

Abstract
Purpose
Mucosal melanoma represents ~1% of all melanomas, frequently having a poor prognosis due to diagnosis at a late stage of disease. Mucosal melanoma differs from cutaneous melanoma not only in terms of poorer clinical outcome but also on the molecular level having e.g. less BRAF and more frequent KIT mutations than cutaneous melanomas. For the majority of mucosal melanomas oncogenic driver mutations remain unknown.
Experimental Design and Results
In our study, 75 tumor tissues from patients diagnosed with mucosal melanoma were analyzed, applying a targeted next generation sequencing panel covering 29 known recurrently mutated genes in melanoma. NF1 and RAS mutations were identified as the most frequently mutated genes occurring in 18.3% and 16.9% of samples, respectively. Mutations in BRAF were identified in 8.4% and KIT in 7.0% of tumor samples.
Conclusions
Our study identifies NF1 as the most frequently occurring driver mutation in mucosal melanoma. RAS alterations, consisting of NRAS and KRAS mutations, were the second most frequent mutation type. BRAF and KIT mutations were rare with frequencies below 10% each. Our data indicate that in mucosal melanomas RAS/NF1 alterations are frequent, implying a significant pathogenetic role for MAPK and potentially PI3K pathway activation in these tumors.
Keywords: mucosal melanoma, NF1, RAS, sequencing, melanoma
INTRODUCTION
Mucosal melanomas arise from melanocytes of the mucosal membrane and represent a rare subgroup of melanoma, accounting for around 1% of all melanomas [1]. Frequently diagnosed at an advanced tumor stage, they generally have a poor prognosis [2]. In contrast to cutaneous melanomas, where exogenous or endogenous risk factors such as UV-exposure or genetic predisposition are well known and studied in a high number of patients [3], no comparable factors have been identified for mucosal melanomas. Additionally, little information is available with regard to the molecular pathogenesis of mucosal melanoma. In general, mucosal melanomas seem to have a lower overall mutational burden as compared to cutaneous melanomas (8193 vs. 86495 somatic single nucleotide variants per tumor) [4, 5] but a higher number of chromosomal aberrations [6]. However, BRAF V600 mutations, the most frequent and therapeutically best-targetable gene alteration in cutaneous melanoma (present in ~50% of cases) is only rarely found in mucosal melanomas (≤ 10% of cases), which limits clinical treatment options [6–8]. Other activating oncogenic events, e.g. gene amplifications or gain-of-function mutations of KIT are more frequently detected in mucosal melanomas. Existing literature reports KIT activation in 15-39% of mucosal melanomas [9–13]. However, KIT-targeting therapies, e.g. with imatinib, have failed to show convincing therapeutic efficiency in mucosal melanoma in larger studies [14, 15]. NRAS mutations are somewhat less frequent in mucosal (10-20%) [6, 8] than cutaneous melanomas (20-30%) [6, 16–18]. Activating mutations in GNAQ and GNA11, which are commonly detected in uveal melanoma [19], have recently been reported to occur in 9.5% of mucosal melanomas [20], a finding not reported in previous studies [21]. Many existing studies have been performed on cohorts with limited sample numbers.
In summary, mucosal melanoma represents a clinically aggressive cancer entity rarely harboring known therapeutically targetable driver mutations. Our study aimed to identify additional oncogenic driver mutations in mucosal melanoma in a larger cohort of patients to recognize additional molecular pathways with the potential to be exploited for establishing future therapeutic strategies.
RESULTS
Patient characteristics
Samples were obtained from 75 patients, 46 females and 29 males. Clinic-pathological characteristics are summarized in Table 1. In 4 cases, sequencing analysis was not possible due to poor sequencing quality; clinico-pathological characteristics of these patients are not included in Table 1.
Table 1. Clinicopathological characteristics of the patients.
| Variable | Total (n=71) | NF1 | WT | p-value | RAS | WT | p-value | BRAF | WT | p-value |
|---|---|---|---|---|---|---|---|---|---|---|
| Median age (range) | 64 (33-84) | |||||||||
| Gender | ||||||||||
| Male | 26 | 3 | 23 | 0.348 | 8 | 18 | 0.024 | 2 | 24 | 1.0 |
| Female | 45 | 10 | 35 | 4 | 41 | 4 | 41 | |||
| Anatomical site | ||||||||||
| Head and neck | 28 | 3 | 25 | 0.386 | 9 | 19 | 0.224 | 2 | 26 | 0.672 |
| Genital area | 25 | 5 | 20 | 3 | 22 | 2 | 23 | |||
| Anorectum | 9 | 3 | 6 | 0 | 9 | 1 | 8 | |||
| Digestive tract | 3 | 1 | 2 | 0 | 3 | 1 | 2 | |||
| Urinary tract | 3 | 0 | 3 | 0 | 3 | 0 | 3 | |||
| Data missing | 3 | 1 | 2 | 0 | 3 | 0 | 3 | |||
| Sample type | ||||||||||
| Primary tumor | 41 | 8 | 33 | 0.798 | 8 | 33 | 0.936 | 5 | 36 | 0.837 |
| Metastasis | 22 | 5 | 17 | 3 | 19 | 1 | 21 | |||
| Recurrence | 3 | 0 | 3 | 0 | 3 | 0 | 3 | |||
| Data missing | 5 | 0 | 5 | 1 | 4 | 0 | 5 |
Targeted next generation sequencing
Mutations were identified in 50 samples (Figure 1, Table 2, Supplementary Table 1). NF1 and RAS were the most frequently mutated genes (Figure 2, Supplementary Figure 1 and 2, Table 2, Supplementary Table 1), with 15 NF1 mutations identified in 13 samples (18.3.%) and 12 RAS mutations identified in 12 samples (16.9%). In 9 out of 13 samples (69.2%) a clearly inactivating NF1 mutation was present resulting in non-sense (synthesis stop) or frameshift mutations. Examples of some inactivating NF1 mutations detected in our cohort are shown in Supplementary Figure 1. Two samples harbored multiple NF1 mutations (Table 2, Supplementary Table 1); one of them having two inactivating mutations, the other one having an inactivating and a D896N missense mutation. RAS gene alterations were found in 12 out of 71 samples (16.9%), including 8 NRAS and 4 KRAS mutations. Sanger sequencing was performed to validate the identified KRAS mutations (Supplementary Figure 3). Six samples (8.4%) harbored BRAF mutations, 5 of which were activating V600 mutations (4 V600E and 1 V600K) and 1 N188S mutation (Supplementary Table 2). The mutation pattern on protein level for the identified NRAS, KRAS and NF1 mutations are shown in Figure 2. KIT mutations were detected in 5 samples (7%), but only 1 was a known activating mutation. Another 4 samples carried known activating TERT-promoter mutations. One GNA11 S267F mutation and 1 GNAQ R183Q mutation were identified. Examples of the most frequent activating mutations identified are illustrated in Supplementary Figure 2. Three samples harboring KRAS mutations also had concurrent NF1 mutations, 2 of which were clearly functionally inactivating. Additionally, 5 TP53, 7 SF3B1, 4 MITF and 3 PTEN mutations were identified. Other less frequent mutations were identified in various genes including SMARC, BAP1, TERT, WT1, PIK3CA, MAP2K2, CDK4, CTNNB1, RAC1, ARID2 and ARID1A. In 21 samples no non-synonymous protein coding mutation were identified in the 29 genes analyzed.
Figure 1. Mutation distribution in mucosal melanomas.
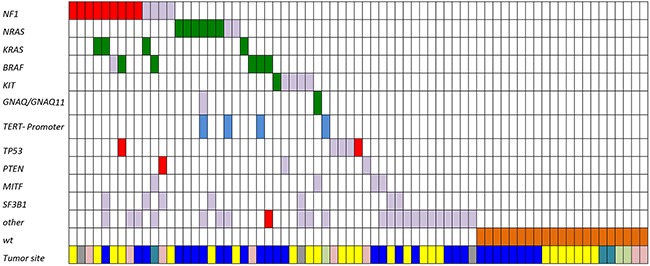
Green: mutations known or assumed to be activating; red: loss of function mutations; blue: mutations in the TERT promoter region; grey: missense mutation (frequently with unknown functional consequences); brown: wild-type samples (showing no mutation in the analyzed gene panel). Tumor location: Yellow, genital area; light pink, anorectum; dark blue, head and neck; light green, urinary tract; petrol, digestive tract; grey, data missing.
Table 2. List of identified mutations.
| Nr. | Type | Location primary | NF1 | RAS | BRAF | Other Mutations |
|---|---|---|---|---|---|---|
| 1 | M | G | E2174fs; L151fs | |||
| 2 | M | DM | R106* | |||
| 3 | M | G | R2258* | KRAS G12D | ||
| 4 | P | A | V1308fs | TERT S663N | ||
| 5 | P | G | G1425fs | V600E | TP53 Q165* | |
| 6 | P | HN | H553fs | PIK3CA E109del | ||
| 7 | P | G | T889fs | N188S | ||
| 8 | M | A | R1306* | |||
| 9 | P | HN | T1184fs; D896N | KRAS G12A | ARID1A V700A; SF3B1 D894N | |
| 10 | M | G | H55R | |||
| 11 | P | A | I183N | PTEN K163fs; SF3B1 R625H | ||
| 12 | P | D | M1376V | V600E | ARID1A R1202Q; ARID2 T1208A; MITF A401S | |
| 13 | P | HN | V1308L | KRAS E63K | SF3B1 R625H | |
| 14 | P | HN | KRAS G12F | |||
| 15 | P | HN | NRAS Q61R | |||
| 16 | P | HN | NRAS Q61R | |||
| 17 | P | HN | NRAS Q61K | |||
| 18 | P | HN | NRAS G13R | TERT Ser1104Thr | ||
| 19 | P | HN | NRAS Q61K | TERT P C228T; RAC1 N92K; GNA11 S267F; | ||
| 20 | U | G | NRAS Q61L | SF3B1 V634A | ||
| 21 | M | HN | NRAS A59D | TERT P C243T; TERT P C252T; SMARCA4 A152T | ||
| 22 | M | V | NRAS I46M | |||
| 23 | M | HN | V600E | TERT P C250T | ||
| 24 | P | HN | V600E | PIK3CA L896fs | ||
| 25 | P | A | V600K | |||
| 26 | M | HN | KIT L576P | |||
| 27 | M | Ur | TERT P C228T; WT1 D497N | |||
| 28 | M | G | GNAQ R183Q; MITF V487I | |||
| 29 | P | G | TP53 P58fs | |||
| 30 | P | G | ARID2 Y612C | |||
| 31 | M | HN | TERT L1002V | |||
| 32 | M | A | PTEN L108R | |||
| 33 | M | G | PIK3R1 T239M | |||
| 34 | P | G | KIT Y553del; MAP2K2 G286R | |||
| 35 | P | G | TERT R819H | |||
| 36 | P | HN | MITF N267K | |||
| 37 | P | A | TP53 C135R | |||
| 38 | R | HN | KIT V50L; PTEN C136R | |||
| 39 | P | G | TP53 P151A | |||
| 40 | U | G | ARID2 M545I; SF3B1 R625H | |||
| 41 | P | G | TERT R819H | |||
| 42 | U | A | MITF V487I; SMARCA4 R1260S | |||
| 43 | R | HN | SF3B1 T916S; CK4 R209C | |||
| 44 | P | HN | TP53 R175G | |||
| 45 | P | G | KIT, L783I | |||
| 46 | M | DM | KIT, I748T; BAP1, G579R: SF3B1, R625L | |||
| 47 | P | HN | BAP1 Y646C | |||
| 48 | P | HN | CTNNB1 Y331C | |||
| 49 | P | HN | TERT S953F | |||
| 50 | M | DM | CK4 V174M |
Green, mutations known to be activating; red, loss of function mutations; black, missense mutation with unknown functional consequences. Abbreviations: Nr. sample number; M metastasis; P primary tumor; R recurrence; U unknown; fs frame shift; * = stop codon (nonsense mutation); HN Head and Neck, G Genital area, A Anorectum, D Digestive tract, Ur Urinary tract, DM data missing.
For more details including allele frequencies and cDNA annotations, see Supplementary Table 1.
Figure 2. Distribution of identified NRAS, KRAS and NF1 mutations.
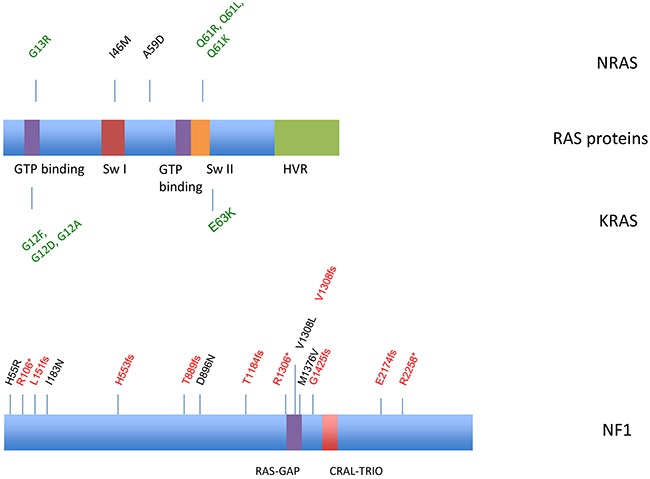
Frameshift and nonsense mutations are annotated in red, activating mutations are demonstrated in green. Missense mutations are black. Switch I, effector/GAP interaction; Switch II, EF interaction; HVR, hypervariable region. NF1 contains a Ras-GAP domain (GTPase-activator protein for Ras-like GTPase) and a CRAL-TRIO domain.
Statistical analysis
We performed a statistical analysis to assess possible associations of clinical parameters such as gender, location of the primary tumor and sample type (primary, metastasis or recurrence) with the NF1, RAS and RAF mutational status. A statistically significant association was determined between RAS mutational status and male sex (p=0.024, Table 1).
DISCUSSION
In this study, 71 mucosal melanoma samples were screened for mutations in known recurrently mutated genes in cutaneous and uveal melanoma. The most frequent mutations were identified in the NF1 gene and RAS gene family members, indicating that mucosal melanomas have a genetic mutation profile which is different from that of cutaneous or uveal melanomas. Our study identified an unexpectedly high number of NF1 mutations. In 13 (18.3,%) out of 71 samples, NF1 mutations were identified. Nine of those (69.2%) harbored clearly inactivating mutations, i.e. nonsense or frameshift mutations.
To our knowledge, there is no previous data demonstrating that mucosal melanomas express such a high frequency of NF1 mutations. Yang et al. recently conducted a targeted sequencing analysis on 15 anorectal melanomas and identified that 3 of these tumors harbored an NF1 mutation [22]. In recent years, NF1 has been recognized as the third most commonly mutated gene (after BRAF and RAS) resulting in activation of the MAPK pathway with a reported mutation frequency of 14% [18]. In cutaneous melanomas, more than half of the mutations reported are loss-of-function (LoF) events. In the mucosal melanomas studied here, 9 out of 13 cases carried NF1 LoF mutations (69.2%). It is known that NF1 is a GTPase-activating protein which downregulates the activity of the RAS protein [18]. As such, LoF mutations in NF1 are an important genetic mechanism for constitutive MAPK pathway activation.
A relevant role for NF1 mutations in cutaneous melanomas lacking conventional (i.e. BRAF or NRAS) activating mutations has been already highlighted by other studies. Wiesner et al. demonstrated that desmoplastic melanomas frequently harbor NF1 mutations [23]. Desmoplastic melanomas are typically associated with high UV-exposure and high mutational loads. Congruently, Krauthammer et al. described NF1 mutations in association with mutations in RASopathy genes (e.g. RASA2, PTPN11, etc.) in cutaneous melanomas with evidence of high sun-exposure [24]. The association of UV-exposure with NF1 mutations observed in cutaneous melanoma is not to be expected in mucosal melanoma [4, 5]. Although mutational mechanisms may differ, these studies and our data support NF1 mutations being highly relevant in melanoma subgroups that rarely harbor BRAF or NRAS mutations.
Twelve out of 71 (16.9%) mucosal melanomas analyzed harbored RAS mutations, 8 in the NRAS and 4 in the KRAS gene. No mutations were identified in HRAS. Previous studies have focused primarily on NRAS, where the mutation frequency ranges from 5% to 15% [6, 8, 25–27]. The frequency of NRAS mutations detected in our study with 11.2% is comparable. KRAS mutations have not been assessed in most previous studies of mucosal melanoma, however, accounted for one third of the mutations in RAS genes observed in our cohort. A slightly significant association of RAS mutation status with male gender was noted (p=0.024). This potential association will need be to assessed in future studies with considerably larger sample numbers.
BRAF was the third most frequently mutated gene identified in our study. Of the 6 mutations (8.4%) identified, 5 were well known V600 activating mutations, consisting of 4 V600E and 1 V600K mutation. The other identified mutation, resulting in an N188S exchange, is of unclear significance and could be a non-relevant passenger mutation. These findings are in accordance with reports stating that activating BRAF mutations in mucosal melanomas are rare with a frequency between 5 and 17% [8, 9, 26, 28, 29]. Only one patient in our cohort received BRAF inhibitor therapy showing a partial response (Supplementary Table 2). The efficacy of BRAF inhibitor therapies in mucosal melanomas will need to be further assessed in larger studies.
Genetic alterations of KIT, including mutations and copy number increases, have been reported to occur in up to 39% of mucosal melanomas [9–13]. In our study, 5 out of 71 (7.0%) samples had a KIT mutation; 2 mutations in tumors of the head and neck region and 2 in vulvar melanomas. Omholt et al. [27] reported KIT mutations in 17% (n=12) of primary mucosal melanomas. They found that 35% (8 out of 23) of vulvar melanomas harbored KIT mutations. In our study, 2 out of 9 (22.2%) vulvar melanomas had a KIT mutation. Generally, our findings support existing literature stating that KIT mutations are more frequent in melanomas of the genital area followed by melanomas of the head and neck and the anorectal area [8, 26, 30, 31].
TERT promoter mutations resulting in increased transcription of the TERT gene have been identified as the most common mutation in cutaneous melanoma [32–34]. In congruence with previous studies reporting low mutation rates in mucosal melanomas [35, 36], the mutation frequency in our cohort was 5.6%. All TERT mutations were C>T mutations. As C>T alterations are classically associated with UV-exposure [32, 37, 38], the lower TERT mutation frequency may be due to the very limited UV-exposure of tumors arising in mucosal locations.
Two alterations were identified in GNAQ and GNA11, mutations that are usually associated with uveal melanomas and blue nevi [19]. Of those, only the GNAQ R183Q mutation is known to be functionally activating, resulting in increased mitogenic signaling in melanocytic tumors and rare vascular diseases such as Sturge-Weber syndrome and phakomatosis pigmentovascularis [39, 40]. The other mutation identified (GNA11 S267F) is not known to be functionally relevant and probably represents a bystander mutation considering the sample also harbored a NRAS Q61K hot-spot mutation. While GNAQ and GNA11 mutations were recently reported to occur in 9.5% of mucosal melanomas [20], our study suggests these mutations are less frequent in this tumor entity.
In out cohort, 31 samples presented mutations in the MAPK pathway (some of them harboring more than one mutation): 13 NF1, 12 RAS, 6 RAF, 5 KIT, 1 GNAQ and 1 GNA11 mutation. In 23 of those samples mutations resulting in MAPK activation were found suggesting that this is a critical event in the pathogenesis of mucosal melanoma. This is similar to cutaneous melanoma, where constitutive activation of the MAPK pathway is a known critical event. The genetic alterations leading to this activation however vary between these melanoma subtypes [18].
Although our study represents the most comprehensive genetic analysis of 75 mucosal melanomas presented to date, it does have some limitations. Mutations occurring in genes not covered by our panel could not be identified. Additionally, our approach did not allow us to reliably detect copy number variations.
Our results underline that mucosal melanomas are genetically distinct from cutaneous and uveal melanomas with frequent inactivating mutations in NF1 and activating mutations in RAS genes. Our findings suggest that similar to cutaneous melanoma, activation of the MAPK pathway is a pivotal event in mucosal melanoma. Taken into consideration that both NF1 and RAS alterations can additionally activate PI3K signaling, this pathway could be of particular significance in mucosal melanomas. It stands to reason that other, so far unidentified, mutations are present in mucosal melanomas and future whole-exome or whole-genome studies of larger tumor cohorts will be required to fully elucidate the landscape of genetic alterations involved.
MATERIALS AND METHODS
Sample selection
Samples of mucosal melanomas were retrieved from the biobank of the Department of Dermatology, Essen, Germany, from the TRIM (Tissue Registry in Melanoma) project of the German DeCOG (Dermatological Cooperative Oncology Group) as well as from patients enrolled into the DeCOG trial ChemoSensMM [ClinicalTrials.gov: NCT00779714 [41]. Samples were considered mucosal melanoma if the tumor originated in a mucosal site, was diagnosed as melanoma histopathologically (confirmed by at least one immunohistochemical marker [Melan-A, HMB-45 or S100]) and no other preexisting, e.g. cutaneous, melanoma was reported (to exclude potential mucosal metastasis). The study was performed with informed patient consent in accordance with the guidelines of the Ethics Committee of the Medical Faculty of the University Duisburg-Essen (ethical approval no. 15-6473-BO, no. 15-6566-BO).
DNA isolation
FFPE tissue was prepared in 10 μm sections and deparaffinized according to standard procedures. In brief, 2 steps of 5 min xylene, 5 min 100% ethanol, 5 min 95% ethanol, 5 in 70% ethanol, 5 min 50% ethanol, rinsing in water. After air drying, the tumor tissue was manually macrodissected from the sections. The tumor content in the area of macrodissection was required to be at least 25%, however, was generally considerably higher (documented values are listed in Supplementary Table 1). Genomic DNA was isolated applying the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Targeted sequencing
A custom designed amplicon-based sequencing panel covering the TERT promoter and the complete coding regions of 29 known recurrently mutated genes in cutaneous and uveal melanoma (Supplementary Table 3) was applied. This panel was initially clinically validated over a period of 3 months, where Sanger and NGS panel sequencing were performed in parallel for all samples analyzed. All known recurrent mutations in melanoma (incl. BRAF V600, NRAS G12, G13 and Q61 and KIT L576, K642 and N822) were repeatedly picked up by our NGS panel which showed a 100% concordance with mutations identified by Sanger sequencing, however demonstrated a higher level of sensitivity. After successful validation, our routine clinical sequencing effort has solely relied on the NGS panel, which in over 2 years has been applied to more than 1300 melanocytic tumors. Our panel sequencing approach is not able to reliably detect copy number variations.
Adapter ligation and barcoding of individual samples occurred applying the NEBNext Ultra DNA Library Prep Mastermix Set and NEBNext Multiplex Oligos for Illumina from New England Biolabs. Sequencing analysis was performed using CLC Cancer Research Workbench from QIAGEN as previously described [42]. In brief, analysis included the following steps: The CLC workflow included adapter trimming and read pair merging before mapping to the human reference genome (hg19). Insertions and deletions as well as single nucleotide variant detection, local realignment and primer trimming followed. Additional information was then obtained regarding potential mutation type, known single nucleotide polymorphisms and conservation scores by cross-referencing various databases (COSMIC, ClinVar, dbSNP, 1000 Genomes Project, HAPMAP and PhastCons_Conservation_scores_hg19). The CLC generated csv files were further analyzed manually with mutations affecting the protein-coding portion of the gene considered if predicted to result in non-synonymous amino acid changes. The average fold coverage was 2585x. To eliminate questionable low frequency background mutation calls, not uncommon in our experience with FFPE amplicon sequencing approaches [43], mutations were reported if overall coverage of the mutation site was ≥30 reads, ≥10 reads reported the mutated variant and the frequency of mutated reads was ≥10 %.
Statistical analysis
The associations of mutation status with clinical parameters such as gender, location of the primary tumor and sample origin (primary, metastasis or recurrence), was investigated using chi-squared tests and Fisher exact tests as appropriate. Statistical analyses were performed using SPSS 23.0 (IBM Corp., Armonk NY, USA), considering a p-value of p≤0.05 as statistically significant.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
Acknowledgments
We thank Nadine Stadler and Mingxia Song (Department of Dermatology, University Hospital Essen) for technical assistance.
Author contributions
Study concept and design was performed by K. Griewank and A. Roesch. Involved in data acquisition were S. Ugurel, L. Zimmer, M. Ziemer, C. Pföhler, J. Utikal, P. Mohr and C. Pfeiffer. Quality control of data was performed by A. Sucker, I. Cosgarea, S. Horn, U. Hillen. Data analysis was covered by I. Cosgarea and K. Griewank. Involved in the statistical analysis was L. Livingstone and I. Cosgarea. Manuscript preparation was covered by I. Cosgarea, K. Griewank, A. Roesch, D. Schadendorf. In charge with manuscript editing were I. Cosgarea, K. Griewank and A. Roesch. Manuscript review was performed by all the authors.
CONFLICTS OF INTEREST
Ioana Cosgarea has received travel support from Novartis, Bristol-Meyers Squibb, TEVA. Selma Ugurel is on the advisory board or has received honoraria from Bristol-Myers Squibb, Merck Sharp & Dohme and Roche, and received grant support or travel support from Bristol-Myers Squibb, medac and Roche. Lisa Zimmer has received honoraria from Roche, Bristol-Myers Squibb, MSD Sharp & Dohme, Merck, GlaxoSmithKline, and Novartis, and travel support from MSD Sharp & Dohme, Novartis and Bristol-Meyers Squibb. Elisabeth Livingstone has received honoraria from Roche, Bristol-Myers Squibb, Amgen, Novartis, Boehringer-Ingelheim, Merck Sharp & Dohme and Merck, and travel support from Amgen, Merck Sharp & Dohme, Bristol-Myers Squibb, Novartis and Boehringer-Ingelheim. Mirjana Ziemer is on the advisory board or has received honoraria from Bristol-Myers Squibb, Merck Sharp & Dohme and Roche, as well as received travel support from Bristol-Myers Squibb. Jochen Utikal is on the advisory board or has received honoraria and travel support from Amgen, BMS, GSK, LeoPharma, MSD, Novartis, Roche. Claudia Pföhler has received honoraria and travel support from Novartis, BMS, Roche, Merck Serono, MSD Merck Sharp & Dohme, Bencard and Celgene and is on the advisory board from Novartis and Merck Serono. Dirk Schadendorf is on the advisory board or has received honararia from Roche, Genentech, Novartis, Amgen, GSK, BMS, Boehringer Ingelheim, and Merck. Klaus Griewank has received travel support from Roche. Alexander Roesch received travel grants and honoraria from Roche, TEVA, Bristol-Myers Squibb, MSD Sharp & Dohme, Amgen, and Novartis and a research grant from Novartis.
All other authors have nothing to declare.
FUNDING
This work was supported in part by Deutsche Stiftung Dermatologie (Stipendium Förderung der Dermatohistologie), the Monika Kutzner Stiftung, the Deutsche Forschungsgemeinschaft (Grant DFG3671/1-3), Deutsche Krebshilfe (Grant 70111402) and the Hiege-Stiftung gegen Hautkrebs.
REFERENCES
- 1.Chang AE, Karnell LH, Menck HR, The American College of Surgeons Commission on Cancer and the American Cancer Society The National Cancer Data Base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. Cancer. 1998;83:1664–78. doi: 10.1002/(sici)1097-0142(19981015)83:8<1664::aid-cncr23>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 2.Manolidis S, Donald PJ. Malignant mucosal melanoma of the head and neck: review of the literature and report of 14 patients. Cancer. 1997;80:1373–86. doi: 10.1002/(sici)1097-0142(19971015)80:8<1373::aid-cncr3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 3.Postow MA, Hamid O, Carvajal RD. Mucosal melanoma: pathogenesis, clinical behavior, and management. Curr Oncol Rep. 2012;14:441–48. doi: 10.1007/s11912-012-0244-x. [DOI] [PubMed] [Google Scholar]
- 4.Furney SJ, Turajlic S, Stamp G, Nohadani M, Carlisle A, Thomas JM, Hayes A, Strauss D, Gore M, van den Oord J, Larkin J, Marais R. Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J Pathol. 2013;230:261–69. doi: 10.1002/path.4204. [DOI] [PubMed] [Google Scholar]
- 5.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, Zhang H, Zeid R, Ren X, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–06. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 7.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 8.Tacastacas JD, Bray J, Cohen YK, Arbesman J, Kim J, Koon HB, Honda K, Cooper KD, Gerstenblith MR. Update on primary mucosal melanoma. J Am Acad Dermatol. 2014;71:366–75. doi: 10.1016/j.jaad.2014.03.031. [DOI] [PubMed] [Google Scholar]
- 9.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340–46. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 10.Rivera RS, Nagatsuka H, Gunduz M, Cengiz B, Gunduz E, Siar CH, Tsujigiwa H, Tamamura R, Han KN, Nagai N. C-kit protein expression correlated with activating mutations in KIT gene in oral mucosal melanoma. Virchows Archiv. 2008;452:27–32. doi: 10.1007/s00428-007-0524-2. [DOI] [PubMed] [Google Scholar]
- 11.Antonescu CR, Busam KJ, Francone TD, Wong GC, Guo T, Agaram NP, Besmer P, Jungbluth A, Gimbel M, Chen CT, Veach D, Clarkson BD, Paty PB, Weiser MR. L576P KIT mutation in anal melanomas correlates with KIT protein expression and is sensitive to specific kinase inhibition. International Journal of Cancer. 2007;121:257–264. doi: 10.1002/ijc.22681. [DOI] [PubMed] [Google Scholar]
- 12.Ashida A, Takata M, Murata H, Kido K, Saida T. Pathological activation of KIT in metastatic tumors of acral and mucosal melanomas. International journal of cancer. 2009;124:862–868. doi: 10.1002/ijc.24048. [DOI] [PubMed] [Google Scholar]
- 13.Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, Pavlick AC, Fusco A, Cane L, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–34. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KB, Alrwas A. Treatment of KIT-mutated metastatic mucosal melanoma. Chin Clin Oncol. 2014;3:35. doi: 10.3978/j.issn.2304-3865.2014.08.02. [DOI] [PubMed] [Google Scholar]
- 15.Carvajal RD. Another option in our KIT of effective therapies for advanced melanoma. J Clin Oncol. 2013;31:3173–75. doi: 10.1200/JCO.2013.50.3144. [DOI] [PubMed] [Google Scholar]
- 16.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–14. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas N, Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–96. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–99. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheng X, Kong Y, Li Y, Zhang Q, Si L, Cui C, Chi Z, Tang B, Mao L, Lian B, Wang X, Yan X, Li S, et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur J Cancer. 2016;65:156–63. doi: 10.1016/j.ejca.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 21.Pappa KI, Vlachos GD, Roubelakis M, Vlachos DE, Kalafati TG, Loutradis D, Anagnou NP. Low mutational burden of eight genes involved in the MAPK/ERK, PI3K/AKT, and GNAQ/11 pathways in female genital tract primary melanomas. Biomed Res Int. 2015;2015:303791. doi: 10.1155/2015/303791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang HM, Hsiao SJ, Schaeffer DF, Lai C, Remotti HE, Horst D, Mansukhani MM, Horst BA. Identification of recurrent mutational events in anorectal melanoma. Mod Pathol. 2017;30:286–96. doi: 10.1038/modpathol.2016.179. [DOI] [PubMed] [Google Scholar]
- 23.Wiesner T, Kiuru M, Scott SN, Arcila M, Halpern AC, Hollmann T, Berger MF, Busam KJ. NF1 Mutations Are Common in Desmoplastic Melanoma. Am J Surg Pathol. 2015;39:1357–62. doi: 10.1097/PAS.0000000000000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, McCusker JP, Ma S, Cheng E, Straub R, Serin M, Bosenberg M, Ariyan S, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet. 2015;47:996–1002. doi: 10.1038/ng.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minor DR, Kashani-Sabet M, Garrido M, O’Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012;18:1457–1463. doi: 10.1158/1078-0432.CCR-11-1987. [DOI] [PubMed] [Google Scholar]
- 26.Omholt K, Grafstrom E, Kanter-Lewensohn L, Hansson J, Ragnarsson-Olding BK. KIT pathway alterations in mucosal melanomas of the vulva and other sites. Clin Cancer Res. 2011;17:3933–3942. doi: 10.1158/1078-0432.CCR-10-2917. [DOI] [PubMed] [Google Scholar]
- 27.Zebary A, Jangard M, Omholt K, Ragnarsson-Olding B, Hansson J. KIT, NRAS and BRAF mutations in sinonasal mucosal melanoma: a study of 56 cases. Br J Cancer. 2013;109:559–64. doi: 10.1038/bjc.2013.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, Ono T, Albertson DG, Pinkel D, Bastian BC. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95:1878–90. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- 29.Turri-Zanoni M, Medicina D, Lombardi D, Ungari M, Balzarini P, Rossini C, Pellegrini W, Battaglia P, Capella C, Castelnuovo P, Palmedo G, Facchetti F, Kutzner H, et al. Sinonasal mucosal melanoma: molecular profile and therapeutic implications from a series of 32 cases. Head Neck. 2013;35:1066–77. doi: 10.1002/hed.23079. [DOI] [PubMed] [Google Scholar]
- 30.Satzger I, Schaefer T, Kuettler U, Broecker V, Voelker B, Ostertag H, Kapp A, Gutzmer R. Analysis of c-KIT expression and KIT gene mutation in human mucosal melanomas. Br J Cancer. 2008;99:2065–69. doi: 10.1038/sj.bjc.6604791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F, 3rd, Azar S, Rubin BP, Muller S, West R, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14:6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 32.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 33.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–59. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ekedahl H, Lauss M, Olsson H, Griewank KG, Schadendorf D, Ingvar C, Jönsson G. High TERT promoter mutation frequency in non-acral cutaneous metastatic melanoma. Pigment Cell Melanoma Res. 2016;29:598–600. doi: 10.1111/pcmr.12500. [DOI] [PubMed] [Google Scholar]
- 35.Griewank KG, Murali R, Puig-Butille JA, Schilling B, Livingstone E, Potrony M, Carrera C, Schimming T, Möller I, Schwamborn M, Sucker A, Hillen U, Badenas C, et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J Natl Cancer Inst. 2014;106:106. doi: 10.1093/jnci/dju246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jangard M, Zebary A, Ragnarsson-Olding B, Hansson J. TERT promoter mutations in sinonasal malignant melanoma: a study of 49 cases. Melanoma Res. 2015;25:185–88. doi: 10.1097/CMR.0000000000000148. [DOI] [PubMed] [Google Scholar]
- 37.Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Pontén J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–28. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–18. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas AC, Zeng Z, Rivière JB, O’Shaughnessy R, Al-Olabi L, St-Onge J, Atherton DJ, Aubert H, Bagazgoitia L, Barbarot S, Bourrat E, Chiaverini C, Chong WK, et al. Mosaic Activating Mutations in GNA11 and GNAQ Are Associated with Phakomatosis Pigmentovascularis and Extensive Dermal Melanocytosis. J Invest Dermatol. 2016;136:770–78. doi: 10.1016/j.jid.2015.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971–79. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ugurel S, Schadendorf D, Pföhler C, Neuber K, Thoelke A, Ulrich J, Hauschild A, Spieth K, Kaatz M, Rittgen W, Delorme S, Tilgen W, Reinhold U, Dermatologic Cooperative Oncology Group In vitro drug sensitivity predicts response and survival after individualized sensitivity-directed chemotherapy in metastatic melanoma: a multicenter phase II trial of the Dermatologic Cooperative Oncology Group. Clin Cancer Res. 2006;12:5454–63. doi: 10.1158/1078-0432.CCR-05-2763. [DOI] [PubMed] [Google Scholar]
- 42.van de Nes J, Gessi M, Sucker A, Möller I, Stiller M, Horn S, Scholz SL, Pischler C, Stadtler N, Schilling B, Zimmer L, Hillen U, Scolyer RA, et al. Targeted next generation sequencing reveals unique mutation profile of primary melanocytic tumors of the central nervous system. J Neurooncol. 2016;127:435–44. doi: 10.1007/s11060-015-2052-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murali R, Chandramohan R, Möller I, Scholz SL, Berger M, Huberman K, Viale A, Pirun M, Socci ND, Bouvier N, Bauer S, Artl M, Schilling B, et al. Targeted massively parallel sequencing of angiosarcomas reveals frequent activation of the mitogen activated protein kinase pathway. Oncotarget. 2015;6:36041–52. doi: 10.18632/oncotarget.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.