

Table of Contents
Ribosome profiling has the power to interrogate — in vivo and on a global scale — what is being translated, how this translation is regulated, and where in the cell the translation of specific sets of proteins occurs.
Preface
Ribosome profiling, the deep sequencing of ribosom-protected mRNA fragments, represents a powerful tool for globally monitoring protein translation in vivo. The method has enabled discovery of the gene expression regulation underlying diverse and complex biological processes, of important aspects of the mechanism of protein synthesis, and even of new proteins, by providing the first systematic approach for experimental coding region annotation. Here we introduce the methodology of ribosome profiling and discuss examples in which this approach has been a key factor in guiding biological discovery, including prominently in identifying thousands of novel translated short ORFs and alternative translation products.
Translation, the process by which a ribosome reads an mRNA template to guide protein synthesis, is a critical step in gene expression. Translation is energetically costly and therefore is tightly regulated to conserve cellular resources, as well as to avoid mistakes that may result in the production of toxic proteins. Indeed a wide range of disease states including neurodegeneration, anemia, and specific developmental defects result when the translational process is compromised (selected refs1–6). Although much is known about the structure and function of the ribosome, our understanding of many aspects of translation regulation has been far more limited.
Efforts to globally monitor gene expression have historically focused on measuring mRNA levels (for example, using microarrays or RNA-seq), although we know that translational control is an essential and regulated step in determining protein expression. Until recently, precisely monitoring protein translation was far more challenging than measuring mRNA levels. This has changed with the development of the ribosome profiling approach, first published in 20097.
Ribosome profiling is a deep sequencing-based tool that enables the detailed measurement of translation globally and in vivo7. At the core of this approach is the observation that a translating ribosome strongly protects about 30 nucleotides of an mRNA from nuclease activity8,9. Sequencing of these ribosome-protected fragments (ribosome footprints [G]) thus provides a precise record of the position of the ribosome at the time when translation was halted. Measuring the density of protected fragments on a given transcript provides a proxy for the rate of protein synthesis. Also, determining the position of the protected fragments makes it possible to empirically measure the identity of translation products (for example, where they begin and end, and even the frame being read). This has led to the discovery of a number of novel or alternative protein products10–19. The distribution of ribosome footprints can provide insights into the mechanism of translational control (for example, it can be used to identify regulatory translational pauses and translated upstream open reading frames (uORFs) [G]). Finally novel adaptations to the ribosome profiling approach make it possible to monitor the translation of subsets of ribosomes based on their physical location in the cell or their interaction partners.
Here we discuss the principles of the ribosome profiling approach, its strengths and limitations, and recent examples in which it has guided biological discovery. We focus on the value of ribosome profiling as a tool to interrogate what is being translated, how this translation is regulated, and where in the cell the translation of specific sets of proteins occurs.
What is ribosome profiling and what can it reveal?
Ribosome profiling exploits the classical molecular method of ribosome footprinting8,9, in which in vitro translated mRNAs are nuclease-treated to destroy the regions that are not protected by the ribosome7,9. Such treatment leaves ‘footprints’ of approximately 30 nucleotides, which can be mapped back to the original mRNA to define the exact location of the translating ribosome. Ribosome profiling expands on this method by mapping and measuring the full complement of in vivo ribosome footprints to quantify new protein synthesis and annotate coding regions globally (Figures 1 and 2; refs7,10–12). Extraordinary advances in sequencing technology20 now make it possible to deeply sample all translating ribosomes. In mammalian cells, for example, which encode ~20,000 proteins with an average mRNA coding region length of ~500 nucleotide triplets, nuclease digestion of all translating ribosome-mRNA complexes yields 10 million possible footprints. The billions of reads that are now accessible with next-generation sequencing enables reliable quantification of the set of footprints tiling across all but the rarest mRNAs, and a recently developed kit facilitates sample preparation21,22. With such easily attainable and quantitative information, ribosome profiling has a range of uses, from a broad proteomic tool to a specific probe of translation in an in vivo setting, and a valuable complement to mRNA sequencing.
Figure 1.
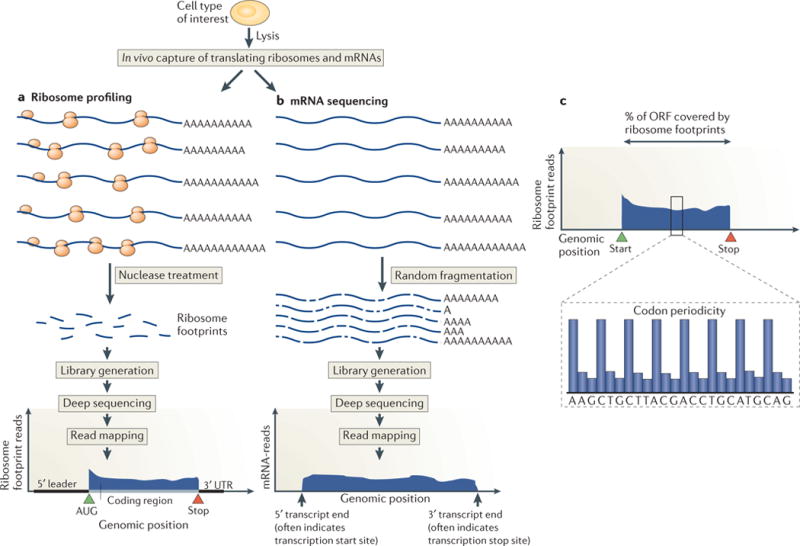
An overview of ribosome profiling. a) Ribosome-bound mRNAs are isolated by size and treated with nonspecific nuclease (typically RNAse I or micrococcal nuclease), resulting in protected mRNA fragments or ‘footprints’. These ribosome footprints are isolated and converted to a library for deep sequencing. The ribosome footprints typically show precise positioning between the start and stop codon of a gene, which enables global and experimental genomic coding region identification. b) By comparison, mRNA sequencing captures random fragments covering the entire mRNA transcript. The positional information determined by standard mRNA sequencing enables approximate determination of transcript boundaries, but is less precise than ribosome profiling due to the loss of 5′ and 3′ ends during the fragment generation method typically used. c) Translated open reading frames (ORFs) house a stereotyped organization of ribosome footprints. Ribosome density over ORFs begins sharply at the start codon, ends sharply at the stop codon, and shows evidence of codon periodicity. True translated regions tend to show ribosome footprint coverage over the majority of the ORF and not typically in the regions before the putative start codon and after the putative stop codon.
Figure 2.
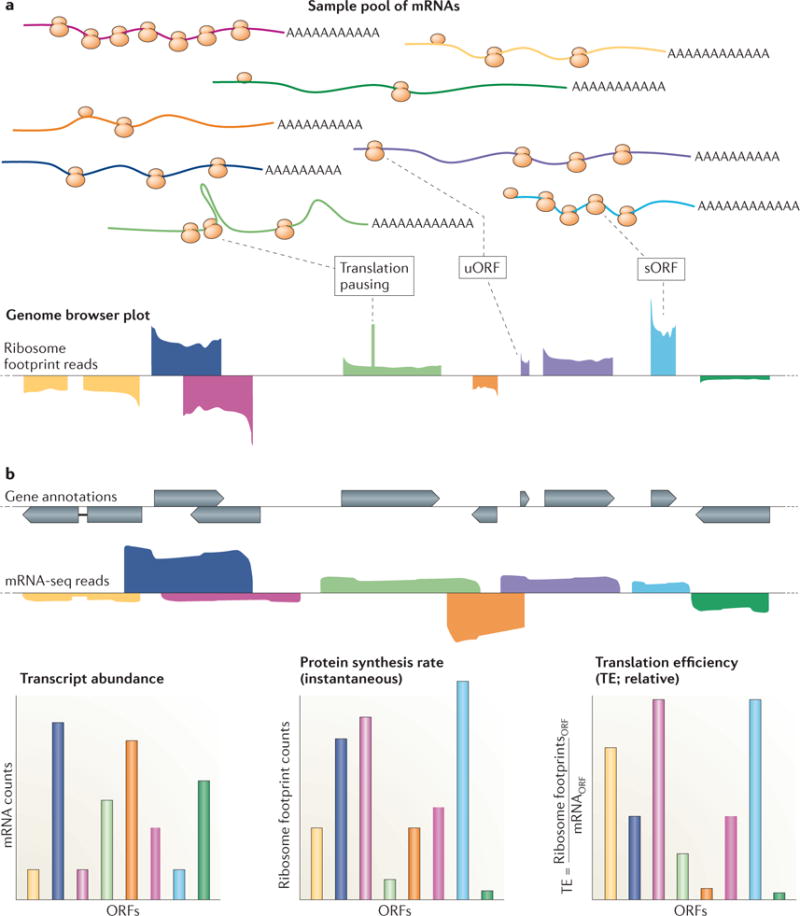
Qualitative and quantitative data provided by ribosome profiling. a) A diverse sample pool of mRNAs, distinguished by color, are shown together with a corresponding representative genome browser plot of ribosome profiling data derived from this pool. Note that ribosome profiling enables experimental determination of translated regions, including short Open Reading Frames (sORFs), which may be an important new source of cellular peptides, and upstream ORFs (uORFs), which are thought to be largely regulatory. Pausing during translation elongation may result in peaks in ribosome footprints within ORFs. b) Overlaid gene annotations and mRNA-seq data for the examples shown in (a). c) Examples of quantitative data derived from b). Note that transcript abundances may not correlate closely with the instantaneous protein synthesis rates. The collection of quantitative data for both transcript abundances and their protein synthesis rates enables inference of the relative translation efficiencies. These can vary over several orders of magnitude within a given organism in a given state. The translation efficiency can also change over time for a given mRNA, reflecting dynamic regulation at the level of translation.
Ribosome profiling requires collection of a physiological sample, inhibition of translation to freeze ribosomes in the act of translation, nuclease digestion to produce ribosome-protected fragments, isolation of ribosomes and, subsequently, of ribosome footprints21. Ribosome footprints are converted to a strand-specific library and subjected to next generation sequencing, and the fragments are then mapped to the appropriate reference genome. Ribosome profiling is typically carried out on a split sample, with parallel libraries constructed for measuring mRNA abundance by mRNA-Seq. Comparison between the rates of protein synthesis and the mRNA abundance makes it possible to determine the transational efficiency (TE) for each mRNA7 (Figures 1a, 1b, 2b, 2c). The common biophysical properties of the ribosome and the lack of genetic manipulation required for this approach make ribosome profiling highly adaptable to cells or tissues from essentially any organism with modest modifications. Organisms interrogated thus far by ribosome profiling include a variety of bacteria, yeast, parasitic protozoa, zebrafish, flies, nematodes, mouse, rat, plant, viruses, and human cells7,10–12,19,23–30. Even mitochondrial translation within human cells has been effectively assayed by this method31 and a similar approach has been applied to chloroplasts in plant cells32. Many of these datasets have been compiled and made readily accessible for data mining and comparison33.
What are the strengths of ribosome profiling?
Despite its recent development, ribosome profiling has rapidly become a widely used tool for understanding diverse and complex biological problems. Three key features, outlined below, have enabled this method’s broad utility.
Sensitivity and precision of quantitation
Ribosome profiling provides a large dynamic range of detection and quantitation of translation in unperturbed cells. The method’s sensitivity, which results from the depth of sampling possible in sequencing studies, enables measurement of even relatively rare translation events, with detection range generally limited only by the counting variability that is seen with very low numbers of sequencing reads. Complementary methods, including pulsed label-based mass spectrometry, analyses of transcript distributions on polysome gradients [G], and 35S methionine-based metabolic labeling, enable sensitive measurement of new protein synthesis, but the highly parallel sequencing readout of all ribosome positions that is provided by ribosome profiling typically yields more quantitative and detailed information than is currently accessible by alternate methods.
Precision of positional information
In addition to its broad dynamic range of detection, ribosome profiling provides uniquely rich and precise positional information. The nearly universal biophysical properties of ribosomes across species yield a characteristic footprint size that enables prediction of the codon in the ribosome P site [G] (the position of peptide bond formation) and the detection of codon periodicity [G]7 (Figure 1c). Analyses of ribosome footprint positions can be used to mechanistically probe aspects of translation, thus far identifying many novel instances of ribosomal frameshifting, stop codon read-through, ribosome pausing, translation initiation at non-AUG codons, and uORF translation (Figure 2a; refs7,10,12,34–37). Furthermore, years after many genomes were originally annotated, the precise positional information from ribosome profiling experiments has provided the first opportunity to experimentally define translated ORFs (Figure 1, 2;refs11–13,38,39), resulting in the identification of new classes of coding regions in diverse organisms.
Instantaneous measurements
A final valuable property of ribosome profiling is the instantaneous nature of the information collected, reflecting a snap-shot of translation. Although mRNA-seq and standard genome-scale mass spectrometry experiments are valuable in following gene expression globally, these widely used measurements report on steady-state levels of mRNA and protein, respectively. This information is important but may not reflect the rapid cellular decision-making that accompanies developmental transitions and environmental responses. Ribosome profiling allows sensitive detection of cellular protein expression changes as they are occurring7,12,40. The common, quantitative output for ribosome profiling and mRNA sequencing data further allows direct comparison of instantaneous protein synthesis and steady state transcript levels, providing an opportunity to quantify in vivo translation efficiencies in detail (Figure 2c).
What are the caveats and weaknesses of the method?
We discuss below notable limitations and caveats to ribosome profiling that should be considered when using the method or interpreting data derived from its use.
Experimentally introduced distortions
The key technical challenge of ribosome profiling is the need to rapidly inhibit translation in order to capture a snapshot of ribosomes in a particular physiological state. The reliability of this step is particularly important for any analyses of translation pausing, given that the fast rate of translation elongation may result in signal blurring or the artificial accumulation of ribosomes at specific positions if inhibition is slow. The use of a translation elongation inhibitor (such as cycloheximide), can be valuable, but it is clear that such inhibitors can alter the local distributions of ribosomes on an mRNA, especially near translation start sites7,18,21,41. Although this does not seem to interfere with global measurements of the density of ribosomes on an mRNA used to determine protein synthesis rates, it can cause spurious peaks of ribosome binding at particular sites. Thus far, flash freezing is the most robust approach in a wide range of diverse organisms, enabling the physiological capture of local and global ribosome distributions21. In general, each experimental step, from cell harvesting to nuclease digestion to library generation, has the potential to cause distortions in the data output. These distortions must be accounted for carefully, as the degree to which any given distortion might be problematic will depend strongly on the questions being addressed and system probed.
The need to infer protein synthesis rates
A caveat to consider when interpreting ribosome profiling data is that rates of synthesis are typically inferred from the average ribosome density along the mRNA in question. The accuracy of this measure depends on the premise that all ribosomes finish translation and that, on average, the ribosome elongation rate is similar between different mRNAs in a cell. These assumptions can be tested and are appropriate for a wide range of conditions, but will not always be the case. Known exceptions42–44, including the build-up of ribosomes at and immediately proximal to the start codon in a partially cycloheximide-dependent manner7 or regulated translational pausing and abortion under starvation conditions45, can be corrected for to increase measurement accuracy, but there may be cases in which these and other, unknown, exceptions pose challenges for proper data analysis.
Contaminating footprint-sized fragments
Another important issue for ribosome profiling experiments is that footprints are inferred on the basis of their size and their association with assembled (80S) ribosomes. Contaminating RNA fragments, including from structured non-coding RNAs or large ribonucleoprotein complexes that co-migrate in a sucrose gradient with the ribosome, may be processed with a ribosome profiling library and provide false readouts of translation (Supplementary Figure). A recent approach, termed FLOSS (fragment length organization similarity score) analysis [G] aims to identify such fragments and remove them post-experimentally (in silico)39. FLOSS analysis is based on the observation that bona fide ribosome footprints have stereotypical distributions of footprint sizes (Supplementary Figure a, b). The distribution of typical 80S footprint sizes used in FLOSS analysis is empirically measured for each experiment based on the size distribution of footprints in that same experiment from known protein coding regions and can be used to computationally remove contaminating fragments. Nonetheless, there are instances in which genuine 80S mRNA footprints that do not conform to the typical size pattern. Two recent cases that highlight interesting biology determined by analysis of alternatively sized ribosome footprints point to effects due to both alternate ribosome conformations46 and alternate mRNA properties41 (see below). Nuclease protection assays can be a useful adjunct control for identifying the full spectrum of ribosome footprint sizes in a new organism or condition, thus informing design of a ribosome profiling experiment to best capture all translating ribosomes in a given system.
Ribosomal RNA fragments commonly result from the nuclease treatment step of ribosome profiling and may significantly decrease ribosome footprint sequencing space in a ribosome profiling experiment7, particularly for conditions in which global translation levels are low. Whereas mRNA-sequencing often uses poly-A selection as an effective method for the isolation of desired sequences, this approach is not possible with ribosome profiling. Selective subtraction of ribosomal fragments, however, is highly effective and recommended, particularly for samples in which a small number of footprint-sized rRNA fragments are seen as contaminants21.
Mapping ambiguous reads
A general challenge of sequencing data analysis is determining the correct alignment position for reads from repetitive or highly similar regions, such as gene families, or from alternate transcript variants. In the case of genome sequencing or mRNA-seq, longer reads or paired-end47 approaches can help to resolve such ambiguities, but the inherently short size of ribosome footprints precludes these experimental approaches. Computational methods developed for mRNA-seq data to assign multiply mapping reads probabilistically based on overall read distributions48 can, however, be applied to ribosome profiling data to mitigate this limitation.
Material quantities
Currently, the major limitation of ribosome profiling relative to mRNA-seq approaches is the requirement for relatively large samples. In contrast to mRNA-seq49, ribosome profiling cannot yet be applied to single cells. This limitation results from the extra processing step required to isolate ribosomes21, as well as the small fraction of any given mRNA molecule that is being translated at any given time and thus recoverable as footprints (Figure 1a). It is likely that the types of technical advances that have greatly enhanced the sensitivity of RNA-seq approaches to small cell numbers will also be applicable in the future to ribosome profiling, though no such major effort has yet been undertaken.
Insights provided by ribosome profiling
With these advantages and disadvantages in mind, the application of ribosome profiling to specific biological questions has recapitulated much of what we know about translation mechanism from decades of elegant structural, biochemical, and genetic studies50. Ribosome profiling has also made it possible to monitor translation with unprecedented depth and precision, providing important—and at times quite surprising— insights. Application of this method to numerous organisms and cellular states has illuminated fundamental aspects of cell biology that were previously challenging to probe experimentally, providing measurements for how much of each protein is synthesized, how translation is regulated, where synthesis starts and stops, and what is being synthesized.
How much? A quantitative view of protein synthesis
The simplest and broadest application of ribosome profiling is as a quantitative proteomics tool to monitor which proteins are being synthesized—and at what levels—thus gaining rich molecular insight into a given cell state. Ribosome footprint density reflects the number of ribosomes at a given position. Assuming that the average translation elongation rate is similar between genes, ribosome profiling provides direct, global, and quantitative measurements of protein synthesis rates, capturing information that has been largely invisible to gene expression measurements based on mRNA levels alone. Mass spectrometry can in principle measure rates of protein synthesis but this is technically difficult, as it typically requires metabolic labeling and multiple measurements per sample. Analysis of the position of mRNAs in polysome gradients provides valuable complementary information to ribosome profiling, but again this method is laborious and typically yields only a qualitative measure of protein synthesis.
In many cases, the ability to observe new protein synthesis globally and quantitatively provides insights that are not apparent from mRNA abundance measurements. Bacterial operons provide a vivid example of the value of being able to directly measure the rates of protein synthesis. As is the case for many protein complexes in bacteria, the 8 different subunits of the FoF1 ATPase are expressed from a single polycistronic mRNA and thus by mRNA level measurements would seem to be all expressed at very similar levels. Ribosomal profiling, however, shows that the individual ORFs that encode the subunits of the FoF1 ATPase operon are translated at a ratio of 1:1:1:1:2:3:3:10. Remarkably these ratios precisely reflect the stoichiometry of these components in the ATP synthetase51,52 (Figure 3a). This property of proportional synthesis, by which subunits of multiprotein complexes are synthesized at rates that are proportional to their stoichiometry in their complex, turns out to be generally true for Escherichia coli and was also observed for some but not all complexes in budding yeast (Saccharomyces cerevisiae). Such measurements of instantaneous protein synthesis rates may prove to be a general tool for exploring how proteins assemble and function together51.
Figure 3.
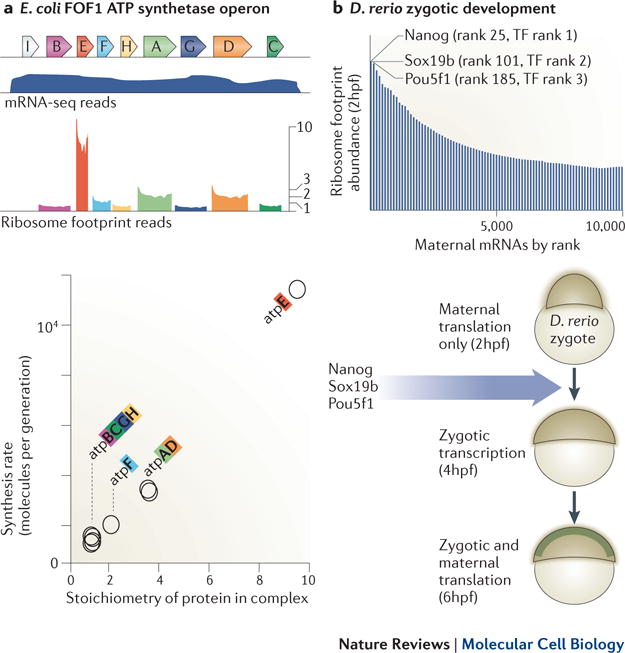
Ribosome profiling enables quantitative proteomic discovery in diverse systems. a) Bacterial cells translate components of multi-member protein complexes in ratios that are proportional to their stoichiometry in these complexes. A notable example is the FoF1 ATP synthetase, which is composed of 8 different proteins (A to H) translated from a single operon. mRNA abundance for each gene is thus similar, but ribosome profiling reveals intricate translational control. Modified with permission from ref51b) Zebrafish zygotic development requires the initiation of zygotic transcription 2 hours post fertilization (hpf), although the specific transcription factors responsible for this transcription have been unclear. Ribosome profiling of embryos at 2 hpf showed the three most highly translated transcription factors (TFs) from maternal messages were Nanog, Sox19b, and Pou5f1 and subsequent experiments confirmed that these three proteins drive zygotic activation. Modified with permission from ref54
Quantitative measurement of protein synthesis rates over multiple time points of a dynamic process can also provide information about specific gene function. For example, hierarchical clustering of patterns of new protein synthesis for each gene over the dynamic process of meiosis in budding yeast resulted in an intricate map of gene expression that provided highly detailed functional information12. In these data, the genes responsible for the complex, conserved, and meiosis-specific processes of homologous recombination and synaptonemal complex (SC) assembly emerged as a single cluster of 46 genes. This observation was surprising because these processes are known to be regulated extensively at the post-translational level, and also because this cluster included nearly every gene found through decades of intensive genetic and cytological screening focused on these processes. This cluster also included several uncharacterized genes, two of which (GMC1 and GMC2) were subsequently shown to have roles in recombination and SC formation12,53.
Another striking recent example of this type of analysis used ribosome profiling to identify the factors responsible for initiation of the zygotic developmental program in zebrafish (Figure 3b)54. The initiation of zygotic development in vertebrates depends heavily on translational control, as maternal mRNAs provide the starting pool of material for translation. Zygotic activation (ZGA) then requires destruction of these maternal mRNAs and transfer of developmental control to the zygote itself. In order to determine the factors that mediate the first wave of zygotic transcription, ribosome profiling data were analyzed for samples collected just prior to this ZGA transition. This study identified Nanog, SoxB1 and Pou5f1 as the three transcription factors that were most heavily translated from the large pool of maternal mRNAs at this stage (Figure 3b). Subsequent morpholino knockdown experiments showed that blocking translation of these three factors specifically resulted in a shutdown in the first-wave of zygotic transcription and development, indicating that they are the key factors responsible for the initiation of the zygotic developmental program54
Other recent studies, in disparate systems, from the Drosophila oocyte to embryo transition55 to the Trypanosome life cycle56 to the mammalian cell cycle57 to plants under hypoxic conditions27, have used ribosome profiling to identify specific proteins that drive these complex processes. Cases in which ribosome profiling data provide dramatically different information than can be obtained by traditional mRNA abundance measurements for gene expression tend to fall into two categories: systems in which transcriptional regulation is minimal26,54,55, and dynamic cellular programs11,12,27,35,57–59. The latter category includes cellular differentiation, organismal development, and dynamic responses to cellular stress, all cases in which the instantaneous and downstream gene expression measurements provided by ribosome profiling are particularly illuminating in understanding molecular control.
How? Insights into the mechanism of translational control
The basic mechanism by which the ribosomal machinery reads codon information in mRNAs to create proteins is conserved, and many features of this process are well understood50. Nonetheless, there are aspects of translational control that are not amenable to recapitulation in vitro and for which genetic approaches alone may be difficult to interpret due to complex secondary effects stemming from cellular adaptation to chronic abnormal protein synthesis. Furthermore, ribosome profiling enables the identification of translation mechanisms that vary across organisms, cellular state, and individual transcripts, as well as the study of roles for specific translation factors. Several important examples of discovery in translation mechanism have been highlighted in previous reviews22,60,61. Here we focus on just two recent studies in which ribosome profiling illuminated important aspects of protein translation.
Dom34 (a homolog of eukaryotic release factor 1) has been shown to help dissociate stalled ribosomes in vitro, but how and where it acted in vivo was unclear. Recent work explored the function of this protein through ribosome profiling of wild-type and dom34Δ budding yeast cells. The authors reasoned that if Dom34 were either dissociating ribosomes on truncated messages or causing multiple ribosomes to stack up due to stalling, then the relevant footprints might be of smaller and larger sizes, respectively, in the absence of Dom34 (Figure 4)41. Indeed, in the case of the HAC1 message, which was previously shown to exist in a truncated form in the cytosol62, ribosome profiling showed that dom34Δ budding yeast cells accumulated ribosomes with abnormal footprint sizes, indicating a defect in ribosome recycling at these sites41.
Figure 4.
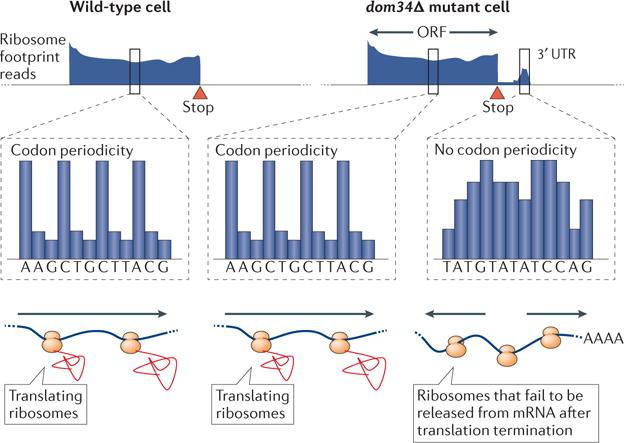
Dom34 facilitates the release of 80S ribosomes from a subset of 3′UTRs. Ribosome footprints indicative of assembled 80S ribosomes are seen in a subset of 3′UTR regions in dom34Δ mutant cells. Unlike 80S ribosome footprints from ORFs, however, these do not show periodicity and represent ribosomes that have failed to properly release following translation termination.
The largest effect revealed by ribosome profiling of dom34Δ cells—the presence of abundant ribosome footprints in 3′ UTRs (UnTranslated Regions) on a subset of mRNAs—was unexpected. In contrast to ribosome footprints in coding regions, the footprints that mapped to 3′ UTRs in the absence of Dom34 were not restricted to a single reading frame (Figure 1c, 4). This observation suggested that they did not represent canonical translating ribosomes, and were instead likely to be a population of ribosomes that failed to be released from mRNAs following translation termination (Figure 4). Together, these data suggest that ribosomes are not always automatically released following stop codon recognition and that Dom34 has a role in freeing ribosomes from truncated messages and 3′UTRs41.
Another important application of ribosome profiling has been the analysis of the mechanism of drugs that target translation. Macrolides, for example, are a class of clinically important antibiotics, which are known to bind in the nascent peptide exit channel of the ribosome. It has long been thought that macrolide activity causes early translational inhibition by blocking nascent peptide egress from the ribosome. However, this view has been overturned by recent ribosome profiling studies63–65, which found that macrolides function primarily by selectively affecting the ability of the ribosome to form peptide bonds in specific sequence contexts. A key observation in this regard was the finding that drug treatment did not inhibit synthesis of all proteins in bacteria treated with high doses of erythromycin or telithromycin, a next-generation macrolide. In fact, telithromycin inhibited the translation of fewer proteins than erythromycin, despite being a more effective antibiotic.64
Application of ribosome profiling to erythromycin- or telithromycin-treated bacterial cells also showed that, even among cases of inhibited translation for a given mRNA, the ribosome did not always stop translating early in the transcript, as predicted by the classic model for macrolide action. Rather, ribosome footprint build-up, indicative of ribosome stalling, could be seen at various regions in the subset of mRNAs that were inhibited. The precise positional information obtained from these experiments made it possible to determine that these points of translation interruption were dependent on specific positively charged sequences (R/KXR/K) present in the peptidyl transfer center of the ribosome. Macrolide- mediated inhibition of translation thus was not occurring primarily through obstruction of the exit channel and instead was a result of ineffective peptide bond formation for certain amino acid strings. This effect could be recapitulated precisely in vitro for some mRNAs but poorly for others, suggesting that additional cellular factors might contribute to macrolide action.63 This improved understanding of macrolide action has direct relevance to the development of newer, more effective antibiotics.
Where? Monitoring localized translation
A hallmark of eukaryotic cells is the presence of intricate subceullar structures that enable compartmentalization of different biological processes. Localized protein synthesis has a crucial role in creating these subcellular structures by allowing protein production at the site of action and in response to local cellular need (see ref66 for review). Because translation is an important amplification step, localization of a single mRNA molecule allows for correctly localized synthesis of hundred of protein molecules. Additionally, local synthesis prevents potentially toxic effects of having proteins present —even if only during transit time—in an inappropriate cellular compartment. Finally, localized translation allows regulation of protein synthesis based on a proximal stimulus, such as that seen in dendrites in response to neuronal stimulation, which is thought to contribute to the learning process66.
Despite the broad importance of localized translation, few gene expression tools are available that faithfully preserve spatial information. Until recently, global approaches for studying subcellular control of protein synthesis have been limited to bulk interrogations that cannot uniquely identify proteins or that require careful biochemical fractionation of the compartment of interest, limiting both the location and resolution of analyses. Proximity specific ribosome profiling, now also enables in vivo measurement of localized translation within cells. The basis of proximity-specific ribosome profiling is selective biotinylation of ribosomes in a manner that depends on their subcellular location in intact, unperturbed cells (Figure 5). The use of in vivo labeling enables the recovery of ribosomes from defined locations, including those that cannot be purified by classical cell fractionation techniques. Combining this purification strategy with ribosome profiling provides identification of locally translated messages and sub-codon monitoring of translation at the site of interest.
Figure 5.
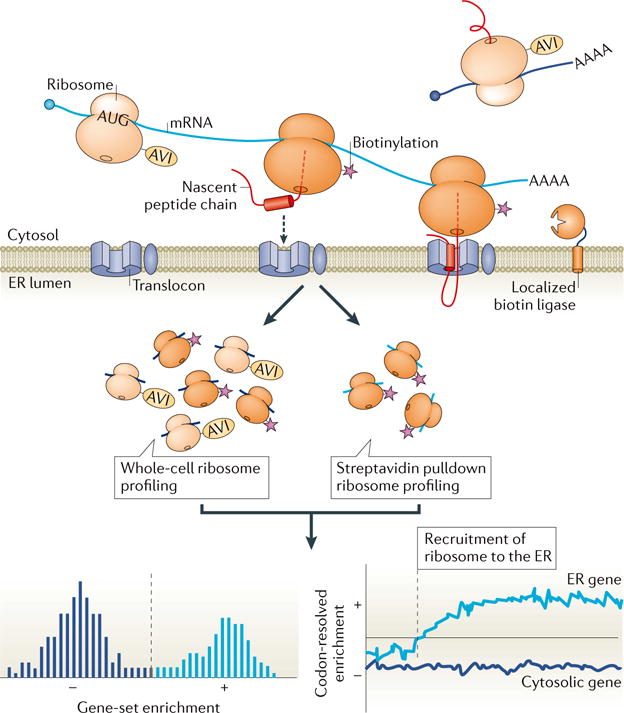
Proximity specific ribosome profiling at the ER. A ribosome subunit is fused to a biotin-acceptor (AVI) tag and BirA biotin ligase is fused to a localization element that spatially restricts its activity, for example, to the ER. Only ribosomes that orient AVI towards the ER surface, as seen during their close association with the ER membrane during translocation, are biotinylated when a controlled pulse of biotin is applied to cells. Cells are then frozen and ribosomes are collected. Ribosome profiling is carried out on all ribosomes and also only on ribosomes pulled down with streptavidin. The pulldown-enriched message population (light blue) represents genes that are greatly enriched for translation at the ER. The positional data from these analyses also reveals the point in the message when a translating ribosome is recruited to the ER. Modified with permission from ref68.
So far, proximity specific ribosome profiling has been used to probe two processes: translocation into the mitochondria and the endoplasmic reticulum (ER), with both studies yielding unexpected results67,68. In the case of the mitochondria, the approach revealed insight into a longstanding question: do mitochondrial proteins begin translocation co-translationally or is the predominant route of mitochondrial translocation post translational? Proximity-specific ribosome profiling showed that the majority of mitochondrial inner membrane proteins—but not proteins targeted to other mitochondrial sites—were co-translationally targeted67. These studies also revealed exquisite specificity in protein trafficking, with the vast majority of translocated proteins identified in these studies being targeted exclusively to either the ER or the mitochondria. A prominent exception was the fumarate reductase Osm1; follow up studies showed that this dual targeting was enabled by the translation of alternate isoforms with distinct targeting signals67.
Monitoring of translation on the ER surface revealed several principles used by cells to coordinate translation with ER targeting (Figure 5; ref68). First, this work showed that co-translational targeting to the ER is pervasive and is principally determined by the location of the hydrophobic targeting sequence within the protein. The observation that co-translationally targeted mRNAs can be translated at the translocon immediately after or even prior to translation of their targeting sequence suggested a critical role for polysomes in retaining mRNAs at the ER. Additionally, distinct translocon [G] complexes engage nascent chains at different points during synthesis. ER-targeted nascent chains typically undergo a conformational rearrangement within the translocon that results in a “looped” conformation of the nascent chains, with their N-termini facing the cytosol. However, proximity specific ribosome profiling revealed that a subset of proteins, whose targeting requires the translocon-associated factor Sec66, engage the translocon only after 120 amino acids have been synthesized, which enables direct adoption of the looped conformation. Finally, monitoring the fate of ER-associated ribosomes following translation termination using pulsed biotinylation experiments showed that any given ribosome can exchange readily between the ER and cytosol, as ribosomes labeled on the ER are able to access the full range of cytosolic mRNAs following at most a few rounds of translation at the ER.68
In principle, proximity specific-ribosome profiling could be applied to any subcellular location for which it is possible to target biotin ligase activity. Additionally, it can be combined with approaches to look at different polysome fractions55,69 or the translating ribosome affinity capture (TRAP)70–76 [G] strategy. Together these techniques could make it possible to explore regulated localized translation in specific neuronal subtypes and in response to learning programs.
What is being made? Defining translation events
Perhaps the most surprising emergent area of discovery enabled by ribosome profiling results from the method’s ability to identify in a systematic manner, the full spectrum of ribosome-translated polypeptides in a cell. Algorithm-based analyses of an organism’s genomic sequence alone can direct identification of likely coding sequences. Such strategies, however, rely on assumptions about what a coding region should look like, including start and stop codon identity, splice junction cues, conservation, and total codon length of an ORF. Such approaches for identifying protein-coding genes could miss functional coding sequences, particularly those that are short and/or species-specific77. These approaches might also miss coding regions that result from translational frame-shifting or stop codon read-through. Furthermore, translation and protein synthesis have effects beyond the production of stable proteins with discrete molecular functions. Polypeptide products from all cellular translation must be degraded, and non-canonical translation products yield unanticipated antigens that may have roles in viral detection or in autoimmunity39,78. Finally, the process of translation can affect the stability of the template message, by triggering co-translational decay pathways including nonsense-mediated decay (NMD) [G]79. Thus knowing what transcripts are translated has important implications for the fate of the mRNA, the ribosome, and the cell. Ribosome profiling provides a unique opportunity to experimentally address this question in a given biological system or cell state of interest.
The use of ribosome profiling in many organisms has largely provided experimental evidence for the translation of ORFs already predicted to encode proteins. Ribosome profiling data have also suggested a diverse set of translated areas outside of canonical coding regions (reviewed in refs60,80). These include, in some cases, ribosome footprints that are not clearly organized within ORFs, most commonly in 5′ leader regions and mammalian long noncoding RNAs. The importance of translation of these regions remains an open question, although the unusual pattern of ribosome footprints within ORFs suggests that they may not reflect regions that are translated into canonical peptide products. In some cases, the translation that produces these footprints may mediate translational regulation, as is the case for translation of regulatory uORFs. Alternatively, some such cases may reflect translation that is used to regulate mRNA stability81.
In addition, however, in diverse organisms and conditions, ribosome footprints are seen that are organized within ORFs that were not previously known to encode proteins in a manner that resembles those in canonical coding regions (as in Figure 1c), indicating that there is greater coding region diversity and flexibility than previously recognized10–13. The translated ORFs defined by such ribosome footprints fall into two broad categories: translated short ORFs (sORFs) [G] in predicted intergenic regions, often on RNAs that had been provisionally characterized as noncoding; and translated ORFs encoding alternate isoforms of known proteins [G]. Both categories could represent major emergent areas of biological importance.
How pervasive is sORF translation?
Algorithms for predicting protein-coding regions typically rely on ORF length. The minimum ORF length of 100 codons that is used by most computational annotation approaches was chosen both to minimize the number of false positive gene calls as well as to reflect the predicted biophysical folding stability of 100 amino acid proteins relative to shorter amino acid strings. Recently, however, several short peptides have been shown to be translated and to have crucial intracellular and extracellular roles in metazoans14,82–84. Concomitant with these findings, ribosome profiling data in several systems, including mouse embryonic stem cells, meiotic yeast cells, hypoxic plants, and virus-infected human fibroblasts, have identified a large number of ribosome footprints that fall outside of canonical coding regions but that cover a short and discrete region between an AUG and a stop codon10–12,16,27,85. These observations suggest that canonical protein coding sequences may represent only a subset of what cells translate.
There are, however, some features of the newly identified translated sORFs that have led to doubts about their authenticity. First, some are present on RNAs that were thought to be noncoding10–12,82,83,86. In many cases, these sORFs are not well conserved13,87,88. They also sometimes seem to be translated in overlapping reading frames10–12,87,89, a feature that has been thought to be unusual among typical eukaryotic genes (although ribosome profiling data has been used recently to identify such cases among canonical genes, as well90). Finally, translated sORF products are difficult to detect systematically by mass spectrometric approaches. The validation or exclusion of these regions as examples of biologically relevant translation has been a major recent focus of interest.
Several analytical approaches to ribosome profiling data allow rigorous testing of the degree to which ribosome footprints over newly predicted translated sORFs match those seen for traditional protein coding sequences (Table 1). These analyses often examine whether translated ORFs predicted by ribosome profiling show footprint organization consistent with the canonical mechanism for translation, such as sharp footprint abundance transitions at known start codons and stop codons, and codon periodicity (Figure 1c; Table 1; refs12,13,16,38,85,87,91; discussed in ref80). Most of these approaches provide support for the predicted widespread translation of short and alternate ORFs (Table 1; refs11–13,15,16,38,67,85,91). Nevertheless, even with ribosome profiling data, reliably identifying the full set of translated ORFs remains a challenge, especially in cases in which protein coding sequences overlap.
Table 1.
Novel translated ORFs identified by ribosome profiling compared to characterized translated ORFs by diverse metrics.
| Table 1 | Characterized noncoding RNAs (snoRNAs, tRNAs, Xist, HOTAIR, etc) | Characterized protein coding sequences | sORFs* identified as translated by ribosome profiling | uORFs* identified as translated by ribosome profiling |
|---|---|---|---|---|
| Association between footprint arrangement and putative start codons7,11,12,* | No general association | Footprint-covered regions usually start precisely at AUG codons, occasionally near-cognate codons | Footprint-covered regions usually start precisely at AUG codons, occasionally near-cognate codons | Footprint-covered regions often start precisely at AUG and near-cognate codons |
| Association between footprint arrangement and putative stop7,* codons | No general association | Footprint-covered regions stop precisely at canonical stop codons | Footprint-covered regions stop precisely at canonical stop codons | Footprint-covered regions stop precisely at canonical stop codons |
| Footprint abundance relative to mRNA abundance7,10,38, | Very low (especially properly-sized footprints) | Low to High, depending on translation efficiency | Low to High, depending on translation efficiency | Low to High, depending on translation efficiency |
| Codon periodicity of footprints7,13,41,90,* | No | Yes | Yes | Often unclear due to generally short length |
| Coding conservation signatures13,38,77,80,98 | No | Often | Sometimes, difficult to assess for very short regions | Unclear, primarily due to short length |
| Identification of protein product by mass spectrometry11,92–97 | No | Often | Sometimes (dependent on length, peptide properties) | Sometimes (dependent on length, peptide properties) |
| Stable physical association of transcript with ribosomes17,39 | Not generally, though may occur in specific cases (eg. tRNAs) | Yes | Yes | Yes |
| Sensitivity of footprints to translation inhibitors39 | No | Yes | Yes | Yes |
| FLOSS (Fragment Length Organization Similarity Score)39,* | High | Low | Low | Low |
| % putative ORF covered by footprints38,* | Low | High | High | Difficult to assess due to frequent uORF overlap |
| Inside/out ratio (local enrichment of footprints within putative ORF)38,* | Low | High | High, Difficult to assess when translated sORFs overlap | Difficult to assess due to frequent uORF overlap |
| Ratio of footprints at putative start codons to footprints at immediately prior codons12,* | Low | High | High | HIgh |
| RRS (Ribosome Release Score)87 | Poor | Good | Sometimes high, but particularly poor in cases of translated sORF overlap | Frequent overlap in uORF translation leads to poor scores, difficult to assess |
| Cellular function determined by genetic or molecular analyses14,82–84 | Sometimes | Sometimes | Rarely, thus far, but important examples exist | Not assayed in many cases, but a subset are regulatory for translation of other ORFs |
| Likelihood based on the above metrics that regions encode functional proteins/peptides | Low | High | High for some, but not for all. Likely to be a heterogenous population with diverse cellular roles. | Unclear. uORF regions predicted to be translated by ribosome profiling likely represent true translation, but resultant peptides may not be stable. |
See Glossary, Figure 1c, Figure 2, and Supplementary Figure for class definitions and examples.
Numerous complementary experimental approaches have aimed to further probe the degree to which newly predicted protein coding sequences represent true cellular translation (Table 1). Thus far, these approaches generally confirm that the reads detected in regions predicted to be translated by ribosome profiling experiments represent translating 80S ribosomes. For example, ribosome footprints over putative translated sORFs tend to respond to translation inhibitors comparably to benchmarked translating ribosomes39. Translated mRNA regions predicted from mouse ribosome profiling data immunoprecipitate with tagged 60S ribosomal subunits in a specific manner, similar to that seen for characterized translated ORFs39. This finding suggests that true translating ribosomes produce the footprints detected by ribosome profiling over ORFs not previously annotated as translated, rather than these mRNA fragments resulting from artifactual protection of mRNA by scanning translation initiation complexes or alternate RNA-protein complexes. An important outstanding question is whether these translated regions produce stable peptides. Suggesting that they may, sORFs identified as being translated by ribosome profiling that have been C-terminally tagged in yeast and Human Cytomegalovirus (HCMV)-infected cells can be seen to accumulate in a regulated manner that mirrors predictions from ribosome profiling data11,39. Meanwhile, specialized mass spectrometry approaches continue to identify a subset of peptides resulting from such sORFs in several systems11,92–97, suggesting that at least some of these sORFs do encode abundant, stable peptides.
Most convincingly, a few sORFs predicted to be translated from polysome association and ribosome profiling data have now been shown to have biological function39,83. In Drosophila, the peptides encoded by two such translated sORFs contained in the sarcolamban locus, have been shown to directly bind a calcium transporter in heart cells and thus regulate normal heart function83 (Figure 6a). In zebrafish, the short protein Toddler was found to drive gastrulation by functioning as a secreted developmental signal14. In mammals, a prominent example is the several translated sORFs predicted from HCMV-infected human foreskin fibroblasts that reside on the β2.7 RNA, which has traditionally been defined as noncoding11. Peptides resulting from the translation of two of these short ORFs have been shown by mass spectrometry to accumulate during CMV infection. Additionally, analysis of sera samples from HCMV-positive and -negative blood bank donors revealed a robust immune response to the peptides produced from several of these β2.7 sORFs, specifically in the HCMV-positive individuals39 (Figure 6b). This result suggests that the ribosome-occupied sORFs are translated, and their products are processed and presented on MHC molecules as functional antigens in humans, thus expanding the range of epitopes displayed during viral infections. The condition-specific translation of many sORFs suggests that they could similarly be used to distinguish cancer cells from normal cells, with important implications for immunomodulatory therapies.
Figure 6.
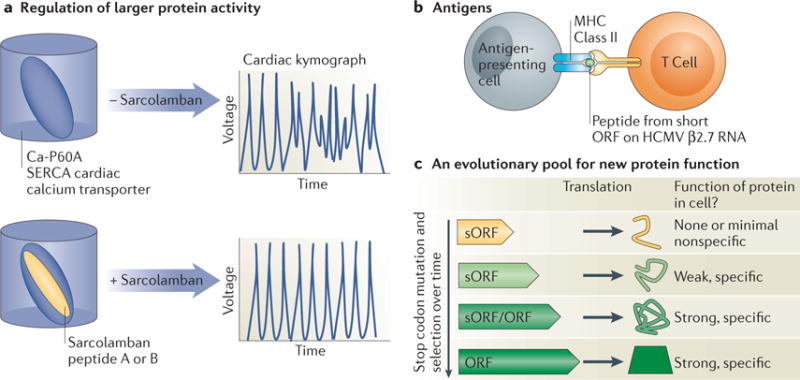
Proposed cellular roles for the peptide products of translated short ORFs identified by ribosome profiling. a) The two Sarcolamban peptides are 28 and 29 amino acids in length, conserved from fruit flies to human, and regulate normal heart function in flies through direct binding to the Ca-P60A SERCA Calcium transporter in cardiac tissue. Modified with permission from ref83. b) Sera from HCMV positive blood donors identified a specific and robust antigenic response against multiple short peptides translated from the β 2.7 RNA, previously thought to act as a noncoding RNA. c) Spurious translation of short regions may produce a pool of peptides with weak or no cellular function. New protein domains may evolve through selection for maintenance of peptides with weak cellular function, followed by stop codon mutation and further selection for increasingly specific and important cellular function over time.
The translation of some sORFs could also help to fuel the evolution of new proteins88. It is possible that transcriptional noise, together with the propensity of the ribosome to translate capped cytosolic RNAs, may allow novel messages to engage the ribosome and allow translational sampling of new, short motifs. Initially these short ORFs may evolve under neutral selection. However, a subset could provide a small fitness advantage, enabling their positive selection and possible stabilization through lengthening over time to resemble canonical long protein coding genes (Figure 6c). Such regions would not necessarily be expected to show signatures of protein coding conservation [G] (as in ref98) and few might produce a robust mutant phenotype when disrupted, making their study challenging.
How plastic is translation? Alternate isoforms abound
Ribosome profiling in yeast and mammals has suggested that many genes may yield two or more protein variants independent of splicing, which indicates that there may be surprising flexibility in both where translation starts and stops in eukaryotes. Such alternate isoforms have been seen and characterized previously; in budding yeast, for example, both ALA1 and GRS1 are tRNA synthetases that have been shown to exist in two isoforms, providing populations of the protein that are either cytosolic or mitochondrial, depending on the presence or absence of an N- terminal in-frame extension99,100. These examples are also detected by ribosome profiling12 and appear to be just a few of many10,12,67,101, supporting a model in which diverse but targeted localization might be achieved for many proteins through sometimes small alterations in the site of translation initiation18,91,101 Conversely, ribosome profiling of several yeast species, and of Drosophila embryos and cultured cells, revealed extensive heterogeneity in translation termination sites15,102,103, resulting from regulated read-through of hundreds of genes. As with the N-terminal extension isoforms, many of these C-terminal extensions are predicted to confer new subcellular localizations to the protein products15,104.
Use of ribosome profiling has also enabled identification of interesting examples of regulated truncated protein isoforms10–12,89. In human cells, a recent study identified a shortened alternate isoform of a protein called MAVS, which is an important player in innate immune signaling89. The alternate MAVS isoform results from translation initiation downstream of the canonical start site to create an N- terminal in-frame truncation, which the authors term ‘miniMAVS’. Whereas full length MAVS induces interferon production, mini-MAVS antagonizes MAVS function by interfering with such production.
The large and diverse set of unconventional regions of translation suggested by ribosome profiling reveals that there is considerably more to translational regulation and cellular content than was previously known. Some of these regions are likely to be translated into functional proteins, but it is likely that some of these regions will not produce stable protein products akin to traditional genes. Rather, subsets of these newly identified regions of translation may serve regulatory, immune, or currently neutral cellular roles. Unraveling the set of functions carried out by translated genomic regions poses a fascinating and daunting challenge.
Perspective
Protein synthesis consumes a large fraction of cellular resources and is central to virtually every function of a cell. Ribosome profiling enables, for the first time, in vivo and global measurement of translation, enabling a precise and quantitative account of what cells are translating, how this translation is regulated, and when and where translation happens. The rich and quantitative nature of ribosome profiling data provides an unprecedented opportunity to explore and model complex cellular processes.
Although it has long been known that translational regulation has a major role in development, cellular responses to stimuli and disease, the limited number of well studied examples of regulation at the level of protein synthesis have generally been identified in an ad hoc manner. When paired with RNA-seq measurements of mRNA levels, ribosome profiling now allows instantaneous measurement of all translational control in a given system, providing a tool for broad discovery in the underlying biology of a cellular process or state of choice. Further, the detailed information yielded by this method provides valuable insight into fundamental aspects of how translation works. Despite the conserved nature of much of the translation machinery, major outstanding mysteries in the mechanism of protein synthesis remain, including the basis for most specificity in translation among different mRNAs and the connections between translation and nascent protein folding.
Finally, by virtue of the precise genomic positional information obtained by ribosome profiling, the protein coding capacity of genomes can now be defined experimentally. This has led to the identification of a broad range of non-canonical translation events, including the translation of novel short open reading frames as well as alternate forms of previously annotated proteins, thereby challenging traditional definitions of protein coding regions and gene diversity. Analytical advances that enable more comprehensive identification of other non-canonical translation events, such as those resulting from frame-shifting and stop codon read- through will continue to expand our understanding of the protein coding capacity of complex genomes. The function for the many novel, short and alternate translated regions identified thus far by ribosome profiling remains an intriguing and largely open question and one whose answer could fundamentally change the way that we think about information encoding in genomes. Newly available CRISPR-based methods105 now make it possible to shut down the expression of any transcript106–109 or introduce nonsense mutations into any open reading frame. As such, these approaches provide a central tool for efforts to define the functional roles for this broad array of newly identified translation products.
Specialized alterations to ribosome profiling that will advance its utility to complex systems include analysis of subsets of ribosomes, either associated with specific factors or protein modifications, or those in increasingly specific cell types or subcellular locations. Additional transformative advances are likely to emerge from progressively more sophisticated and creative analysis of the rich datasets generated from ribosome profiling experiments, enabling major surprises to be revealed, even in systems that were thought to be well characterized.
Supplementary Material
Online Summary.
Ribosome profiling is a deep sequencing-based tool that enables the detailed measurement of translation globally and in vivo.
The method provides quantitation of levels of new protein synthesis, as well as information about ribosome positions that can be used to infer details about translation mechanism or to identify translated Open Reading Frames (ORFs).
Ribosome profiling enables instantaneous rather than steady-state measurement and is thus a particularly valuable tool for study of gene expression over dynamic processes.
Proximity specific ribosome profiling is based on localized labeling of ribosome populations within cells and enables in vivo measurement of translation at specific organelles or subcellular structures.
Ribosome profiling is the first tool available for experimental annotation of translated ORFs and has allowed discovery of a wide range of new translation products. These include novel short peptides and alternate isoforms of characterized proteins, the vast majority of which are currently of unknown function.
Acknowledgments
We wish to thank Calvin Jan and Elçin Ünal for helpful comments on this manuscript and Nick Ingolia for development of the original ribosome profiling protocol and helpful discussions. This work was partially supported by the Winkler Family Biological Sciences Award to GAB and Howard Hughes Medical Institute and Center for RNA Systems Biology funding to JSW.
Glossary
- Ribosome footprints
~30 nucleotide mRNA fragments that result from nuclease treatment of translating ribosomes. These are mRNA regions that are protected by the ribosome as the mRNA is decoded to a protein sequence
- upstream open reading Frames (uORFs)
ORFs in the 5′ leader region of a characterized mRNA transcript. uORF translation may regulate translation of a downstream ORF. Ribosome profiling allows the empirical identification of all translated uORFs in vivo under a condition of interest. Although uORFs are short, here we do not include them in the class of sORFs, which are on an mRNA that was not previously thought to encode a protein
- Polysome gradients
A method for fractionating ribosomes that are bound to mRNAs by density centrifugation of cell extract on sucrose gradients, allowing separation of mRNAs associated with one ribosome (monosome) and those being translated by multiple ribosomes (polysome). Sucrose gradient fractionation allows qualitative analysis of the translation status of cells
- Ribosome P site
The site within an actively translating ribosome that is usually associated with the tRNA attached to the growing peptide chain
- Codon periodicity
The three nucleotide pattern of ribosome occupancy reflecting mRNA translocation in the ribosome by codon as translation occurs
- FLOSS (fragment length organization similarity score) analysis
A metric for determining the likelihood that ribosome footprints over a given region (or set of regions) result from translation. This analysis involves comparing size distributions of footprints over a query region and over validated coding regions, based on the concept that the biophysical properties of translating ribosomes result in characteristic signatures in ribosome footprint sizes
- Translocon
The proteinaceous tunnel through which nascent proteins cross the ER membrane
- Translating ribosome affinity capture (TRAP)
A method that allows identification of translated mRNAs based on their in vivo association with a tagged ribosomal subunit that is expressed in a cell-type specific manner. This method is a valuable tool for assaying tissue-specific translation in animal and plant systems
- nonsense-mediated decay (NMD)
mRNA degradation, traditionally thought to result from stop codons that terminate translation more 5′ than “normal” on an mRNA
- short ORFs (sORFs)
Open Reading Frames of less than 100 codons on mRNAs that are not known to encode a canonical (long) protein. sORFs have not traditionally been a class of ORFs that were thought to be frequently translated, although ribosome profiling and other approaches have recently validated translation of thousands of sORFs in a variety of organisms
- ORFs encoding alternate isoforms of known proteins
Open Reading Frames that differ in either start codon or stop codon position from another ORF at the same locus but share the same reading frame. Translation of these ORFs may result in, for example, different subcellular targeting for a similar protein
- signatures of protein coding conservation
Purifying evolutionary selection results in higher levels of nonsynonymous (ns) than synonymous (s) substitutions specifically among homologous coding sequences. The pattern of ns to s differences among homologous regions compared in a phylogenetic group can be used to predict the likelihood that a genomic locus encodes a translated ORF
Biographies
Gloria Brar received her undergraduate biology degree from University of California at Berkeley. She worked with Angelika Amon at the Massachusetts Institute of Technology, where she obtained a Ph.D. in biology, studying the regulation of meiotic chromosome segregation. Dr. Brar joined Jonathan Weissman’ s lab as a postdoctoral fellow supported by the American Cancer Society. In the Weissman lab, Dr. Brar used ribosome profiling to define the complex regulation of gene expression that underlies meiosis. She is currently an Assistant Professor of Molecular and Cell Biology at UC-Berkeley.
Gloria Brar’s lab probes the dynamic and non-canonical gene regulation that allows cells to complete the comprehensive cellular remodeling that accompanies meiosis. Towards this end, the Brar lab studies specializations to the meiotic ribosome, the role of stress response pathways in meiotic differentiation and organelle remodeling, and the roles of uORF-mediated translational regulation and pervasive translation of short Open Reading Frames in meiotic cells.
Gloria Brar’s lab web page is: http://www.brarlab.org
Jonathan Weissman received his undergraduate physics degree from Harvard College. After obtaining a Ph.D. in physics from the Massachusetts Institute of Technology, where he worked with Peter Kim, Dr. Weissman pursued postdoctoral fellowship training in Arthur Horwich’s laboratory at Yale University School of Medicine. He has received the Raymond and Beverly Sackler International Prize in Biophysics and the National Academy of Sciences Award for Scientific Discovery. Dr. Weissman is a member of the National Academy of Sciences and a faculty member at the University of California at San Francisco.
Jonathan Weissman’s lab studies how cells ensure that proteins fold into their correct shape, as well as the role of protein misfolding in disease and normal physiology. The Weissman lab is also developing experimental and analytical approaches for exploring the organizational principles of biological systems and globally monitoring protein translation through ribosome profiling. A broad goal of his work is to bridge large-scale approaches and in depth mechanistic investigations to reveal the information encoded within genomes.
Jonathan Weissman’s lab web page is: http://weissmanlab.ucsf.edu/
Footnotes
Competing Interests Statement: JSW is an inventor on patent application for ribosome profiling.
Subject categories
Biological sciences / Molecular biology / Translation / Ribosome [URI /631/337/574/1789]
Biological sciences / Molecular biology / Proteomics [URI /631/337/475]
Biological sciences / Biological techniques / Proteomic analysis [URI /631/1647/2067]
Techniques
Life sciences techniques, High-throughput screening [Gene expression profiling]
Life sciences techniques, Protein techniques [High-throughput screening assays]
Life sciences techniques, Gene expression analysis [Gene expression profiling]
Life sciences techniques, Genomic analysis [RNA sequencing]
Bibliography
- 1.McCann KL, Baserga SJ. Genetics. Mysterious ribosomopathies. Science. 2013;341:849–850. doi: 10.1126/science.1244156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cleary JD, Ranum LPW. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum Mol Genet. 2013;22:R45–51. doi: 10.1093/hmg/ddt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis SR. Nucleolar stress in Diamond Blackfan anemia pathophysiology. Biochim Biophys Acta. 2014;1842:765–768. doi: 10.1016/j.bbadis.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 4.Trainor PA, Merrill AE. Ribosome biogenesis in skeletal development and the pathogenesis of skeletal disorders. Biochim Biophys Acta. 2014;1842:769–778. doi: 10.1016/j.bbadis.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolze A, et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 2013;340:976–978. doi: 10.1126/science.1234864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kondrashov N, et al. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145:383–397. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. This work defined the ribosome profiling method and detailed its specificity, precision, and utility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolin SL, Walter P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBOJ. 1988;7:3559–3569. doi: 10.1002/j.1460-2075.1988.tb03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steitz JA. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature. 1969;224:957–964. doi: 10.1038/224957a0. [DOI] [PubMed] [Google Scholar]
- 10.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stern-Ginossar N, et al. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brar GA, et al. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–557. doi: 10.1126/science.1215110. References 11 and 12 applied ribosome profiling to physiological dynamic cellular processes, the HCMV infection cycle of human cells and meiosis in budding yeast, respectively. In these disparate systems, both studies identified numerous new examples of translational control, uORF translation, and the translation of numerous short and alternate ORFs in genomic regions thought to be noncoding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bazzini AA, et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014;33:981–993. doi: 10.1002/embj.201488411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pauli A, et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343 doi: 10.1126/science.1248636. Epub 1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunn JG, Foo CK, Belletier NG, Gavis ER, Weissman JS. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife. 2013;2:e01179. doi: 10.7554/eLife.01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aspden JL, et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. eLife. 2014;3:e03528. doi: 10.7554/eLife.03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith JE, et al. Translation of Small Open Reading Frames within Unannotated RNA Transcripts in Saccharomyces cerevisiae. Cell Rep. 2014;7:1858–1866. doi: 10.1016/j.celrep.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S, et al. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A. 2012;109:E2424–2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreev DE, et al. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. eLife. 2014;4:e03971. doi: 10.7554/eLife.03971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet TIG. 2014;30:418–426. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuersten S, Radek A, Vogel C, Penalva LOF. Translation regulation gets its ‘omics’ moment. Wiley Interdiscip Rev RNA. 2013;4:617–630. doi: 10.1002/wrna.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh E, et al. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell. 2011;147:1295–1308. doi: 10.1016/j.cell.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arias C, et al. KSHV 2.0: a comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014;10:e1003847. doi: 10.1371/journal.ppat.1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stadler M, Fire A. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA N Y N. 2011;17:2063–2073. doi: 10.1261/rna.02890211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336:233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juntawong P, Girke T, Bazin J, Bailey-Serres J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc Natl Acad Sci U S A. 2014;111:E203–212. doi: 10.1073/pnas.1317811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen BC, et al. Extensive stage-regulation of translation revealed by ribosome profiling of Trypanosoma brucei. BMC Genomics. 2014;15:911. doi: 10.1186/1471-2164-15-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caro F, Ahyong V, Betegon M, DeRisi JL. Genome-wide regulatory dynamics of translation in the Plasmodium falciparum asexual blood stages. eLife. 2014;3:e04106. doi: 10.7554/eLife.04106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schafer S, et al. Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat Commun. 2015;6:7200. doi: 10.1038/ncomms8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rooijers K, Loayza-Puch F, Nijtmans LG, Agami R. Ribosome profiling reveals features of normal and disease-associated mitochondrial translation. Nat Commun. 2013;4:2886. doi: 10.1038/ncomms3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zoschke R, Watkins KP, Barkan A. A rapid ribosome profiling method elucidates chloroplast ribosome behavior in vivo. Plant Cell. 2013;25:2265–2275. doi: 10.1105/tpc.113.111567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michel AM, et al. GWIPS-viz: development of a ribo-seq genome browser. Nucleic Acids Res. 2014;42:D859–864. doi: 10.1093/nar/gkt1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Jiang H, Gu Z, Roberts JW. High-resolution view of bacteriophage lambda gene expression by ribosome profiling. Proc Natl Acad Sci U S A. 2013;110:11928–11933. doi: 10.1073/pnas.1309739110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerashchenko MV, Lobanov AV, Gladyshev VN. Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc Natl Acad Sci U S A. 2012;109:17394–17399. doi: 10.1073/pnas.1120799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li GW, Oh E, Weissman JS. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woolstenhulme CJ, Guydosh NR, Green R, Buskirk AR. High-Precision Analysis of Translational Pausing by Ribosome Profiling in Bacteria Lacking EFP. Cell Rep. 2015;11:13–21. doi: 10.1016/j.celrep.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chew GL, et al. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Dev Camb Engl. 2013;140:2828–2834. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ingolia NT, et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014;8:1365–1379. doi: 10.1016/j.celrep.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreev DE, et al. Oxygen and glucose deprivation induces widespread alterations in mRNA translation within 20 minutes. Genome Biol. 2015;16:90. doi: 10.1186/s13059-015-0651-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guydosh NR, Green R. Dom34 rescues ribosomes in 3′ untranslated regions. Cell. 2014;156:950–962. doi: 10.1016/j.cell.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shalgi R, et al. Widespread regulation of translation by elongation pausing in heat shock. Mol Cell. 2013;49:439–452. doi: 10.1016/j.molcel.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han Y, et al. Ribosome profiling reveals sequence-independent post-initiation pausing as a signature of translation. Cell Res. 2014;24:842–851. doi: 10.1038/cr.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu B, Han Y, Qian SB. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol Cell. 2013;49:453–463. doi: 10.1016/j.molcel.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramaniam AR, Zid BM, O’Shea EK. An integrated approach reveals regulatory controls on bacterial translation elongation. Cell. 2014;159:1200–1211. doi: 10.1016/j.cell.2014.10.043. This work probed position-specific changes in ribosome distribution between multiple cellular conditions, concluding that tRNA abundances do not account for elongation rates for most codons, and that pausing of ribosomes during starvation may result in translation abortion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lareau LF, Hite DH, Hogan GJ, Brown PO. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 2014;3:e01257. doi: 10.7554/eLife.01257. This work identified a class of short ribosome footprints that may be enriched by translation elongation inhibitor treatment and that likely represent a distinct conformation of the ribosome at a specific stage of the elongation cycle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siegel AF, van den Engh G, Hood L, Trask B, Roach JC. Modeling the feasibility of whole genome shotgun sequencing using a pairwise end strategy. Genomics. 2000;68:237–246. doi: 10.1006/geno.2000.6303. [DOI] [PubMed] [Google Scholar]
- 48.Roberts A, Schaeffer L, Pachter L. Updating RNA-Seq analyses after re-annotation. Bioinforma Oxf Engl. 2013;29:1631–1637. doi: 10.1093/bioinformatics/btt197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saliba AE, Westermann AJ, Gorski SA, Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014;42:8845–8860. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Green R, Noller HF. RIBOSOMES AND TRANSLATION. Annu Rev Biochem. 1997;66:679–716. doi: 10.1146/annurev.biochem.66.1.679. [DOI] [PubMed] [Google Scholar]
- 51.Li GW, Burkhardt D, Gross C, Weissman JS. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aris JP, Klionsky DJ, Simoni RD. The Fo subunits of the Escherichia coli F1Fo-ATP synthase are sufficient to form a functional proton pore. J Biol Chem. 1985;260:11207–11215. [PubMed] [Google Scholar]
- 53.Humphryes N, et al. The Ecm11-Gmc2 complex promotes synaptonemal complex formation through assembly of transverse filaments in budding yeast. PLoS Genet. 2013;9:e1003194. doi: 10.1371/journal.pgen.1003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee MT, et al. Nanog, Pou5f1 and SoxB1 activate zygotic gene expression during the maternal-to-zygotic transition. Nature. 2013;503:360–364. doi: 10.1038/nature12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kronja I, et al. Widespread Changes in the Posttranscriptional Landscape at the Drosophila Oocyte-to-Embryo Transition. Cell Rep. 2014;7:1495–1508. doi: 10.1016/j.celrep.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vasquez JJ, Hon CC, Vanselow JT, Schlosser A, Siegel TN. Comparative ribosome profiling reveals extensive translational complexity in different Trypanosoma brucei life cycle stages. Nucleic Acids Res. 2014;42:3623–3637. doi: 10.1093/nar/gkt1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D. The translational landscape of the mammalian cell cycle. Mol Cell. 2013;52:574–582. doi: 10.1016/j.molcel.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stadler M, Fire A. Conserved translatome remodeling in nematode species executing a shared developmental transition. PLoS Genet. 2013;9:e1003739. doi: 10.1371/journal.pgen.1003739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu B, Qian SB. Translational reprogramming in cellular stress response. Wiley Interdiscip Rev RNA. 2014;5:301–305. doi: 10.1002/wrna.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15:205–213. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- 61.Michel AM, Baranov PV. Ribosome profiling: a Hi-Def monitor for protein synthesis at the genome-wide scale. Wiley Interdiscip Rev RNA. 2013;4:473–490. doi: 10.1002/wrna.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 63.Kannan K, et al. The general mode of translation inhibition by macrolide antibiotics. Proc Natl Acad Sci U S A. 2014;111:15958–15963. doi: 10.1073/pnas.1417334111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kannan K, Vázquez-Laslop N, Mankin AS. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 2012;151:508–520. doi: 10.1016/j.cell.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 65.Davis AR, Gohara DW, Yap MNF. Sequence selectivity of macrolide-induced translational attenuation. Proc Natl Acad Sci U S A. 2014;111:15379–15384. doi: 10.1073/pnas.1410356111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jung H, Gkogkas CG, Sonenberg N, Holt CE. Remote control of gene function by local translation. Cell. 2014;157:26–40. doi: 10.1016/j.cell.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams CC, Jan CH, Weissman JS. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science. 2014;346:748–751. doi: 10.1126/science.1257522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jan CH, Williams CC, Weissman JS. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science. 2014;346:1257521. doi: 10.1126/science.1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arava Y, et al. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heiman M, Kulicke R, Fenster RJ, Greengard P, Heintz N. Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP) Nat Protoc. 2014;9:1282–1291. doi: 10.1038/nprot.2014.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inada T, et al. One-step affinity purification of the yeast ribosome and its associated proteins and mRNAs. RNA N Y N. 2002;8:948–958. doi: 10.1017/s1355838202026018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zanetti ME, Chang IF, Gong F, Galbraith DW, Bailey-Serres J. Immunopurification of polyribosomal complexes of Arabidopsis for global analysis of gene expression. Plant Physiol. 2005;138:624–635. doi: 10.1104/pp.105.059477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mustroph A, Zanetti ME, Girke T, Bailey-Serres J. Isolation and analysis of mRNAs from specific cell types of plants by ribosome immunopurification. Methods Mol Biol Clifton NJ. 2013;959:277–302. doi: 10.1007/978-1-62703-221-6_19. [DOI] [PubMed] [Google Scholar]
- 75.Thomas A, et al. A versatile method for cell-specific profiling of translated mRNAs in Drosophila. PloS One. 2012;7:e40276. doi: 10.1371/journal.pone.0040276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Housley MP, et al. Translational profiling through biotinylation of tagged ribosomes in zebrafish. Dev Camb Engl. 2014;141:3988–3993. doi: 10.1242/dev.111849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reinhardt JA, Jones CD. Two rapidly evolving genes contribute to male fitness in Drosophila. J Mol Evol. 2013;77:246–259. doi: 10.1007/s00239-013-9594-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Starck SR, et al. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science. 2012;336:1719–1723. doi: 10.1126/science.1220270. [DOI] [PubMed] [Google Scholar]
- 79.Rebbapragada I, Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol. 2009;21:394–402. doi: 10.1016/j.ceb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 80.Pauli A, Valen E, Schier AF. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. BioEssays News Rev Mol Cell Dev Biol. 2015;37:103–112. doi: 10.1002/bies.201400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ulitsky I, Bartel D. P lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kondo T, et al. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329:336–339. doi: 10.1126/science.1188158. This paper identified key roles in fly development for several short peptides from 11 to 32 amino acids, translated from sORFs on a transcript that was previously thought to be noncoding. [DOI] [PubMed] [Google Scholar]
- 83.Magny EG, et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 2013;341:1116–1120. doi: 10.1126/science.1238802. [DOI] [PubMed] [Google Scholar]
- 84.Anderson DM, et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015;160:595–606. doi: 10.1016/j.cell.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ruiz-Orera J, Messeguer X, Subirana JA, Alba MM. Long non-coding RNAs as a source of new peptides. eLife. 2014;3:e03523. doi: 10.7554/eLife.03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu Y, Ganem D. Making sense of antisense: seemingly noncoding RNAs antisense to the master regulator of Kaposi ’ s sarcoma-associated herpesvirus lytic replication do not regulate that transcript but serve as mRNAs encoding small peptides. J Virol. 2010;84:5465–5475. doi: 10.1128/JVI.02705-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–251. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carvunis AR, et al. Proto-genes and de novo gene birth. Nature. 2012;487:370–374. doi: 10.1038/nature11184. Here the authors present evidence to support the proto-gene hypothesis, by which new proteins can eve through selection and elongation of ORFs encoding peptides translated from putative intergenic transcripts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brubaker SW, Gauthier AE, Mills EW, Ingolia NT, Kagan JC. A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell. 2014;156:800–811. doi: 10.1016/j.cell.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Michel AM, et al. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res. 2012;22:2219–2229. doi: 10.1101/gr.133249.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Noderer WL, et al. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Mol Syst Biol. 2014;10:748. doi: 10.15252/msb.20145136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schwaid AG, et al. Chemoproteomic discovery of cysteine-containing human short open reading frames. J Am Chem Soc. 2013;135:16750–16753. doi: 10.1021/ja406606j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Slavoff SA, et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ma J, et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 2014;13:1757–1765. doi: 10.1021/pr401280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crappé J, et al. Combining in silico prediction and ribosome profiling in a genome-wide search for novel putatively coding sORFs. BMC Genomics. 2013;14:648. doi: 10.1186/1471-2164-14-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Menschaert G, et al. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol Cell Proteomics MCP. 2013;12:1780–1790. doi: 10.1074/mcp.M113.027540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vanderperre B, et al. Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PloS One. 2013;8:e70698. doi: 10.1371/journal.pone.0070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinforma Oxf Engl. 2011;27:i275–282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang HY, Tang HL, Chao HY, Yeh LS, Wang CC. An unusual pattern of protein expression and localization of yeast alanyl-tRNA synthetase isoforms. Mol Microbiol. 2006;60:189–198. doi: 10.1111/j.1365-2958.2006.05083.x. [DOI] [PubMed] [Google Scholar]
- 100.Chang KJ, Wang CC. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. J Biol Chem. 2004;279:13778–13785. doi: 10.1074/jbc.M311269200. [DOI] [PubMed] [Google Scholar]
- 101.Wan J, Qian S-B. TISdb: a database for alternative translation initiation in mammalian cells. Nucleic Acids Res. 2014;42:D845–850. doi: 10.1093/nar/gkt1085. This work presents a database of alternate translation initiation sites identified by ribosome profiling in mammalian cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Artieri CG, Fraser HB. Evolution at two levels of gene expression in yeast. Genome Res. 2014;24:411–421. doi: 10.1101/gr.165522.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jungreis I, et al. Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 2011;21:2096–2113. doi: 10.1101/gr.119974.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schueren F, et al. Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. eLife. 2014;3:e03640. doi: 10.7554/eLife.03640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zalatan JG, et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160:339–350. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gilbert LA, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.