

Summary
Autophagy traditionally sustains metabolism in stressed cells via promoting intracellular catabolism and nutrient recycling. Here, we demonstrate that in response to stresses requiring increased glycolytic demand, the core autophagy machinery also facilitates glucose uptake and glycolytic flux by promoting cell surface expression of the glucose transporter Glut1/Slc2a1. During metabolic stress, LC3+ autophagic compartments bind and sequester the RabGAP protein TBC1D5 away from its inhibitory interactions with the retromer complex, thereby enabling retromer recruitment to endosome membranes and Glut1 plasma membrane translocation. In contrast, TBC1D5 inhibitory interactions with the retromer are maintained in autophagy-deficient cells, leading to Glut1 mis-sorting into endolysosomal compartments. Furthermore, TBC1D5 depletion in autophagy deficient cells rescues retromer recruitment to endosomal membranes and Glut1 surface recycling. Hence, TBC1D5 shuttling to autophagosomes during metabolic stress facilitates retromer-dependent Glut1 trafficking. Overall, our results illuminate key interconnections between the autophagy and endosomal pathways dictating Glut1 trafficking and extracellular nutrient uptake.
Keywords: autophagy, Glut1, glycolysis, retromer
ETOC
Roy et al. demonstrate that in response to increased glycolytic demand, autophagy induction coordinates glucose uptake from the extracellular milieu by promoting the retromer-dependent cell surface trafficking of the key nutrient transporter, Glut1.
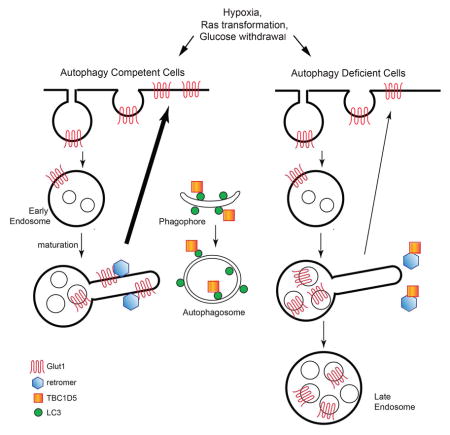
Introduction
Autophagy, a catabolic process involved in the degradation of cellular constituents and organelles, sustains core metabolic functions and nutritional requirements during starvation or stress. Autophagy also supports the anabolic demands of rapidly proliferating tumor cells; for example, in cancers driven by oncogenic Ras activation, genetic autophagy inhibition leads to multiple defects in mitochondrial metabolism, including decreased production of tricarboxylic acid (TCA) cycle intermediates, and reduced oxidative phosphorylation (Guo et al., 2011; Yang et al., 2011). In addition to supporting mitochondrial metabolism, autophagy has been demonstrated to promote glycolysis during oncogenic transformation. Genetic deletion of multiple core autophagy regulators, including ATG3, ATG5, ATG7 and FIP200 impairs oncogenic Ras-driven glycolysis in mouse fibroblasts (Lock et al., 2011; Wei and Guan, 2012). Similar results are observed in human triple negative breast cancer cells and in a transgenic model of polyoma middle T (PyMT) mammary cancer (Lock et al., 2011; Wei et al., 2011). Moreover, ATG7 knockdown in chronic myeloid leukemia cells impairs glucose uptake and glycolysis, sensitizing cells to tyrosine kinase inhibitor induced cell death (Karvela et al., 2016). Nevertheless, the precise mechanisms through which the autophagy pathway facilitates glycolysis remain unclear. Here, we uncover that the core autophagy machinery promotes glycolytic metabolism by augmenting intracellular glucose uptake into both normal and oncogenic cells.
Glucose uptake, a critical metabolic control point in glycolysis, is mediated by the Glut (SLC2) family of integral membrane transporters (Mueckler and Thorens, 2013). Glut1 (SLC2A1), one of the most well-studied members of the SLC2 family, exhibits a wide tissue distribution; cell surface expression of this transporter is increased during hypoxia and in diverse cancers exhibiting high levels of aerobic glycolysis (Warburg effect) (Carvalho et al., 2011). The kinetics of Glut1 internalization, endosomal sorting and recycling back to the plasma membrane, play principal roles in maintaining glucose homeostasis in both normal and cancerous cells (Eyster et al., 2009; Wieman et al., 2009; Wu et al., 2013). Importantly, the retromer complex, a protein assembly orchestrating the export of transmembrane proteins from early endosomes to the trans-Golgi network (TGN) or plasma membrane, is critical for Glut1 recycling (Steinberg et al., 2013). In addition to promoting Glut1 export to the cell surface, the retromer counteracts the trafficking and subsequent degradation of this nutrient transporter in the endolysosomal compartment.
Here, we demonstrate that autophagy facilitates Glut1 trafficking to the plasma membrane surface in a retromer-dependent manner and averts Glut1 sorting into late endosomes and lysosomes. Finally, we uncover that autophagy is responsible for shuttling TBC1D5, a Rab GTPase-activating protein that interacts with both the retromer and LC3 (Popovic et al., 2012; Seaman et al., 2009), away from endosomal retromer complexes and toward LC3+ compartments. Relief of TBC1D5-mediated inhibition of the retromer at the early endosome via autophagosome induction promotes Glut1 recycling and export to the cell surface. Therefore, in response to diverse metabolic and oncogenic stresses, autophagy not only drives intracellular degradation to sustain core metabolic needs, but also coordinates glucose uptake from the extracellular milieu by instructing the cell surface trafficking of a key nutrient transporter, Glut1.
Results
Autophagy Facilitates Glucose Uptake by Promoting Glut 1 Cell Surface Expression
Previous studies of HRASV12 transformed MEFs, human and mouse breast cancer cells collectively demonstrate that autophagy promotes glycolysis, which drives cancer cell proliferation and adhesion-independent transformation. Notably, these phenotypes genetically require multiple ATGs, corroborating a general role for autophagosome formation in glycolysis (Lock et al., 2011; Wei and Guan, 2012). Since the mechanisms underlying these glycolytic phenotypes were unclear, we sought to further dissect how autophagy supports glucose metabolism, initially scrutinizing the role of the core autophagy regulator ATG5 in HRASV12 transformed fibroblasts. Upon HRASV12 transformation, autophagy-competent MEFs exhibit higher levels of glucose-derived lactate compared to autophagy null counterparts (Figure S1A, left) (Lock et al., 2011). Accordingly, we first tested whether autophagy directly regulated lactate dehydrogenase A (LDHA), which mediates the conversion of pyruvate to lactate and sustains aerobic glycolysis in cancer cells. We labeled atg5+/+ and atg5−/− MEFs expressing either an empty vector (BABE) or HRASV12, with 3-13C-pyruvate and assessed 3-13C-lactate formation using NMR spectroscopy. Unlike 3-13C-lactate production from 1-13C-glucose (Figure S1A, left), no significant differences in 3-13C-lactate derived from 3-13C-pyruvate were observed among the four cell types (Figure S1A, right). Thus, LDHA function was unaffected by either oncogenic Ras or autophagy status. In parallel, we utilized radioisotopic labeling with 1-14C-glucose to assess the activity of the oxidative pentose phosphate pathway (PPP), a key side-branch of the glycolytic pathway that promotes antioxidant response and biosynthetic capacity. Similar to the increase in glycolysis, PPP activity was higher in HRasV12 atg5+/+ cells versus atg5−/− cells, evidenced by increased 14CO2 production from 1-14C-glucose (Figure S1B). We next performed NMR analysis of the PPP by labeling cells with 2-13C-glucose which yields 2-13C lactate through glycolysis or 3-13C-lactate through the oxidative PPP (Figure S1C). The ratio of 2-13C-lactate to 3-13C-lactate provides an estimate of the relative PPP flux from glucose. Although PPP activity, based on 14CO2 production, was higher in HRasV12 atg5+/+ cells, the 3-13C-lactate/2-13C-lactate ratio was equivalent between these cell lines (Figure S1C). Thus, relative PPP flux was unaffected by autophagy; rather, the increase in the PPP in autophagy competent HRasV12 MEFs was secondary to increased glycolysis. These results also motivated the hypothesis that autophagy modulates a proximal step in glycolysis upstream of the PPP bifurcation, such as glucose uptake. In support, HRasV12 atg5+/+ cells exhibited increased glucose uptake, based on uptake of 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-D-glucose (2-NBDG), compared to atg5−/− cells (Figure S1D).
To extend these findings beyond oncogenic Ras transformation, we examined non-transformed cells during hypoxia, a metabolic stress that upregulates both glycolysis and autophagy. Although autophagy status did not affect glucose-derived lactate production during normoxia, autophagy deficient MEFs had significantly reduced lactate production upon exposure to hypoxia compared to autophagy competent cells (Figures 1A–B). During hypoxia, 2-NBDG uptake was also severely compromised in both atg5−/− and atg7−/− MEFs compared to controls, further supporting that autophagy is required for glucose uptake in non-transformed cells subject to this metabolic stress (Figures 1C–D). Enhanced degradation of p62/SQSTM1 confirmed autophagy induction during hypoxia in control MEFs (Figure 1E). The transcription factor HIF1α controls the metabolic switch from oxidative phosphorylation to glycolysis (Pasteur effect) in response to hypoxia (Seagroves et al., 2001). However, reduced glycolytic flux in autophagy deficient cells was not due to impaired induction of HIF1α or key HIF1α glycolytic targets, including hexokinase II (HKII), LDHA, and Glut1 (SLC2A1) (Figures 1E–F). In contrast, immunofluorescence for endogenous Glut1, a key hypoxia-induced glucose transporter, demonstrated that hypoxic autophagy competent cells exhibited high levels of Glut1 surface expression at the plasma membrane, whereas in autophagy-deficient cells, Glut1 was mostly intracellular (Figure 1G). To rigorously quantify Glut1 surface expression, we ectopically expressed Glut1 containing a Flag epitope tag in the first exofacial loop (Wieman et al., 2009). Autophagy ablation significantly compromised hypoxia-induced Flag-Glut1 surface expression (Figures 1H–I). Similar to normal cells exposed to hypoxia, Glut1 is crucial for glucose uptake and the Warburg effect in Ras-transformed cells (Yun et al., 2009). Ras-transformed autophagy deficient and competent cells exhibited comparable steady state protein levels of Glut1, HIF1α and other glycolytic targets, similar to our results with hypoxia (Figure 1SE); nevertheless, HRasV12 atg5+/+ cells exhibited higher Glut1 surface expression compared to HRasV12 atg5−/− cells (Figures S1F–G). Consistent with this inherent defect in cell surface expression of Glut1 in Ras-transformed autophagy deficient cells, the stable overexpression of green fluorescent protein-tagged Glut1 (GFP-Glut1) in HRasV12-transformed atg5−/− MEFs was not sufficient to rescue its growth or oncogenic transformation potential (Figure S1H).
Figure 1. Autophagy facilitates glucose uptake and lactate production in hypoxic cells by promoting cell surface expression of Glut1.

(A and B) MEFs of indicated genotypes were cultured in normoxia (N, 21% O2) or exposed to hypoxia (H, 1% O2) in 1-13C-glucose containing media for 48h; extracellular 3-13C-lactate levels measured by NMR spectroscopy (mean ± SEM, n=3 independent experiments). (C and D) MEFs exposed to H for 24h were labeled with 2-NBDG (100 μM) for 3h to measure glucose uptake (mean ± SEM, n=3). (E) Cells exposed to H for the indicated times were lysed and immunoblotted for indicated proteins. (F) Relative mRNA expression of indicated transcripts in atg5 +/+ and atg5−/− MEFs following exposure to H for 18h (mean ± SEM, n=3). (G) Indicated cell types were exposed to H for 24h and immunostained for Glut1 protein. Scale bars, 25μm. (H and I) Cells transiently expressing Flag-Glut1 were exposed to H for 24h; cell surface expression of Flag-Glut1 was FACS quantified (mean ± SEM, n=3). (J) 2-NBDG uptake in wild-type MEFs exposed to H in the presence of WZB117 (10μM) for 24h (mean ± SEM, n=4). P-values calculated using ANOVA followed by Tukey’s HSD test. ***P<0.001, **P<0.01, *P<0.05; ns, not significant. Related to Figure S1 and S2.
We next asked whether other autophagy inducers were broadly able to promote the plasma membrane expression of Glut1. Treatment of cells with the mTORC1 inhibitor Rapamycin (Rap) caused robust autophagy induction, as evidenced by p62 degradation (Figure S2A), but no changes in Glut1 protein levels (Figure S2B). Nevertheless, in contrast to hypoxia or HRasV12 transformation, there was no increase in glucose uptake (Figure S2C) and no evidence of increased Glut1 surface expression (Figure S2D) upon Rap treatment in both atg5+/+ and atg5−/− cells. Thus, despite autophagy induction, rapamycin failed to mediate any change in Glut1 trafficking and glucose uptake. These results suggest that the autophagy-dependent increase in Glut1 plasma membrane expression is specifically observed in response to metabolic stresses requiring increased glucose demand, such as during hypoxia.
To further establish the functional importance of Glut1 during metabolic stress in autophagy competent cells, MEFs were treated with the Glut1 pharmacological inhibitor, WZB117, and 2NBDG uptake was evaluated. The glucose uptake potential of autophagy competent cells was significantly compromised with WZB117 (Figures 1J, S2E–F), underscoring the role of Glut1 in promoting autophagy-dependent glucose uptake during metabolic stress. Overall, these results support that autophagy facilitates glycolysis during both hypoxia and oncogenic Ras transformation via the control of Glut1 surface expression and glucose uptake.
Autophagy Promotes Glut1 Recycling to the Plasma Membrane
Glut1 undergoes continuous endocytosis and recycling to membrane surface. To elucidate how autophagy facilitates the trafficking of Glut1 to the plasma membrane, we performed pulse chase assays to directly measure the internalization or recycling of Flag-Glut1 protein in live cells. We observed similar kinetics of Flag-Glut1 internalization between autophagy competent and deficient cells (Figures 2A–B). By contrast, the recycling rate of internalized Flag-Glut1 back to the plasma membrane surface was profoundly impeded in both atg5−/− and atg7−/− MEFs relative to controls (Figure 2C–D). To assess whether defective Flag-Glut1 recycling represented a more general deficit in the early endocytic pathway, we incubated cells with fluorescently labeled transferrin (Tfn), which is taken up by receptor-mediated endocytosis, sorted to early endosomes, and recycled back to the plasma membrane. In contrast to Flag-Glut1 recycling and consistent with previous results (Murrow et al., 2015), transferrin (Tfn) recycling was identical in atg5−/− and atg5+/+ cells, indicating early endocytic function was intact (Figure 2E).
Figure 2. Autophagy promotes Glut1 recycling to the plasma membrane.

(A and B) Indicated cell types transiently expressing Flag-Glut1 incubated with α-Flag, washed and cultured at 37°C for indicated times prior to staining with Alexa 488 secondary antibody to determine the fraction of surface label remaining by flow cytometry (mean ± SEM, n=3 independent experiments). (C and D) Cells transiently expressing Flag-Glut1 incubated with α-Flag at 37°C in full media, washed and chased in glucose-free media for indicated times, followed by staining with Alexa 488 secondary antibody to determine the kinetics of surface Flag-Glut1 re-emergence by flow cytometry (mean ± SEM, n=3). (E) Cells stained with Alexa Fluor 488-conjugated transferrin (Tfn) at 37°C in serum-free media, and chased in full media (mean ± SEM, n=3). P-values calculated using unpaired t-test. *P<0.05 (F) Confocal live-cell images of atg5+/+ and atg5−/− MEFs stably expressing GFP-Glut1 following 2h of glucose starvation for the indicated times. Insets: Magnification of the boxed regions. In top insets, arrows delineate GFP-Glut1 containing vesicular structures migrating towards the plasma membrane in atg5+/+ MEFs. Scale bar, 10μm. Related to Movie S1 and S2.
To further scrutinize the Glut1 trafficking defect in autophagy deficient cells, we performed live-cell imaging of MEFs stably expressing GFP-Glut1 and monitored the dynamics of GFP-Glut1 trafficking in response to acute glucose starvation, which potently stimulates Glut1 plasma membrane translocation (von der Crone et al., 2000). Upon glucose withdrawal, GFP-Glut1 exhibited a striking tubulovesicular pattern in autophagy competent cells (Figure 2F, Movie S1), which resembled tubulovesicular endocytic carriers previously described in receptor recycling to the plasma membrane (Grant and Donaldson, 2009). We also observed GFP-Glut1 migration from these tubulovesicular structures toward the membrane surface in autophagy competent cells (Figure 2F, insets, Movie S1). In contrast, such tubulovesicular structures were not readily observable in autophagy-deficient cells; instead, GFP-Glut1 was predominantly found in a perinuclear punctate pattern. During glucose starvation, these puncta failed to migrate towards the plasma membrane over progressive time points (Figure 2F, insets, Movie S2). Overall, these studies reveal that genetic autophagy ablation impaired recycling of Glut1 to the plasma membrane surface, resulting in aberrant subcellular localization of Glut1 away from a tubulovesicular compartment. Importantly, these results broached that autophagy does not solely function as an intracellular salvage pathway during metabolic stress; it also exerts upstream control over glucose uptake from the extracellular milieu by facilitating the cell surface trafficking of Glut1.
Autophagy Deficiency Results in Glut1 Mis-sorting into Endolysosomal Compartments
Because autophagy deficiency resulted in prominent vesicular trafficking aberrations preventing Glut1 recycling back to the cell surface, we next dissected how genetic autophagy ablation impacted the intracellular localization of Glut1. After reaching early endosomes, endocytosed transmembrane proteins are either sorted for degradation in lysosomes or recycled to the plasma membrane or the TGN (Bonifacino and Traub, 2003). The prominent vesicular pattern we observed with both endogenous Glut1 immunofluorescence and GFP-Glut1 imaging (Figures 1G and 2F) suggested that Glut1 was mis-sorted into the late endocytic pathway in autophagy deficient cells. Indeed, in atg5−/− cells, we observed significantly increased co-localization of endogenous Glut1 with LAMP1 vesicles in both control and hypoxic conditions, indicating that Glut1 was primarily located in late endosomes or lysosomes (Figures 3A–B). In contrast, wild-type controls showed prominent cell surface Glut1 staining in response to hypoxia (Figures 1G and 3A).
Figure 3. Autophagy deficiency causes Glut1 mis-sorting to late endolysosomes.

(A) atg5+/+ and atg5−/− MEFs exposed to hypoxia (H, 1% O2) for 24h and immunostained for Glut1 and LAMP1. (B) Pearson’s correlation coefficient for Glut1-LAMP1 colocalization (mean ±SEM, n=30 cells from 3 independent experiments). (C) Cells stably expressing GFP-Glut1 exposed to H for 24h or subject to acute glucose starvation (-glc) for 18h and probed for GFP and LAMP1. (D) Pearson’s correlation coefficient for GFP-LAMP1 colocalization (mean ±SEM, n=45 cells from 3 independent experiments). Cont, Control (full media, 20% O2); H, hypoxia (1% O2), -glc, glucose deficient media. P values calculated using ANOVA followed by Tukey’s HSD test. ***P<0.001, **P<0.01, *P<0.05; ns, not significant. Scale bars 10μm. Related to Figure S3.
To extend these results, we analyzed cells stably expressing GFP-Glut1 following hypoxia and acute glucose deprivation; Glut1 exhibited a tubulovesicular and cell surface expression pattern in autophagy competent cells, whereas in autophagy deficient cells, a significant fraction of intracellular GFP-Glut1 was found in the LAMP1 positive compartment (Figure 3C–D). Moreover, in response to both stresses, the GFP-Glut1 fraction in the late endolysosomal pool was significantly reduced in atg5+/+ cells, while in atg5−/− cells, Glut1 persisted in this LAMP1+ late endolysosomal compartment (Figure 3D). Furthermore, despite genetic ablation of autophagy, the degradation kinetics of endogenous surface Glut1 was increased in atg5−/− MEFs compared to atg5+/+ cells (Figure S3A). Previous studies demonstrate that the C-terminal PDZ binding motif in Glut1 plays an essential role in Glut1 recycling; deletion of this motif results in the profound mis-sorting of Glut1 into the late endocytic pathway (Wieman et al., 2009). We corroborated this finding in autophagy-competent fibroblasts; deletion of these last four amino acids in Glut1 (GFP-Glut1Δ4) caused it to be exclusively localized in LAMP1 positive structures with no evidence of surface expression in response to both metabolic stresses (Figures S3B–C), a pattern phenocopying the punctate distribution of wild-type GFP-Glut1 in autophagy null cells. Therefore, during hypoxia and glucose withdrawal, the autophagy pathway is required for Glut1 recycling back to the cell surface and averting its degradation by the endolysosomal pathway.
Glut1 Recycling in Autophagy Competent cells is Retromer-Dependent
The retromer complex serves as a major sorting station that recycles cargo to the TGN or directly to the plasma membrane. The core vacuolar protein sorting trimer (Vps26/29/35) of the retromer complex mediates cargo binding and selectivity (Klinger et al., 2015). Recent studies demonstrate that the retromer assembly mediates Glut1 cell surface recycling. This pathway proceeds via direct retromer-dependent trafficking of cargo from tubulovesicular endosomes to the plasma membrane, bypassing the more classical route involving the TGN (Klinger et al., 2015). Thus, we interrogated whether the differential Glut1 trafficking in autophagy competent versus deficient cells arose from any specific differences in retromer function. First, we confirmed that Glut1 did not traffic to the TGN via the classical retromer pathway, evidenced by the lack of colocalization of GFP-Glut1 with TGN46 in autophagy competent cells (Figure S4A). Furthermore, autophagy status did not impact the steady state protein expression of Vps35, Vps26, or Vps29 (Figure 4A) or their mutual interaction (Figure S4B), indicating that the assembly of the core retromer complex was intact in autophagy-deficient cells. However, transient depletion of Vps35 in atg5+/+ MEFs (Figure 4B) led to the disappearance of the tubulovesicular distribution of GFP-Glut1, with a concomitant increase in its localization in LAMP1+ compartments (Figures 4C–D). These results demonstrated that Glut1 was mis-sorted into an endolysosomal compartment when retromer function was impaired in autophagy competent cells, a defect resembling that observed in autophagy deficient cells. In functional assays, Vps35 knockdown significantly compromised Flag-Glut1 recycling to the plasma membrane surface in autophagy competent cells, but minimally impacted recycling in autophagy deficient cells (Figures 4E–F).
Figure 4. Autophagy mediated Glut1 trafficking is retromer-dependent.

(A) atg5+/+ and atg5−/− MEFs stably expressing GFP-Glut1 were exposed to hypoxia (H) for 24h or glucose starved (-glc) for 18h and immunoblotted for Vps35, Vps26 and Vps29. (B–D) atg5+/+ MEFs expressing GFP-Glut1 were transfected with non-targeting control (CTL) or Vps35 siRNAs; 24h following transfection, cells were exposed to hypoxia (H) for 24h or glucose starved (-glc) for 18h. (B) Cells were lysed and immunoblotted with indicated antibodies. (C) MEFs immunostained for GFP and LAMP1. (D) Pearson’s correlation coefficient for GFP-LAMP1 colocalization. P values calculated using ANOVA followed by Tukey’s HSD test (n=27 for CTL siRNA cells and n=45 for Vps35 siRNA cells imaged over 3 independent experiments). (E and F) Cells were first transfected with siRNAs against non-targeting control (CTL) or Vps35 and subsequently with Flag-Glut1 24h later. Kinetics of Flag-Glut1 recycling over indicated times measured by flow cytometry (mean ± SEM, n=3). (G) Cells were glucose starved for 18h and immunostained for Vps35. (H) The number of Vps35 puncta per cell were quantified (n=60 cells per condition from 3 independent experiments). (I) Cells were exposed to hypoxia for 48h and immunostained for MCT1 and LAMP1. Scale bars 25μM. P values calculated using ANOVA followed by Tukey’s HSD test. ***P<0.001, *P<0.05. Related to Figure S4.
The recruitment of retromer subunits to the endosomal membranes is critical for its function and can be visualized via immunofluorescence as punctate structures containing Vps35 and Vps26; in previous studies, the quantification of such puncta has been utilized as a measure of retromer activity (Seaman et al., 2009). Remarkably, upon glucose starvation, there was a significant increase in the number of Vps35 and Vps26 puncta in autophagy competent cells; in contrast, no significant increase in these puncta was observed in autophagy-deficient cells starved of glucose (Figure 4G–H & S4C). These results provide important evidence that endosomal recruitment of the retromer assembly is indeed downstream of autophagy.
Finally, we postulated that other plasma membrane transporters that undergo retromer-dependent recycling would be impacted by autophagy status. We focused on MCT1 (monocarboxylate transporter 1), a lactate transporter, which exhibits increased surface localization during hypoxia, and has been identified through surface proteomic studies as a plasma membrane transporter dependent on the retromer complex for endosomal recycling and surface abundance (Steinberg et al., 2013). Congruent with previous studies, we observed increased levels of endogenous MCT1 at the plasma membrane of wild-type cells subjected to hypoxia. Strikingly, the majority of MCT1 within hypoxic autophagy deficient cells was sequestered in endolysosomes, similar to trafficking defects observed for Glut1 in the absence of a functional autophagy pathway (Figure 4I).
Overall, these results establish that the autophagy and retromer pathways are both required for Glut1 recycling to the plasma membrane. They also demonstrate that an intact autophagy pathway is required for the membrane recruitment of retromer components, which enables the efficient translocation of Glut1 and other membrane transporters to the plasma membrane.
TBC1D5 Interactions with Vps35 and LC3 Regulate Glut1 Recycling
TBC1D5 is a RabGAP family protein that functions as a negative regulator of retromer complex (Seaman et al., 2009) and was recently reported to relocate from the retromer assembly to LC3+ autophagosomes during nutrient starvation (Popovic et al., 2012) Therefore, we postulated that upon autophagy induction in response to metabolic stress, TBC1D5 would similarly shuttle away from its inhibitory interaction with the retromer complex and toward autophagosomes. First, we confirmed equivalent levels of TBC1D5 protein expression between atg5+/+ and atg5−/− MEFs, which did not change upon hypoxia or glucose deprivation (Figure S4D). TBC1D5 knockdown significantly augmented Flag-Glut1 recycling in atg5−/− cells (Figure 5A–B), supporting that TBC1D5 impairs Glut1 recycling in the context of autophagy deficiency. To scrutinize the molecular interactions between Vps35, TBC1D5 and LC3, we performed co-immunoprecipitation assays in HEK293T cells. First, we confirmed that TBC1D5 strongly interacts with LC3A and to a lesser extent with LC3C. (Figure S4E). Based on these results, we chose LC3A for further co-immunoprecipitation analysis in response to glucose withdrawal. Glucose starvation diminished the interaction between TBC1D5 and Vps35, while concomitantly increasing TBC1D5 association with LC3A (Figure 5C). By immunofluorescence, we observed increased co-localization of TBC1D5 with LC3 puncta following glucose starvation (Figure 5D). By contrast, the interactions between TBC1D5 and Vps35 remained intact in glucose starved ATG7 or ATG12 deleted cells (Figure 5E & S4F). Finally, to assess the effects of TBC1D5 on retromer function in autophagy deficient cells, we quantified the effects of TBC1D5 depletion on Vps35 membrane recruitment. Compared to controls, TBC1D5 depletion significantly increased the number of Vps35 puncta per cell in atg5−/− cells subject to glucose starvation (Figure 5F–G). Overall, these results support that autophagy is important for titrating TBC1D5 away from the retromer, resulting in increased retromer functional activity and efficient Glut1 surface trafficking.
Figure 5. Autophagy-dependent shuttling of TBC1D5 from the retromer to LC3+ compartments controls Glut1 trafficking.

(A) atg5−/− MEFs were transfected with siRNAs against non-targeting control (CTL) and TBC1D5, glucose starved for 18h and lysates were immunoblotted for TBC1D5 and retromer subunits. (B) atg5−/− MEFs were first transfected with CTL or TBC1D5 siRNAs, and subsequently transfected with Flag-Glut1 24h later. Kinetics of Flag-Glut1 recycling over indicated times measured by flow cytometry (mean ± SEM, n=3). P values calculated using unpaired t-test. (C) HEK293T cells were transiently transfected with Flag-TBC1D5, V5-Vps35 and Myc-LC3A. Cells were glucose starved for 8h. Lysates were immunoprecipitated with α-Flag (left) or α-Myc (right), resolved on SDS-PAGE and immunoblotted as indicated. (D) HEK293T cells transiently overexpressing Myc-LC3A and Flag-TBC1D5 were glucose starved for 6h and immunostained with α-Flag and α-Myc. Scale bar, 10μm. (E) HEK293T cells with or without ATG7 deletion were transiently transfected with Flag-TBC1D5 and V5-Vps35; cells were glucose starved and lysates were immunoprecipitated with α-Flag and immunoblotted as indicated. Right: Quantification of co-immunoprecipitated V5-Vps35 (mean±SEM, n=3 independent experiments). P values calculated using ANOVA followed by Tukey’s HSD test. (F) atg5−/− MEFs were transfected with CTL or TBC1D5 siRNAs, glucose starved for 18h and immunostained for Vps35. Scale bar, 25 μm. (G) Quantification of the number of Vps35 puncta per cell (n=60 cells from 3 independent experiments). P values calculated using unpaired t-test. ***P<0.001, *P<0.05; ns, not significant. Related to Figure S4.
Discussion
Macroautophagy is traditionally viewed as a lysosomal degradation pathway in which autophagy-derived catabolic intermediates provide substrates for biosynthesis and energy production to promote cell survival. Here, we identify a role for autophagy in promoting glucose uptake from the extracellular environment via retromer-dependent cell surface trafficking of Glut1. In contrast, autophagy does not impact downstream glycolytic events or the relative flux of carbon into the oxidative PPP, an early proximal side-branch of glycolytic metabolism. Our results also substantiate a general role for the core ATG machinery and for autophagosome formation in facilitating glucose metabolism, rather than a specialized function mediated by individual autophagy regulators. Recently, the autophagy regulator ULK1/2 was reported to directly control the activities of several glycolytic enzymes independently of the core autophagic machinery during amino acid starvation (Li et al., 2016). However, these studies were predominantly conducted using saline-induced amino acid starvation, which causes a severe reduction in glycolytic activity; in this context, ULK loss-of-function led to a further drop in glycolysis. In contrast, we now corroborate previous work demonstrating that in situations of high glycolytic demand, such as oncogenic Ras transformation and hypoxia, multiple ATGs are genetically required for increased glycolytic flux by promoting Glut1 surface expression, thereby augmenting glucose uptake. Remarkably, autophagy status does not significantly impact glycolysis in non-transformed cells grown in nutrient replete or normoxic conditions nor in response to rapamycin-mediated mTORC1 inhibition, thus illuminating a distinct requirement for autophagy in augmenting glycolytic phenotypes during specific metabolic and oncogenic contexts.
Glut1 internalization proceeds through both clathrin-dependent (CDE) and independent (CIE) endocytosis pathways (Eyster et al., 2009; Wu et al., 2013). Thereafter, Glut1-containing vesicles rapidly fuse with EEA1-positive vesicles, which are either delivered to the lysosome for degradation or recycled back to the membrane surface via tubular endosomes through the actions of multiple GTPases including Arf6 (Radhakrishna and Donaldson, 1997) and Rab22 (Weigert et al., 2004). We demonstrate here that genetic autophagy inhibition does not significantly impact Glut1 internalization; instead, it distinctly compromises Glut1 recycling back to the plasma membrane surface. Moreover, using live-cell imaging, we observe Glut1 in tubulovesicular structures that traffic to the plasma membrane in autophagy-competent cells; in contrast, Glut1 is trapped in the late endolysosomal compartment of autophagy deficient cells. The early endocytic compartment, composed of thin tubular extensions and large vacuolar regions, serve as a major sorting hub for membrane proteins. However, transferrin receptor (TnFR) recycling via the early endocytic pathway is unaffected by genetic autophagy ablation, indicating that autophagy does not broadly impact early endosomal recycling pathways (Murrow et al., 2015).
Instead, our results reveal that autophagy controls the membrane association of the cargo selective SNX-BAR retromer assembly (comprised of the Vps35/29/26 trimer in association with SNX1/2 and SNX5/6), which has been demonstrated to promote Glut1 surface recycling. Accordingly, Glut1 localization in tubulovesicular intermediates requires both core ATGs and components of the retromer complex. Although best characterized for cargo retrieval from endosome to TGN, recent work unveils a direct role of the retromer in endosome-to-plasma membrane recycling through its association with SNX27. In fact, a recent proteomic analysis showed that over 100 cell surface proteins, including Glut1, interact with SNX27 through the PDZ domain; in conjunction with the WASH complex, they are recycled to the membrane surface in a retromer-dependent manner (Steinberg et al., 2013). In agreement with the important role of PDZ in retromer mediated Glut1 recycling, Glut1 mutants lacking the C-terminal PDZ motif are mis-sorted into LAMP1 positive vesicles, a phenotype similar to both autophagy and Vps35 deficient cells.
Finally, we demonstrate that autophagy-dependent shuttling of TBC1D5 serves as a switch directing retromer association with endosomal membranes and retromer-dependent Glut1 trafficking to the plasma membrane. TBC1D5 functions as a RabGAP for Rab7 (Seaman et al., 2009). Since Rab7 is critical for the recruitment of the cargo-selective Vps35/29/26 complex to the endosomal membrane (Rojas et al., 2008), TBC1D5 thus serves as an important restraint on retromer function. TBC1D5 also has multiple interactions with the autophagy pathway including the ability to physically interact with LC3 as well as to regulate ATG9 trafficking (Popovic and Dikic, 2014). Recent work demonstrates that TBC1D5 can shuttle between the retromer complex and LC3+ autophagosomes during starvation (Popovic et al., 2012), but the physiological functions for this Rab-GAP in endosomal-autophagy crosstalk have not been fully delineated. Here, we find that TBC1D5 strongly interacts with the retromer complex in autophagy-competent cells cultured in full nutrient conditions, but dissociates upon glucose starvation, coincident with the increased interaction of TBC1D5 with LC3+ autophagosomal membranes. This interaction between Vps35 and TBC1D5 remains intact upon glucose starvation in autophagy-deficient cells, effectively inhibiting the retromer complex. Though TBC1D5 has primarily been demonstrated to inhibit retromer-mediated endosome-to-TGN transport, our studies implicate TBC1D5 in plasma membrane recycling functions of the retromer, because depletion of TBC1D5 significantly rescues Glut1 recycling to the plasma membrane in autophagy-null cells. Notably, in addition to Glut1, we demonstrate that hypoxic autophagy competent cells exhibited increased MCT1 expression at the plasma membrane in comparison to autophagy-deficient cells, where this lactate transporter and retromer target is mis-sorted to late endolysosomes. Further analysis of how autophagy impacts the plasma membrane trafficking of these and other retromer-dependent cargos remains an important area for future study.
Overall, our studies reveal that the autophagy pathway functions in extracellular nutrient uptake during metabolic stress; specifically, it augments glucose consumption via enabling retromer-dependent cell surface trafficking of Glut1. They also illustrate the exquisite crosstalk between the autophagic machinery and endosomal trafficking pathways in the control of glucose metabolism, which has profound implications in disease states, such as cancer, where one or both of these machineries are perturbed (Goldenring, 2013).
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Jayanta Debnath (jayanta.debnath@ucsf.edu)
Experimental Model and Subject Detail
The sources of all cell lines utilized in this study are reported in the reagent and resource table.
Method Details
Cell culture and treatments
Dr. Noboru Mizushima (University of Tokyo, Japan) provided atg5+/+ and atg5−/− MEFs and Dr. Masaaki Komatsu (Nigata University, Japan) provided atg7+/+ and atg7−/− MEFs. MEFs were maintained in DMEM high glucose media supplemented with 10% FBS and Penicillin-streptomycin. HEK293T cells were obtained from ATCC and maintained in DMEM supplemented with 10% FBS, penicillin-streptomycin, sodium pyruvate and HEPES. Cells were cultured for less than 3 months following resuscitation, tested for Mycoplasma contamination using the Mycoalert detection kit (Lonza LT07) and authenticated using STR profiling (Almeida et al., 2016); MEFs were also authenticated via immunoblotting for ATG protein levels. For analysis of cells undergoing hypoxia (H), cells were incubated in 1% O2 and 5% CO2 at 37°C in a Biospherix hypoxia chamber for 24h or 48h. Normoxic controls (N) were cultured at 21% O2 and 5% CO2 at 37°C in an Autoflow NU-8500 tissue culture incubator (NuAire). Glucose starvation (-glc) was achieved by incubating cells in glucose free DMEM, supplemented with 2% FBS for 18h.
RNA interference
For siRNA-mediated knockdown, cells were electroporated using an Amaxa Nucleofector 2b device (Program U-020) and Nucleofector kit V according to the manufacturer’s instructions.
CRISPR/Cas9 deletion of ATGs
Human ATG7 guide sequence (CTTGAAAGACTCGAGTGTGT), ATG12 guide sequence (GAAACTGCAGCGGAAGACGG) or scramble sequence (GCACTACCAGAGCTAACTCA) were ligated into pSpCas9(BB)-2A-Puro (PX459) plasmid using the BbsI site. HEK293T cells were transfected with plasmid DNA using Lipofectamine 3000. Cells were selected using 1μg/ml puromycin and seeded in 96 well plate. Monoclonal wells were identified, expanded, and analyzed.
cDNA constructs and generation of stable lines
To clone 3xFLAG-TBC1D5 (NM_001134381), myc-LC3A (NM_032514), Myc-LC3B (NM_022818), Myc-LC3C (NM_001004343), Myc-Vps26a (NM_004896) and Myc-Vps29 (NM_016226), mRNA isolated from MCF10A cells was reverse transcribed using AccuScript High Fidelity Reverse Transcriptase (Agilent) and cDNA amplified using PfuUltra II Hot Start DNA polymerase and gene specific primers (TBC1D5 Fwd: agtcggatccatgtatcattccttatctgaaact, Rev: agtcctcgagtcagatgtccaggggactca; LC3A Fwd: agtcggatccatgccctcagaccggcct, Rev: gactctcgagtcagaagccgaaggtttcct; LC3B Fwd: agtcggatccatgccgtcggagaagacct, Rev: gactctcgagttacactgacaatttcatcccg; LC3C Fwd: agtcggatccatgccgcctccacagaaaat, Rev: gactctcgagctagagaggattgcagggtc; Vps26a Fwd: agctggatccatgagttttcttggaggctttt, Rev: agct ctcgagtcacatttcaggctgttcgg; Vps29 Fwd: agctggatccatgttggtgttggtattaggat, Rev: agcttcgagttaaggttttttgtattcgattcg). Subsequently, the cDNAs were cloned into pcDNA3 between the BamHI and XhoI restriction sites downstream of an N-terminal 3xFlag-tag and Myc-tag, respectively, and the vectors verified by sequencing. pLenti6/V5/DEST-Vps35 was purchased from Addgene. An exofacial-tagged rGlut1, constructed by inserting a tandem repeat of a Flag epitope tag (5′-GACTACAAAGACGATGACGACAAG-3′) into the first extracellular loop (between 55 and 56 amino acids) of rat Glut1 by pEF6/V5-His TOPO cloning, was generously provided by J. Rathmell (Vanderbilt University). An N-terminal green fluorescent protein (GFP)-tagged rGlut1 construct was a gift from J. Rathmell (Vanderbilt University) and was subcloned from pcDNA3.1 GFP-Glut1 into 5′ BsaI and 3′ EcoRI sites of the retroviral vector pBabepuro. The generation of pBABEpuro-H-RasV12 has been described previously (Debnath et al., 2002). For retroviral transduction, VSV-G–pseudotyped retroviruses were generated, and MEFs were infected and selected as previously described (Debnath et al., 2003).
Quantitative RT-PCR
Following 24h exposure to hypoxia (1% O2), cells were harvested and total RNA was extracted, reverse-transcribed and PCR amplified (Step One Plus System, Applied Biosystems) using the following TaqMan gene expression probes (Glut1: Mm00441480_m1; HKII: Mm00443385_m1; LDHA: Mm01612132_g1). ACTINB (β-actin: Mm00607939_s1) was used to normalize expression.
Nuclear Magnetic Resonance spectroscopy
To label with 1-13C-glucose, MEFs were maintained in DMEM containing equal concentrations (12.5 mmol/L) of 1-13C-glucose and unlabeled glucose for 24- 48h as indicated, after which medium was collected for NMR analysis and cells were harvested for protein quantification. To label with 3-13C-pyruvate, cells were maintained in DMEM containing 1mmol/L of 3-13C pyruvate for 24h. To acquire 13C spectra, the media samples were run on a 600-MHz INOVA spectrometer (Varian; Palo Alto, CA) using a 30° flip angle, 3.5-s repetition time, and broadband proton decoupling. Analysis of the NMR spectra was performed using Bruker Topspin3 software. The concentrations of metabolites were determined with respect to an external reference, 2,2,3,3-tetradeutero-3-(trimethylsilyl)propionic acid (TSP), and normalized to protein content.
Analysis of oxidative pentose phosphate pathway
To measure oxidative PPP, the indicated cell types were plated onto 6 cm dishes, grown for 2d, washed twice with PBS, and the tissue culture media was incubated with media containing 0.8μCi of 1-14C-glucose (Perkin Elmer) for 5h. To capture gaseous 14CO2, Whatman filter paper was taped to the inside of culture dish lid and the plate was then sealed with Parafilm; the experiment was terminated by adding 500μL of perchloric acid to the cells; the plate was resealed and kept at room temperature overnight. Filter papers were then carefully removed and placed into vials containing Scintisafe Plus 50% cocktail scintillation fluid (Fisher Scientific) and 14CO2 activity was measured on a Beckman LS6000 Liquid Scintillation Analyzer. Notably, the incubation of cells with 6-14C-glucose, used to correct for 14CO2 production from the citric acid cycle, yielded minimal 14CO2 activity. To measure relative PPP via NMR, cells were maintained in DMEM containing 2-13C glucose for 18h. The next day, media was collected and 13C spectra acquired on above-mentioned 600 Mz INOVA spectrometer.
Immunoblotting
Cells were lysed in RIPA buffer supplemented with protease inhibitor cocktail, 1mM Na3VO4, 10 mM NaF, 0.5 mM PMSF, 10mM β-glycerophosphate, 10 nM calyculin, and 10 μg/ml pepstatin A. Lysates were boiled in 6X sample buffer, resolved on SDS-PAGE and transferred to PVDF membranes. Membranes were blocked in 5% non-fat milk, incubated overnight in primary antibodies, washed in PBST, incubated with HRP-conjugated secondary antibodies and developed via enhanced chemiluminescence (Thermo Scientific).
Immunoprecipitation
Cells were lysed in non-denaturing lysis buffer (25 mM Tris HCl pH 8.0, 150 mM NaCl, 1% NP-40, 5% glycerol, 2mM EGTA pH 8.0, 2mM EDTA) plus protease inhibitor cocktail (Sigma-Aldrich), 10 mM NaF, 10 mM β-glycerophosphate, 1 mM Na3VO4, 0.5 mM PMSF, and 10 mM sodium pyrophosphate. Lysates were precleared with protein A/G beads (Santa Cruz Biotechnology, Inc.) at 4°C and incubated overnight with primary antibody at 4°C. Immune complexes were captured by incubation with protein A/G beads for 4h at 4°C, washed four times with lysis buffer, eluted in 2X sample buffer, and analyzed by immunoblotting.
Immunofluorescence and live-cell imaging
Unless otherwise indicated, cells were fixed in 4% paraformaldehyde at room temperature for 15 min and permeabilized with 0.5% TritonX-100 for 10 min. Subsequently, cells were rinsed with PBS-glycine, blocked (10% goat serum and 0.2% Triton X-100 in PBS), and incubated overnight with primary antibodies at 4°C in blocking buffer. Then, cells were washed, incubated with Alexa Fluor-488 or -568 secondary antibodies (Life Technologies, 1:300) for 60 min at room temperature, washed, and mounted using Prolong Gold Anti-Fade mounting medium (Life Technologies). For colocation analysis of HEK293T cells expressing Myc-LC3A and Flag-TBC1D5, cells were glucose starved for 6h; Bafilomycin A (20nM) added for the last 3h of starvation; cells were fixed and permeabilized as above, and immunostained with anti-Flag and anti-Myc.
Confocal microscopy was performed at ambient temperature using 60X or 100X (NA, 1.49; oil) objective (CFI Apochromat TIRF; Nikon) on a Nikon inverted microscope (TE-2000 PFS) equipped with Borealis-modified Yokagawa CSU10 spinning disc confocal unit (Spectral Applied Research), solid state 488 (for Alexa Fluor 488), and 561 lasers (for Alexa Fluor 568) and a CoolSNAP MYO cooled scientific-grade CCD camera (Photometrics). For live cell imaging, cells were grown on glass bottom dishes and incubated in glucose free media for 2h. Intracellular GFP-tagged Glut1 dynamics in live cells were imaged at 37°C in an environmentally controlled chamber at 37°C for the indicated times using the 60X objective (NA: 1.49, oil) of the Nikon inverted spinning disc confocal microscope described above. All microscope hardware and image acquisition were controlled by NIS-Elements software (Nikon) and images were analyzed using Fiji image processing software (v2). For analysis of all microscopy images, raw image data were used.
Glucose uptake
Cells were incubated with 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-D-glucose (2NBDG, Invitrogen) for 3h in low glucose DMEM. The cells were then harvested, washed with PBS three times and FACS analyzed using FACS caliber (BD Biosciences). For hypoxia exposure, all buffers were pre-equilibrated overnight in the hypoxia chamber before use.
Surface Glut1 measurement
Cells transiently overexpressing Flag-Glut1 were exposed to hypoxia for 24h and then harvested using accutase (Gibco), blocked with 5% serum containing PBS for 30 min, incubated with anti-Flag M2 antibody at 4°C, washed with assay buffer (PBS, 5% FBS), incubated with Alexa 488-conjugated secondary antibody and analyzed via flow cytometry using a FACSCalibur (BD biosciences) and CellQuestPro software. To control for transfection efficiency, the surface expression in each group was normalized to the total cellular Flag-Glut1 expression, obtained by acquiring the fluorescence from fixed (2% PFA for 10 min) and permeabilized (0.1% TritonX-100 for 10 min) cells.
Glut1 internalization and recycling
To measure Glut1 internalization kinetics, cells expressing Flag-Glut1 were glucose-deprived for 2h, incubated with anti-Flag antibody at 4°C for 30 mins, washed with ice cold assay buffer and chased in full media at 37°C for indicated times. The cells were then incubated with secondary antibody and FACS analyzed. For Glut1 recycling assays, Flag-Glut1 transfected cells were incubated with primary antibody in full medium at 37°C for 30 min, then washed and chased in serum and glucose free media for indicated times. The cells were subsequently washed and incubated with secondary antibody and FACS analyzed. In both internalization and recycling assays, the mean fluorescence of cell surface Glut1 expression at each time point was normalized to its corresponding starting value (time= 0 minutes).
Transferrin recycling
Cells were serum-starved in DMEM + 0.5% BSA + 20 mM HEPES for 1h at 37°C, incubated with 25 μg/ml Alexa Fluor 488-conjugated transferrin (Tfn; Life Technologies) for 15 min at 37°C in serum-free media, and chased in full media for up to 60 min.
Glut1 degradation assay
Surface proteins were biotinylated with 1mg/ml of EZlink sulfo-NHS-SS-biotin (Pierce/Thermo) at 4°C for 30 mins. Cells were washed with ice-cold 5mM Tris-HCl and then chased in DMEM media at 37°C for the indicated times. Cells were lysed, biotinylated proteins were captured by incubating lysates with streptavidin ultralink resin (Pierce/Thermo) overnight. Captured proteins were recovered by boiling the beads in 2× SDS sample buffer and biotinylated proteins were resolved by SDS-PAGE and immunoblotted for Glut1. Degradation of Glut1 was quantified as the fraction of signal intensity remaining compared to time point 0h.
Statistical Analyses
All statistical analyses were done using Graphpad Prism 6. Experimental groups were compared using unpaired Student’s t test or one-way ANOVA followed by Tukey’s HSD test for multiple comparisons. Pearson’s correlation coefficients were calculated using Fiji image processing software (v2) to quantify the degree of co-localization between two indicated fluorescent markers.
Data and software availability
The original raw data for the immunoblotting and immunofluorescence studies have been deposited in Mendeley data and are available at http://dx.doi.org/10.17632/2fbtcphv5y.1
Supplementary Material
Highlights.
Autophagy facilitates glucose uptake by promoting Glut 1 cell surface expression.
Autophagy enables retromer-driven translocation of Glut1 to the plasma membrane.
Autophagy deficiency causes Glut1 mis-sorting into endolysosomal compartments.
Shuttling of TBC1D5 into LC3+ autophagosomes relieves inhibition of the retromer.
Acknowledgments
We thank Drs. Noboru Mizushima, Masaaki Komatsu and Jeffrey Rathmell for generously providing reagents, Drs. Emin Maltepe and Aras Mattis (UCSF) for access to hypoxia chambers. Confocal microscopy was performed in the Biological Imaging Development Center and FACS analysis in the Parnassus flow cytometry core at UCSF. Grant support includes the NIH (CA126792, CA201849 to JD, CA172845 to SMR), the DOD BCRP (W81XWH-11-1-0130 to JD) and Samuel Waxman Cancer Research Foundation (to J.D.). A.M.L. is supported by a Banting Postdoctoral Fellowship from the Government of Canada.
Footnotes
Author Contributions: S.R and J.D. conceived the study and designed the experiments. S.R.M helped design the NMR spectroscopy experiments. S.R., J.Y. and A.M.L. performed the experiments. S.R. analyzed the data. S.R. and J.D. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almeida JL, Cole KD, Plant AL. Standards for Cell Line Authentication and Beyond. PLoS biology. 2016;14:e1002476. doi: 10.1371/journal.pbio.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annual review of biochemistry. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Carvalho KC, Cunha IW, Rocha RM, Ayala FR, Cajaiba MM, Begnami MD, Vilela RS, Paiva GR, Andrade RG, Soares FA. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics. 2011;66:965–972. doi: 10.1590/S1807-59322011000600008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Eyster CA, Higginson JD, Huebner R, Porat-Shliom N, Weigert R, Wu WW, Shen RF, Donaldson JG. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic. 2009;10:590–599. doi: 10.1111/j.1600-0854.2009.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Molecular biology of the cell. 2008;19:797–806. doi: 10.1091/mbc.E07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenring JR. A central role for vesicle trafficking in epithelial neoplasia: intracellular highways to carcinogenesis. Nature reviews Cancer. 2013;13:813–820. doi: 10.1038/nrc3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nature reviews Molecular cell biology. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes & development. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvela M, Baquero P, Kuntz EM, Mukhopadhyay A, Mitchell R, Allan EK, Chan E, Kranc KR, Calabretta B, Salomoni P, et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy. 2016;12:936–948. doi: 10.1080/15548627.2016.1162359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger SC, Siupka P, Nielsen MS. Retromer-Mediated Trafficking of Transmembrane Receptors and Transporters. Membranes. 2015;5:288–306. doi: 10.3390/membranes5030288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. The Journal of cell biology. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Li TY, Sun Y, Liang Y, Liu Q, Shi Y, Zhang CS, Zhang C, Song L, Zhang P, Zhang X, et al. ULK1/2 Constitute a Bifurcate Node Controlling Glucose Metabolic Fluxes in Addition to Autophagy. Molecular cell. 2016;62:359–370. doi: 10.1016/j.molcel.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Molecular biology of the cell. 2011;22:165–178. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34:121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrow L, Malhotra R, Debnath J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nature cell biology. 2015;17:300–310. doi: 10.1038/ncb3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D, Akutsu M, Novak I, Harper JW, Behrends C, Dikic I. Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Molecular and cellular biology. 2012;32:1733–1744. doi: 10.1128/MCB.06717-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D, Dikic I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO reports. 2014;15:392–401. doi: 10.1002/embr.201337995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishna H, Donaldson JG. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. The Journal of cell biology. 1997;139:49–61. doi: 10.1083/jcb.139.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJ, Raposo G, van der Sluijs P, Bonifacino JS. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. The Journal of cell biology. 2008;183:513–526. doi: 10.1083/jcb.200804048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, Laderoute K, Johnson RS. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Molecular and cellular biology. 2001;21:3436–3444. doi: 10.1128/MCB.21.10.3436-3444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN, Harbour ME, Tattersall D, Read E, Bright N. Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. Journal of cell science. 2009;122:2371–2382. doi: 10.1242/jcs.048686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg F, Gallon M, Winfield M, Thomas EC, Bell AJ, Heesom KJ, Tavare JM, Cullen PJ. A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nature cell biology. 2013;15:461–471. doi: 10.1038/ncb2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Crone S, Deppe C, Barthel A, Sasson S, Joost HG, Schurmann A. Glucose deprivation induces Akt-dependent synthesis and incorporation of GLUT1, but not of GLUT4, into the plasma membrane of 3T3-L1 adipocytes. European journal of cell biology. 2000;79:943–949. doi: 10.1078/0171-9335-00118. [DOI] [PubMed] [Google Scholar]
- Wei H, Guan JL. Pro-tumorigenic function of autophagy in mammary oncogenesis. Autophagy. 2012;8:129–131. doi: 10.4161/auto.8.1.18171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes & development. 2011;25:1510–1527. doi: 10.1101/gad.2051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert R, Yeung AC, Li J, Donaldson JG. Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Molecular biology of the cell. 2004;15:3758–3770. doi: 10.1091/mbc.E04-04-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieman HL, Horn SR, Jacobs SR, Altman BJ, Kornbluth S, Rathmell JC. An essential role for the Glut1 PDZ-binding motif in growth factor regulation of Glut1 degradation and trafficking. Biochem J. 2009;418:345–367. doi: 10.1042/BJ20081422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Molecular cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes & development. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.