016;166:126–139. doi: 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie Y, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-) : cystine supplier and beyond. Amino acids. 2012;42:231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell metabolism. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Molecular and cellular biology. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea J, Buendia I, Parada E, Navarro E, Rada P, Cuadrado A, Lopez MG, Garcia AG, Leon R. Melatonin-sulforaphane hybrid ITH12674 induces neuroprotection in oxidative stress conditions by a ‘drugprodrug’ mechanism of action. British journal of pharmacology. 2015;172:1807–1821. doi: 10.1111/bph.13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasol E, Jimenez-Vidal M, Chillaron J, Zorzano A, Palacin M. Membrane topology of system xc- light subunit reveals a re-entrant loop with substrate-restricted accessibility. The Journal of biological chemistry. 2004;279:31228–31236. doi: 10.1074/jbc.M402428200. [DOI] [PubMed] [Google Scholar]
- Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)-cystine transporter: a new action for an old drug. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2001;15:1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- Halland N, Schmidt F, Weiss T, Saas J, Li Z, Czech J, Dreyer M, Hofmeister A, Mertsch K, Dietz U, et al. Discovery of N-[4-(1H-Pyrazolo[3,4-b]pyrazin-6-yl)-phenyl]-sulfonamides as Highly Active and Selective SGK1 Inhibitors. ACS medicinal chemistry letters. 2015;6:73–78. doi: 10.1021/ml5003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. The Journal of cell biology. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic acids research. 2015;43:D5520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Lorberg A. TOR regulation of AGC kinases in yeast and mammals. The Biochemical journal. 2008;410:19–37. doi: 10.1042/BJ20071518. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Lamming DW. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell metabolism. 2016;23:990–1003. doi: 10.1016/j.cmet.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kanai Y, Chairoungdua A, Cha SH, Matsuo H, Kim DK, Inatomi J, Sawa H, Ida Y, Endou H. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochimica et biophysica acta. 2001;1512:335–344. doi: 10.1016/s0005-2736(01)00338-8. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Sato M, Kasakoshi T, Tsutsui T, Sugimoto M, Osaki M, Okada F, Igarashi K, Hiratake J, Homma T, et al. Cystathionine is a novel substrate of cystine/glutamate transporter: implications for immune function. The Journal of biological chemistry. 2015;290:8778–8788. doi: 10.1074/jbc.M114.625053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, Sabatini DM. A Central role for mTOR in lipid homeostasis. Cell metabolism. 2013;18:465–469. doi: 10.1016/j.cmet.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of cell science. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, et al. The Cystine/Glutamate Antiporter System x(c)(-) in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxidants & redox signaling. 2012 doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM, et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benz o[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. Journal of medicinal chemistry. 2010;53:7146–7155. doi: 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nature communications. 2012;3:876. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. The Journal of cell biology. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui K, Cavenee WK, Mischel PS. mTORC2 and Metabolic Reprogramming in GBM: at the Interface of Genetics and Environment. Brain pathology. 2015a;25:755–759. doi: 10.1111/bpa.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell metabolism. 2013;18:726–739. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui K, Tanaka K, Ikegami S, Villa GR, Yang H, Yong WH, Cloughesy TF, Yamagata K, Arai N, Cavenee WK, et al. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proceedings of the National Academy of Sciences of the United States of America. 2015b;112:94069411. doi: 10.1073/pnas.1511759112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa Y, Coe FL. A modified cyanide-nitroprusside method for quantifying urinary cystine concentration that corrects for creatinine interference. Clinica chimica acta; international journal of clinical chemistry. 1999;289:57–68. doi: 10.1016/s0009-8981(99)00159-x. [DOI] [PubMed] [Google Scholar]
- Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nature protocols. 2006;1:2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell. 2015;162:259–270. doi: 10.1016/j.cell.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell metabolism. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nature reviews Molecular cell biology. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, Warren PP, White DM, Reid MA, Eschbacher JM, et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Science translational medicine. 2015;7:289ra286. doi: 10.1126/scitranslmed.aaa8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current biology : CB. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schweppe DK, Rigas JR, Gerber SA. Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. Journal of proteomics. 2013;91:286–296. doi: 10.1016/j.jprot.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin CS, Mishra P, Watrous JD, Carelli V, D’Aurelio M, Jain M, Chan DC. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nature communications. 2017;8:15074. doi: 10.1038/ncomms15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi S, Wada K, Toyooka T, Shinomiya N, Shimazaki H, Nakanishi K, Nagatani K, Otani N, Osada H, Uozumi Y, et al. Increased xCT Expression Correlates with Tumor Invasion and Outcome in Patients with Glioblastoma. Neurosurgery. 2012 doi: 10.1227/NEU.0b013e318276b2de. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B, Dang J, Zhu S, Yang H, De Jesus J, et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer discovery. 2011;1:524–538. doi: 10.1158/2159-8290.CD-11-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, Hu M, Chan DA, Ethier SP, van ‘t Veer LJ, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer cell. 2013;24:450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchihashi K, Okazaki S, Ohmura M, Ishikawa M, Sampetrean O, Onishi N, Wakimoto H, Yoshikawa M, Seishima R, Iwasaki Y, et al. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(-) Cancer research. 2016;76:2954–2963. doi: 10.1158/0008-5472.CAN-15-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt C. UniProt: a hub for protein information. Nucleic acids research. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella-Bach M, Nuzzi P, Fang Y, Chen J. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin-associated protein kinase activity and G1 progression. The Journal of biological chemistry. 1999;274:4266–4272. doi: 10.1074/jbc.274.7.4266. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic acids research. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer research. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nature reviews Cancer. 2016;16:650–662. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, Mohammed S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. Journal of proteome research. 2013;12:260–271. doi: 10.1021/pr300630k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The full list of proteins identified from the SILAC experiment was shown.
The top 10 enriched pathways and corresponding gene list obtained from the DAVID gene ontology analysis performed using the list of 125 candidate xCT binding proteins was shown.