

Abstract
The use of Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) in neuroscience has rapidly expanded in rodent studies but has lagged behind in nonhuman primate (NHP) experiments, slowing the development of this method for therapeutic use in humans. One reason for the slow adoption of DREADD technology in primates is that the pharmacokinetic properties and bioavailability of clozapine-n-oxide (CNO), the most commonly used ligand for human muscarinic (hM) DREADDs, are not fully described in primates. We report an extensive pharmacokinetic study using subcutaneous (SC) administration of CNO in five adult rhesus monkeys. CNO reached maximal plasma and cerebrospinal fluid (CSF) concentrations within 2 h after injection, with an observed dose-dependent increase in levels following a 3 and 10 mg/kg SC dose. Since CSF concentrations were below values predicted from unbound plasma concentrations, we investigated whether CNO was restricted from the CNS through active transport at the blood–brain barrier. In vitro assessment demonstrated that CNO is a substrate for P-glycoprotein (Pgp; efflux ratio, 20), thus providing a likely mechanism limiting CNO levels in the CNS. Furthermore, CNO is metabolized to the psychoactive compounds clozapine and n-desmethylclozapine in monkeys. The concentrations of clozapine detected in the CSF are sufficient to activate several types of receptor (including the hM-DREADDs). Our results suggest that CNO metabolism and distribution may interfere with reproducibility and interpretation of DREADD-related experiments in NHPs and calls for a re-evaluation of the use of CNO in DREADD-related experiments in NHPs along with the need to test alternative compounds.
Keywords: Rhesus macaque, DREADDs, pharmacokinetics, blood–brain barrier, clozapine
Graphical Abstract
INTRODUCTION
Chemogenetic techniques allow researchers to manipulate the activity of specific populations of neurons by activating engineered molecules that are genetically inserted into the target neurons and can subsequently be modulated by systemically administered pharmacological agents.1 The relatively low invasiveness of the technique makes it attractive for clinical use. The most widely used chemogenetic method is based on Designer Receptors Activated by Designer Drugs (DREADDs); one common type of DREADD involves the use of artificial G protein-coupled receptors, which are synthetic variants of human muscarinic receptors (hM-DREADD).2–4 These synthetic receptors are not activated by acetylcholine (the endogenous ligand of muscarinic receptors) and are not constitutively active. Instead, DREADDs are exclusively activated with high efficacy by exogenous ligands. The most commonly used hM-DREADD ligand is clozapine-N-oxide (CNO), a metabolite of the antipsychotic drug clozapine.
The available hM-DREADDs include receptors that are coupled to Gi/o and Gq/11 proteins (hM4Di and hM3Dq, respectively, are the most commonly used of each type3,5) providing tools that can increase or decrease the activity of transfected neurons, depending on the intracellular pathway activated. The expression of DREADDs can be targeted to particular neurons with specific promoters using transgenic approaches or viral transfection.3,4,6
While DREADD technology has been widely used in vivo in rodents,5,7 there are only three published reports on the use of hM-DREADDs in primates.8–10 One of the challenges preventing more widespread application of hM-DREADDs in monkeys is our limited knowledge about the metabolism and disposition of systemically administered CNO in monkeys. Considering the existing literature related to the biotransformation of CNO in rodents and humans,11–13 two key areas of investigation remain with regards to this designer drug’s metabolism and disposition: (1) the detailed pharmacokinetics (PK) of CNO following extravascular administration and (2) the extent of metabolism-activated formation of the psychoactive compounds, clozapine and N-desmethylclozapine (NDMC), in nonhuman primates (NHP).
The goals of this study are to provide a thorough characterization of the in vivo pharmacokinetics of CNO at doses demonstrated to activate hM-DREADDs in monkeys, as well as to quantitate the reductive-metabolism of CNO to clozapine and NDMC after extravascular administration. Our study complements and expands the data currently available to the hM-DREADD community regarding the pharmacokinetics of CNO in NHPs, which is based on sparse PK sampling.8,10 To our knowledge, our study represents the first report of active transport/efflux of CNO in vitro by P-glycoprotein (Pgp), a disposition protein expressed at the blood–brain barrier that effectively limits CNS exposure of xenobiotics. These data also represent a significant development and liability for CNO as a hM-DREADD ligand in vivo, potentially impeding the use of hM-DREADDs in NHPs and limiting the clinical translation of this technique for use in humans.
RESULTS AND DISCUSSION
Pharmacokinetics of CNO in NHPs
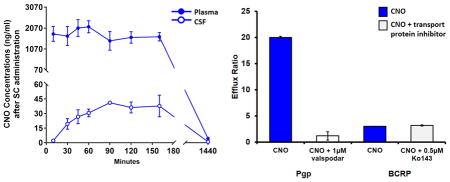
Rhesus monkeys (n = 5) received subcutaneous (SC) administration of CNO (3 or 10 mg/kg) with blood and CSF samples collected at predetermined intervals in order to build corresponding PK profiles of the CNO and its pertinent metabolites. In some instances, blood draws were conducted in awake animals trained for conscious venipuncture procedures (see Methods). Following a 10 mg/kg SC dose, CNO reached a mean maximal plasma concentration (Cmax) of 2528 ng/mL (7 μM) with a corresponding Tmax of 45 min (Figure 1a and Table 1). CNO plasma concentrations remained elevated for approximately 2 h postinjection, dropping to <5 ng/mL by the 24 h blood collection. CNO was below the limit of quantitation at the 48 h blood collection. Importantly, we observed no significant differences in the plasma PK of CNO between the awake and anesthetized (Telazol, 3–5 mg/kg, intramuscular) conditions, suggesting that neither the state of wakefulness nor the anesthetic affected the disposition of the ligand; an important consideration for hM-DREADD-related studies in which CNO is delivered in conscious animals. CNO displayed dose-dependency between the 3 and 10 mg/kg SC dose experiments; Cmax at 3 mg/kg was 1092 ng/mL as compared to 2528 ng/mL at 10 mg/kg (compare Figure 1, panels A and B; see also Table 1). Similarly, the area-under-the-curve (AUC) values produced at 3 mg/kg (AUC, 519 ng/(mL·h)) were approximately 65% of that obtained following the 10 mg/kg dose (AUC, 795 ng/(mL·h); see Table 1).
Figure 1.

Time–concentration profiles of CNO and its metabolites after SC administration to NHPs. Plasma (A,B) and CSF (C,D) concentrations of clozapine-N-oxide (CNO; blue circles), clozapine (purple squares), and n-desmethylclozapine (NDMC; green triangles) following SC administration of CNO at 10 mg/kg (A,C) and at 3 mg/kg (B,D). In panel A, dotted lines represent blood samples obtained from conscious animals, whereas solid lines represent blood or CSF obtained under anesthesia. The data are represented as the mean and standard error of the mean (M ± SEM, n = 5); not all animals’ CSF data were included in the profiles (see Methods).
Table 1.
Pharmacokinetic Parameters of CNOa
| dose | substrate | CNO | clozapine | N-desmethylclozapine |
|---|---|---|---|---|
| AUC (ng/mL·hr) | ||||
| 10 mg/kg | plasma (0 min to 48 h) | 795.68 | 45.19 | 81.93 |
| CSF (10 min to 24 h) | 9.80 | 4.68 | 4.06 | |
| 3 mg/kg | plasma (0 min to 24 h) | 519.45 | 11.18 | 10.22 |
| CSF (30 min to 24 h) | 5.94 | NA | 3.38 | |
| Cmax (ng/mL) | ||||
| 10 mg/kg | plasma | 2528.00 | 49.26 | 50.40 |
| CSF | 41.15 | 11.22 | 13.53 | |
| 3 mg/kg | plasma | 1092.09 | 13.18 | 10.28 |
| CSF | 10.89 | 5.45 | 3.56 | |
| Cmax (nM) | ||||
| 10 mg/kg | plasma | 7374.13 | 150.73 | 161.13 |
| CSF | 120.03 | 34.33 | 43.25 | |
| 3 mg/kg | plasma | 3185.60 | 40.33 | 32.86 |
| CSF | 31.77 | 16.68 | 11.37 | |
| Tmax | ||||
| 10 mg/kg | plasma | 45 min | 6 h | 12 h |
| CSF | 90 minb | 160 minb | 24 hb | |
| 3 mg/kg | plasma | 45 minb | 160 minb | 24 hb |
| CSF | 90 minb | 160 minb | 24 hb | |
| fu | ||||
| PPB | 0.43 | 0.05 | 0.11 | |
Mean plasma and cerebrospinal fluid (CSF) values for two doses of clozapine-N-oxide (CNO; 10 and 3 mg/kg). AUC = area under the curve; Cmax = maximal concentration; Tmax = time at maximal concentration; fu = fraction unbound; PPB = plasma protein binding.
CSF samples after 10 and 3 mg/kg CNO and plasma samples after 3 mg/kg CNO were collected less frequently than plasma samples after 10 mg/kg CNO (see Methods); therefore Tmax for these samples was determined on fewer time points.
To evaluate the CNO brain exposure in animals, we collected CSF samples (via cisterna magna, under anesthesia) at predetermined times following a single SC administration of CNO. Following the 10 mg/kg-dose, CNO reaches a maximal concentration (41 ng/mL, 120 nM) in CSF within 90 min of the SC administration (Table 1, Figure 1C). These data are comparable to those obtained in a previous study following an intramuscular administration of CNO (IM, 10 mg/kg) to monkeys;8 but SC administration was distinct in that its Tmax was right shifted to 90 min, compared to a Tmax of 60 min following IM dosing.8 Additionally, SC dosing resulted in a greater average concentration (41 ng/mL) compared to IM dosing (9 ng/mL).8
Following SC administration of the 3 mg/kg CNO dose, CNO in CSF samples displayed a similar PK as observed after 10 mg/kg, and dose dependency, as seen in plasma samples (Figure 1C,D). The 3 mg/kg CNO dose produced a Cmax of ~11 ng/mL (31 nM, Figure 1D), approximately a log order lower than that previously observed by Nagai and colleagues10 following an intravenous (IV) dose of 3 mg/kg CNO to monkeys. These data would indicate a lower overall SC-bioavailability of CNO, as compared to an IV administration, and thus represent an additional factor for consideration when dosing for maximal CNS effect in hM-DREADD-related experiments.10
At both the 10 and 3 mg/kg doses, the CNO concentrations in CSF were substantially lower than the plasma concentrations (Table 1), and those predicted based on a calculated unbound concentration of CNO in the plasma. This observation is unrelated to the dosing route, as similar CSF exposures were reported in animals receiving both IV and IM administration. 8,10
Despite the low concentrations reached in the 10 mg/kg dose (30–41 ng/mL or 89–120 nM, see Table 1, Figure 1C), this may be sufficient to activate hM-DREADDs (based on studies of PI hydrolysis and Ca2+ mobilization in immortalized human pulmonary artery smooth muscle cells expressing hMDREADDs2). However, in vitro electrophysiological experiments have consistently used higher concentrations of CNO (1–10 μM) to modulate the activity in hM-DREADD-expressing neurons,2,8,14 suggesting that the nanomolar concentration of CNO detected in the CSF in our experiments may be at the lower end of an effective concentration range to activate hM-DREADDs after viral transfection. Alternatively, CNO metabolites could be responsible for hM-DREADD activation, as further discussed below.
Bidirectional Transporter of CNO in MDCK Cells in Vitro
Given the relatively high fraction unbound determined for CNO in vitro (fu plasma 0.43, Table 1), we had expected CNO to freely diffuse across the blood–brain barrier (BBB) and permeate the CNS.15,16 Our CSF PK analysis indicated otherwise, producing an unbound plasma concentration that was incongruent with the CSF concentration. In order to determine the mechanism behind the poor distribution of CNO to the CNS, we investigated the potential contribution of two drug efflux proteins that are localized at the BBB, P-glycoprotein (Pgp or MDR1) and breast cancer resistance protein (BCRP).16–18 To this end, we employed Madin–Darby canine kidney (MDCK) cells that were stably transfected with either human Pgp (MDCK-MDR1 or MDCK-Pgp) or BCRP (MDCK-BCRP).17,18 Briefly, the A-B and B-A transport of CNO across the MDCK cell monolayers was determined in the absence or presence of a specific Pgp or BCRP inhibitor (Figure 2). The permeability of CNO across a MDCK-Pgp monolayer was low, displaying A-B and B-A permeability of 0.05 PappAB (0.05 × 10−6 and 1 × 10−6 cm/s, respectively) and an efflux ratio (B-A/A-B) of 20, indicating that CNO is a substrate of Pgp. Importantly, fortification of the in vitro bidirectionality assay with a specific inhibitor of Pgp, valspodar, resulted in the reduction of the efflux ratio to approximately 1.2, thus confirming the role of Pgp in the active transport of CNO across the MDCK-Pgp monolayer. The permeability of CNO across a MDCK-BCRP monolayer was equally low (A-B, 0.07 × 10−6 cm/s; B-A, 0.21 × 10−6 cm/s), subsequently producing an efflux ratio of approximately 3. However, the BCRP-specific inhibitor, K0143, displayed no significant effect on the bidirectional transport of CNO (+ K0143, efflux ratio ~3.2; Figure 2). Collectively, these data indicate that CNO is a substrate for Pgp-mediated efflux at the BBB in vitro. Our results are in agreement with previous studies demonstrating low blood–brain permeability of CNO19 and may account for its limited CNS distribution (i.e., exposure) in vivo in NHP.
Figure 2.

Bidirectional transport of CNO across MDCK monolayers and the impact of specific inhibitors on active efflux. Bidirectional transport of CNO (5 μM) across MDCK-Pgp/MDCK-MDR1 or BCRP-MDCK cell monolayers in the absence and presence of valspodar (1 μM) and Ko143 (0.5 μM). Open bars, A-B transport; closed bars, B-A transport. Data are presented as the mean ± SEM.
In principle, the Pgp-mediated efflux of CNO would suggest that Pgp specific inhibitors20 could be used in DREADD-related studies to effectively increase CNS exposure of CNO following a parenteral or extravascular administration. However, the resulting outcomes from DREADDs models employing additional pharmacological inhibitors (e.g., Pgp inhibitor) require additional experimentation.21,22 Particularly noteworthy is a consideration of the disruption of the regulation and expression of Pgp in varied neuropathologies.21
CNO Conversion to Clozapine and NDMC after SC Administration
CNO is susceptible to reductive metabolism to clozapine and, indirectly, to NDMC via a putative oxidative N-demethylation.23 Considering both clozapine and NDMC are capable of binding to a variety of neurotransmitter receptors, inducing severe sedation (among other negative side effects),24,25 we investigated if CNO is biotransformed to these active metabolites after SC administration to monkeys. Clozapine and NDMC concentrations were determined in the plasma and CSF from animals receiving SC administration of CNO (10 and 3 mg/kg). Analysis of the CNO dosing solutions demonstrated the relative stability of CNO, since clozapine and NDMC were not present in the CNO formulations at the time of administration (data not shown).
Following a single SC dose (10 mg/kg) of CNO, we observed the in vivo metabolism of CNO to clozapine (N-oxide reduction) and NDMC in the corresponding rhesus monkey plasma and CSF (Table 1 and Figure 1). Concentrations of the metabolites are not detectable in the CSF prior to the 60 and 150 min sample for 10 and 3 mg/kg CNO doses, respectively (Figure 1C,D).
After systemic CNO administration, the maximal concentrations (Cmax) of clozapine and NDMC detected in the CSF were in a concentration range capable of eliciting a pharmacological effect in vivo (34 nM for clozapine and 43 nM for NDMC; Table 1). In particular, these concentrations may be sufficient to activate a variety of endogenous neurotransmitter receptors in the CNS, such as dopaminergic and serotoninergic receptors.24–27 Thus, as observed in rodents and humans, the conversion of CNO to its psychoactive compounds is a relevant consideration for hM-DREADD related studies in NHPs. In rodents for example, CNO has been demonstrated to produce behavioral effects in naive animals;13 anecdotal evidence of the same has been reported in primates.8
Conversion of CNO to clozapine would be of importance not only in terms of the potential side effects resulting from the presence of clozapine. Instead it has to be considered that clozapine at the present CSF concentrations (Figure 1C and Table 1) is capable of activating the DREADDs hM3Dq and hM4Di.2 Coupled with the fact that clozapine distributes to the CNS to a greater extent than CNO, it is possible that activation of hM-DREADDs could be preferentially mediated by clozapine rather than CNO.
In summary, our results indicate that CNO is systemically bioavailable following a single SC administration to rhesus monkeys. While CNO (and metabolites) is detected in the CNS following systemic exposure, the overall distribution of this hM-DREADD ligand to the CNS is restricted (CSF exposure in low nanomolar range) in monkeys. Data obtained from transwell membrane bidirectional assessments (MDCK-MDR1, -BCRP), also indicate that CNO is a substrate for the drug efflux protein, Pgp, and this represents a likely mechanism by which CNO is mostly restricted to systemic circulation.
Importantly, a limitation of this study is that CSF drug levels are considered a surrogate of drug distribution in the brain parenchyma, but this is an oversimplification, since CSF samples only reveal the drug concentrations through the blood–cerebrospinal fluid barrier.28 Given the limitations imposed by in vivo work, CSF drug levels, however, remain a useful (although indirect) reflection of drug distribution in the CNS.
Although the CNO concentrations in CNS may be sufficient to activate hM-DREADDs, it is also a likely scenario that the in situ formation of clozapine may be responsible for a partial activation of these hM-DREADD receptors, as well as other off-target receptors. Although higher doses (and resulting exposures) could be used to overcome disposition enzymes and enhance hM-DREADDs activation by CNO, an unintended consequence would be the increased exposure of the psychoactive metabolites clozapine and NDMC, which would be a concern, especially if long-term CNO dosing regimens are contemplated (as would likely be the case as a therapeutic approach in humans). Considering the present limitations and the potential for interfering pharmacology (via active metabolites) and irreproducibility, a continued pursuit of CNO as a preferred hM-DREADD ligand may contribute to the slow progress of the adoption of this technique in NHP models of neuropharmacology. Recent reports have highlighted the promise of alternatives to CNO, with compounds displaying high affinities for hM-DREADDs.29 Such compounds may be advantageous over CNO, after thorough characterization of their pharmacokinetic and disposition properties in NHPs.
METHODS
Subjects
Five adult rhesus monkeys (Macaca mulatta, 3 males [9–14.5 kg], 2 females [7 and 7.5 kg]) from the Yerkes National Primate Research Center (YNPRC, Atlanta GA, USA) were used in this study. The animals were housed under a 12 h light/dark cycle with ad lib access to water and food (5037 Monkey Chow, LabDiet, Land O’Lakes, Inc., USA). All procedures were approved by the Institutional Animal Care and Use Committee of Emory University and were carried out in accordance with the Guide for the Care and Use of Laboratory Animals, eighth edition (National Research Council, 2011), and the United States Department of Agriculture (USDA) Animal Welfare Act.
Clozapine-N-oxide Preparation
CNO powder (NIMH C-929, RITI International) was stored at −20 °C, protected from light. CNO solutions were freshly prepared on each experimental day, by first dissolving the drug at 66 mg/mL in 100% dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis MO, USA) and then diluting with calcium- and magnesium-free phosphate-buffered saline (PBS, Corning, Corning, NY, USA) to a final concentration of 10 mg/mL in 15% DMSO. To prevent degradation due to light exposure, vials and syringes that contained CNO solution were wrapped in aluminum foil. In some experiments, immediately following administration, an aliquot of the CNO was placed in a sterile Eppendorf tube, frozen on dry ice, and kept at −80 °C until assayed.
CNO Challenges in Conscious Monkeys
Animals were trained to voluntarily present their leg for awake blood collection from the saphenous vein, following established protocols at the YNPRC.30 All CNO challenges were conducted at the same time of day, starting between 9:00 and 10:00 AM. The animals received a subcutaneous injection of CNO (10 mg/kg body weight) in two separate sessions (6 months apart). In the first session, blood samples were collected at 0 (immediately before CNO injection), 10, 30, 45, 60, 90, 120, and 1440 min postinjection, and in the second session, samples were obtained at 0, 5, 180, 360, 720, 2160, and 2880 min postinjection. Assay results from these two challenges were combined for data analysis.
CNO Challenges under Anesthesia
In other experiments, CNO challenges were conducted under anesthesia to collect cerebrospinal fluid (CSF) samples. Animals received a subcutaneous injection of CNO (10 or 3 mg/kg) and were then sedated with tiletamine HCl and zolazepam HCl (Telazol, 3–5 mg/kg, i.m.). Once the animals were anesthetized, the cervical spine area was shaved and disinfected with betadine solution and alcohol. Each CSF sample was obtained from the cisterna magna, using a sterile 23-ga bevel-tipped needle by pressure difference and collected by gravity.31 CSF samples were collected in prechilled sterile Eppendorf tubes, immediately frozen on dry ice, and stored at −80 °C until the time of the assay.
Serial CSF taps were performed in sessions separated by 2 weeks. To reduce the risks associated with repeated spinal taps, and given the time restrictions imposed by the anesthetized state, in each session, only 2–3 serial CSF taps were conducted per animal, so that not all time points were collected for all monkeys.
Thus, after subcutaneous injection of CNO 10 mg/kg, five individual CSF samples were available for analyses at 120 min postinjection, four at 60 min postinjection, three at 10, 30, and 45 min postinjection, and two at 90, 160, and 1440 min. One 90 min postinjection CSF sample was discarded due to blood contamination. Six months later, serial CSF taps were again performed after a subcutaneous injection of CNO, 3 mg/kg, in sessions separated by 2 weeks. After the 3 mg/kg CNO injection, five individual CSF samples were available for analyses at 1440 and 160 min postinjection, four at 90 min postinjection, and three at 30 min postinjection.
Blood samples were collected from the femoral vein immediately following each CSF tap. All blood samples were collected in prechilled 2 mL tubes containing EDTA (3.5 mg) and immediately placed on ice. Samples were centrifuged at 3000 rpm for 15 min in a refrigerated centrifuge (at 4 °C). Plasma was pipetted off and stored at −80 °C until assayed.
Biological Sample and CNO Dosing Solution Assays
All plasma, CSF, and CNO dosing solution samples were assayed for CNO, clozapine, and NDMC by Sano Informed Prescribing (Franklin, TN). Liquid chromatography–tandem mass spectroscopy (LC-MS/MS) analyses were performed via reverse phase chromatography using a Shimadzu Nexera X2 UPLC (Columbia, MD) coupled with a QTrap 5500 (Sciex, Framinghman, MA, USA). For plasma samples, 20 μL aliquots of each standard, quality control sample, plasma sample, blank, and double blank are transferred to a labeled 96-well plate. Next, 120 μL of 50 ng/mL internal standard (carbamazepine in acetonitrile) was added to standards, quality controls, plasma samples, and blanks, while 120 μL of acetonitrile was added to the double blank, then the plate was centrifuged for 5 min at 3000 rcf. Then, 75 μL of supernatant was transferred into a labeled 96-well plate, and 75 μL of water was added to all samples. Finally, the plate was heat-sealed for analysis. The calibration range was 1–10000 ng/mL for CNO and n-desmethyl-clozapine (NDMC) and 0.5–5000 ng/mL for clozapine.
For CSF samples, 50 μL aliquots of each standard, quality control sample, plasma sample, blank, and double blank were transferred to a labeled 96-well plate. Next 150 μL of 50 ng/mL internal standard (carbamazepine in acetonitrile) was added to standards, quality controls, plasma samples, and blanks, while 150 μL of acetonitrile was added to the double blank. Then, the plate was centrifuged for 5 min at 3000 rcf, and 75 μL of water was added to all samples. Finally, the plate was heat-sealed for analysis. The calibration range was 0.5–500 ng/mL for CNO, 1–500 ng/mL for NDMC, and 5–500 ng/mL for clozapine.
Plasma Protein Binding Assay:32
The protein binding of each compound was determined in plasma via equilibrium dialysis employing RED Plates (ThermoFisher Scientific, Rochester, NY). Clozapine, CNO, or NDMC (200 μL of 1 μM each) in plasma was transferred to the cis chamber (red) of the RED plate, with an accompanying 350 μL of phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated for 4 h at 37 °C with shaking. At completion, 50 μL aliquots from each chamber were diluted 1:1 (50 μL) with either blank plasma (cis) or buffer (trans) and transferred to a new 96 well plate, at which time internal standard (50 ng/mL carbamazepine in acetonitrile) (2 volumes) was added. The plate was centrifuged (3000 rcf, 10 min), and supernatants were transferred and diluted 1:1 (supernatant/water) into a new 96 well plate, which was then sealed in preparation for LC-MS/MS analysis. Each compound was assayed in triplicate within the same RED plate. Fraction unbound was determined using the following equation:
Liquid chromatography-tandem mass spectroscopy (LC-MS/MS) analyses were performed via reverse phase chromatography using a Shimadzu Nexera X2 UPLC (Columbia, MD) coupled with a QTRAP 5500 (Sciex, Framinghman, MA, USA). Samples were subjected to a gradient elution using a C18 column (2.1 × 50 mm2, 1.7 μm; Phenomenex, Torrance, CA) that was thermostated at 50 °C. Mobile phase A was 10 mM ammonium acetate in water, and the mobile phase B was 0.1% formic acid in acetonitrile. A 30% B gradient was held for 0.2 min and was linearly increased to 60% B over 0.8 min, followed by a step to 95% B with an isocratic hold for 0.4 min, prior to re-equilibrating at 30% B for 0.6 min. The total run time was 2.0 min, and the HPLC flow rate was 0.5 mL/min. The source temperature was set at 500 °C, and mass spectral analyses were performed using multiple reaction monitoring (MRM) of transitions specific for the analytes and utilizing a Turbo-Ionspray source in positive ionization mode (5.0 kV spray voltage). All data were collected using Sciex Analyst 1.6.2 software and analyzed with MultiQuant 3.0 software.
Transporter Assay
The P-glycoprotein (Pgp or MDR1) and breast cancer resistance protein (BCRP) substrate assessment was performed by Absorption Systems (Exton, PA, USA). MDR1-MDCK cell monolayers were established on a 12-well Costar Transwell plate. The permeability assay buffer was Hanks’ balanced salt solution (HBSS) containing 10 mM HEPES and 15 mM glucose at a pH of 7.4. The buffer in the receiver chamber also contained 1% bovine serum albumin. The test article dosing solution (CNO, 5 μM) was prepared in assay buffer (with or without 1 μM valspodar, Pgp inhibitor). Cells were first preincubated for 30 min with HBSS containing valspodar. Cell monolayers were then dosed on the apical side (A-to-B) or basolateral side (B-to-A) and incubated at 37 °C with 5% CO2 in a humidified incubator. Samples were taken from the donor and receiver chambers at 120 min. Each determination was performed in duplicate. The flux of lucifer yellow was also measured postexperimentally for each monolayer to ensure no damage was inflicted to the cell monolayers during the flux period. All samples were assayed by LCMS/MS as described (vide supra). The apparent permeability (Papp) and percent recovery were calculated as follows:
where dCr/dt is the slope of the cumulative concentration in the receiver compartment versus time in μM s−1, Vr is the volume of the receiver compartment in cm3, Vd is the volume of the donor compartment in cm3, A is the area of the inset (1.13 cm2 for 12-well Transwell), CA is the average of the nominal dosing concentration and the measured 120 min donor concentration in μM; CN is the nominal concentration of the dosing solution in μM, is the cumulative receiver concentration in μM at the end of the incubation period, and is the concentration of the donor in μM at the end of the incubation period. The efflux ratio (ER) is defined as Papp(B-to-A)/Papp(A-to-B).
BCRP-MDCK cell monolayers were also established on 12-well Costar Transwell plates. Details of the plates and their certification are shown below. The permeability assay buffer was Hanks’ balanced salt solution (HBSS) containing 10 mM HEPES and 15 mM glucose at a pH of 7.4. The buffer in the receiver chamber also contained 1% bovine serum albumin. The test article dosing solution (CNO, 5 μM) was prepared in assay buffer (with or without 0.5 μM Ko143, BCRP inhibitor). Cells were first preincubated for 30 min with HBSS with or without 0.5 μM Ko143. Cell monolayers were then dosed on the apical side (A-to-B) or basolateral side (B-to-A) and incubated at 37 °C with 5% CO2 in a humidified incubator. Samples were taken from the donor and receiver chambers at 120 min. Each determination was performed in duplicate. The flux of lucifer yellow was also measured postexperimentally for each monolayer to ensure no damage was inflicted to the cell monolayers during the flux period. All samples were assayed by LC-MS/MS as described (vide supra). The apparent permeability (Papp) and percent recovery were calculated as follows:
where dCr/dt is the slope of the cumulative concentration in the receiver compartment versus time in μM s−1, Vr is the volume of the receiver compartment in cm3, Vd is the volume of the donor compartment in cm3, A is the area of the inset (1.13 cm2 for 12-well Transwell), CA is the average of the nominal dosing concentration and the measured 120 min donor concentration in μM; CN is the nominal concentration of the dosing solution in μM, is the cumulative receiver concentration in μM at the end of the incubation period, and is the concentration of the donor in μM at the end of the incubation period. The efflux ratio (ER) is defined as Papp(B-to-A)/Papp(A-to-B).
Statistical Analyses
Determination of CNO, clozapine, and N-desmethyl-clozapine pharmacokinetic parameters, area under the curve (AUC), maximal plasma concentration (Cmax), and time of maximal plasma concentration (Tmax) was done using Microsoft Office Excel (Microsoft Corporation, Redmond, USA), and GraphPad Prism 7.01 (GraphPad Software Inc. La Jolla, USA) was used to graph the results. The data are reported as means ± SEM. The molar mass used for calculations was 342.82 g/mol for CNO, 326.82 g/mol for clozapine, and 312.80 g/mol for n-desmethylclozapine (Pubchem ID 2819, 2818, and 2820 respectively; https://pubchem.ncbi.nlm.nih.gov/compound).
Acknowledgments
Funding
This work was supported through grants from National Institutes of Health (NIH) Office of Research Infrastructure Programs (ORIP; P51-OD011132) and the National Institute of Neurological Disorders and Stroke (NINDS; P50-NS098685, Udall Center of Excellence in Parkinson’s disease).
The authors thank Rebecca Richardson, M.A. for her assistance with the data collection.
Footnotes
Author Contributions
J.R. and A.G. designed the study, collected and analyze data, and wrote the manuscript. R.D.M. and J.S.D. assisted in the design of the study, assayed samples, and wrote the manuscript. L.H., J.B., and T.W. assisted in the writing and editing of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Farrell MS, Roth BL. Pharmacosynthetics: Reimagining the pharmacogenetic approach. Brain Res. 2013;1511:6–20. doi: 10.1016/j.brainres.2012.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roth BL. DREADDs for Neuroscientists. Neuron. 2016;89:683–694. doi: 10.1016/j.neuron.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith KS, Bucci DJ, Luikart BW, Mahler SV. DREADDS: Use and application in behavioral neuroscience. Behav Neurosci. 2016;130:137–155. doi: 10.1037/bne0000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wess J, Nakajima K, Jain S. Novel designer receptors to probe GPCR signaling and physiology. Trends Pharmacol Sci. 2013;34:385–392. doi: 10.1016/j.tips.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urban DJ, Roth BL. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol. 2015;55:399–417. doi: 10.1146/annurev-pharmtox-010814-124803. [DOI] [PubMed] [Google Scholar]
- 7.Rogan SC, Roth BL. Remote control of neuronal signaling. Pharmacol Rev. 2011;63:291–315. doi: 10.1124/pr.110.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eldridge MA, Lerchner W, Saunders RC, Kaneko H, Krausz KW, Gonzalez FJ, Ji B, Higuchi M, Minamimoto T, Richmond BJ. Chemogenetic disconnection of monkey orbitofrontal and rhinal cortex reversibly disrupts reward value. Nat Neurosci. 2016;19:37–39. doi: 10.1038/nn.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grayson DS, Bliss-Moreau E, Machado CJ, Bennett J, Shen K, Grant KA, Fair DA, Amaral DG. The Rhesus Monkey Connectome Predicts Disrupted Functional Networks Resulting from Pharmacogenetic Inactivation of the Amygdala. Neuron. 2016;91:453–466. doi: 10.1016/j.neuron.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagai Y, Kikuchi E, Lerchner W, Inoue KI, Ji B, Eldridge MA, Kaneko H, Kimura Y, Oh-Nishi A, Hori Y, Kato Y, Hirabayashi T, Fujimoto A, Kumata K, Zhang MR, Aoki I, Suhara T, Higuchi M, Takada M, Richmond BJ, Minamimoto T. PET imaging-guided chemogenetic silencing reveals a critical role of primate rostromedial caudate in reward evaluation. Nat Commun. 2016;7:13605. doi: 10.1038/ncomms13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang WH, Lin SK, Lane HY, Wei FC, Hu WH, Lam YW, Jann MW. Reversible metabolism of clozapine and clozapine N-oxide in schizophrenic patients. Prog Neuro-Psychopharmacol Biol Psychiatry. 1998;22:723–739. doi: 10.1016/s0278-5846(98)00035-9. [DOI] [PubMed] [Google Scholar]
- 12.Jann MW, Lam YW, Chang WH. Rapid formation of clozapine in guinea-pigs and man following clozapine-N-oxide administration. Arch Int Pharmacodyn Ther. 1994;328:243–250. [PubMed] [Google Scholar]
- 13.MacLaren DA, Browne RW, Shaw JK, Krishnan Radhakrishnan S, Khare P, Espana RA, Clark SD. Clozapine N-Oxide Administration Produces Behavioral Effects in Long-Evans Rats: Implications for Designing DREADD Experiments. eNeuro. 2016;3 doi: 10.1523/ENEURO.0219-16.2016. ENEURO.0219-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL. Recurrent network activity drives striatal synaptogenesis. Nature. 2012;485:646–650. doi: 10.1038/nature11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridges TM, Morrison RD, Byers FW, Luo S, Scott Daniels J. Use of a novel rapid and resource-efficient cassette dosing approach to determine the pharmacokinetics and CNS distribution of small molecule 7-transmembrane receptor allosteric modulators in rat. Pharmacol Res Perspect. 2014;2:e00077. doi: 10.1002/prp2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Chen C, Smith BJ. Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther. 2008;325:349–356. doi: 10.1124/jpet.107.130294. [DOI] [PubMed] [Google Scholar]
- 17.Xia CQ, Milton MN, Gan LS. Evaluation of drug-transporter interactions using in vitro and in vivo models. Curr Drug Metab. 2007;8:341–363. doi: 10.2174/138920007780655423. [DOI] [PubMed] [Google Scholar]
- 18.Xiao G, Black C, Hetu G, Sands E, Wang J, Caputo R, Rohde E, Gan LS. Cerebrospinal fluid can be used as a surrogate to assess brain exposures of breast cancer resistance protein and P-glycoprotein substrates. Drug Metab Dispos. 2012;40:779–787. doi: 10.1124/dmd.111.043703. [DOI] [PubMed] [Google Scholar]
- 19.Hellman K, Aadal Nielsen P, Ek F, Olsson R. An ex Vivo Model for Evaluating Blood-Brain Barrier Permeability, Efflux, and Drug Metabolism. ACS Chem Neurosci. 2016;7:668–680. doi: 10.1021/acschemneuro.6b00024. [DOI] [PubMed] [Google Scholar]
- 20.Bauer B, Hartz AM, Fricker G, Miller DS. Modulation of p-glycoprotein transport function at the blood-brain barrier. Exp Biol Med (Maywood) 2005;230:118–127. doi: 10.1177/153537020523000206. [DOI] [PubMed] [Google Scholar]
- 21.Miller DS. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol Sci. 2010;31:246–254. doi: 10.1016/j.tips.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Y, Chen X, Benet LZ. Reliability of In Vitro and In Vivo Methods for Predicting the Effect of P-Glycoprotein on the Delivery of Antidepressants to the Brain. Clin Pharmacokinet. 2016;55:143–167. doi: 10.1007/s40262-015-0310-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bickel MH. The pharmacology and biochemistry of N-oxides. Pharmacol Rev. 1969;21:325–355. [PubMed] [Google Scholar]
- 24.Lameh J, Burstein ES, Taylor E, Weiner DM, Vanover KE, Bonhaus DW. Pharmacology of N-desmethylclozapine. Pharmacol Ther. 2007;115:223–231. doi: 10.1016/j.pharmthera.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Roth BL, Driscoll J. Psychoactive Drug Screening Program. National Institute of Mental Health; Bethesda, MD: 2014. [Google Scholar]
- 26.Canton H, Verriele L, Colpaert FC. Binding of typical and atypical antipsychotics to 5-HT1C and 5-HT2 sites: clozapine potently interacts with 5-HT1C sites. Eur J Pharmacol. 1990;191:93–96. doi: 10.1016/0014-2999(90)94100-c. [DOI] [PubMed] [Google Scholar]
- 27.Seeman P, Van Tol HH. Dopamine receptor pharmacology. Trends Pharmacol Sci. 1994;15:264–270. doi: 10.1016/0165-6147(94)90323-9. [DOI] [PubMed] [Google Scholar]
- 28.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1959–1972. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Choo H, Huang XP, Yang X, Stone O, Roth BL, Jin J. The first structure-activity relationship studies for designer receptors exclusively activated by designer drugs. ACS Chem Neurosci. 2015;6:476–484. doi: 10.1021/cn500325v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raper J, Wilson M, Sanchez M, Machado CJ, Bachevalier J. Pervasive alterations of emotional and neuroendocrine responses to an acute stressor after neonatal amygdala lesions in rhesus monkeys. Psychoneuroendocrinology. 2013;38:1021–1035. doi: 10.1016/j.psyneuen.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raper J, Stephens SB, Henry A, Villarreal T, Bachevalier J, Wallen K, Sanchez MM. Neonatal amygdala lesions lead to increased activity of brain CRF systems and hypothalamic-pituitaryadrenal axis of juvenile rhesus monkeys. J Neurosci. 2014;34:11452–11460. doi: 10.1523/JNEUROSCI.0269-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalvass JC, Maurer TS, Pollack GM. Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p-glycoprotein efflux ratios. Drug Metab Dispos. 2007;35:660– 666. doi: 10.1124/dmd.106.012294. [DOI] [PubMed] [Google Scholar]