

Abstract
SN-38, the active metabolite of irinotecan, and histone deacetylase inhibitors (HDACis) such as belinostat, vorinostat and panobinostat, have all been shown to be deactivated by glucuronidation via UGTs. Since they all compete for UGTs for deactivation, we aimed to investigate the inhibitory effect of various HDACis on the glucuronidation of SN-38. This inhibitory effect was determined by measuring the formation rate of SN-38 glucuronide after SN-38 incubation with human recombinant UGT1A isoforms (1A1, 1A6, 1A7 and 1A9) and pooled human liver microsomes (HLM, wild type, UGT1A1*1*28 and UGT1A1*28*28 allelic variants), with and without HDACis. The data showed that belinostat at 100 and 200 µmol/L inhibited SN-38 glucuronidation via UGT1A1 in a dose-dependent manner, causing significant decrease in Vmax and CLint (p < 0.05) from 12.60 to 1.95 pmol/min/mg and 21.59 to 4.20 μL/min/mg protein respectively. Similarly, in HLMs, Vmax dropped from 41.13 to 10.54, 24.96 to 3.77 and 6.23 to 3.30 pmol/min/mg, and CLint reduced from 81.25 to 26.11, 29.22 to 6.10 and 5.40 to 1.34 µL/min/mg protein for the respective wild type, heterozygous and homozygous variants. Interestingly, belinostat at 200 µmol/L that is roughly equivalent to the average Cmax, 183 µmol/L of belinostat at a dose of 1,400 mg/m2 given intravenously once per day on days 1 to 5 every 3 weeks, was able to inhibit both heterozygous and homozygous variants to same extents (~64%). This highlights the potential clinical significance, as a large proportion of patients could be at risk of developing severe toxicity if irinotecan is co-administered with belinostat.
Keywords: drug-drug interactions, irinotecan, SN-38, belinostat, UGT1A1
INTRODUCTION
Combination therapy is often used for cancer patients in current clinical practice, as it has been shown to be more efficacious than monotherapy in combating cancer [1]. However, drug-drug interactions (DDIs) are an ongoing concern due to the narrow therapeutic windows of most chemotherapeutic agents and their potential for severe toxicity; irinotecan is one such agent whose interactions with other drugs may lead to severe toxicities such as diarrhoea and neutropenia [2, 3].
Irinotecan is an effective antitumour agent, displaying a broad range of clinical activities against neoplastic disorders such as colorectal, gynaecological and lung cancers [4, 5]. This drug is predominantly converted by carboxylesterases (CES) in the liver to SN-38, an active metabolite that is 100-1000 times more potent than irinotecan [6–9]. The potent SN-38 is then primarily deactivated by glucuronidation catalyzed by UGT1A1 to SN-38 glucuronide (SN-38G) [10–12]. Other UGT1A isoforms such as UGT1A6, UGT1A7 and UGT1A9, have also been reported to play a role in the formation of SN-38G [2, 13–15].
An illuminating example of toxicity caused by accumulation of SN-38 can be attributed to the functional polymorphism of UGT1A1. Genetic polymorphisms in UGT1A1 have been demonstrated to have great impact on SN-38 toxicity through influencing enzyme activity, leading to inter-individual differences in drug response [13, 16–18]. Notably, when compared to wildtype genotype, patients expressing the UGT1A1*28 allele experienced significant irinotecan treatment-related toxicity (p < 0.001) due to decreased (1.3 to 3.9 fold lower) enzymatic activity in liver microsomes. This resulted in reduced SN-38G formation and higher levels of SN-38 [3]. Similarly, any potential DDI with irinotecan that increases the serum levels of SN-38 could also cause severe undesirable toxicity. For instance, co-administration of ketoconazole and irinotecan to cancer patients resulted in a significant increase in the formation of SN-38, at least partially due to the inhibitory effect of ketoconazole on UGT1A1 [19,20]. Thus, it is important to investigate the probable interaction of irinotecan with other anti-cancer agents and to prevent such treatment-related toxicity.
Hydroxamic acid histone deacetylase inhibitors have emerged as a promising class of anti-cancer drugs in recent years [21,22]. They enhance histone acetylation and increase the expression of tumour suppressor genes, thereby inducing growth arrest and apoptosis of cancer cells [23–25]. It is also believed that they are capable of sensitising drug resistant cancer cells to anticancer drugs in combination therapy [26, 27]. The most commonly used HDACis in the hydroxamate class include belinostat, vorinostat and panobinostat. It has been shown that all 3 HDACis undergo intensive metabolism via phase II glucuronidation [28–30]. In particular, belinostat, like irinotecan, has recently been discovered to utilise the same phase II metabolic pathway involving the highly polymorphic enzyme, UGT1A1 [28]. Therefore, we hypothesise that HDACis may inhibit the UGT1A isoforms in phase II metabolism, resulting in reduced conversion of SN-38 to SN-38G and greater accumulation of SN-38.
Our objective was to investigate the potential presence of glucuronidation-mediated DDI in combination therapies of irinotecan with HDACis. Belinostat was approved for peripheral T-cell lymphoma (PTCL) by FDA recently [31], whereas vorinostat is FDA-approved for cutaneous T-cell lymphoma (CTCL) [32]. This investigation would then play a vital role in helping clinicians make more informed decisions regarding possible combination chemotherapy. Moreover, our results would also provide vital information to formulation scientists if these combinations of drugs are considered to be formulated together.
In this study, we assessed the inhibitory effects of belinostat on SN-38 glucuronidation in vitro using UGT1A1 enzymes, and its inhibitory effects were further confirmed using 3 types of pooled human liver microsomes (HLMs) (50 donor pool, UGT1A1*1*28 and UGT1A1*28*28 allelic variants). Significant associations were observed between the UGT1A1*28 polymorphisms and SN-38G formation rates in the absence and presence of belinostat. Besides, we also studied the possible inhibitory effects of other HDACis, including vorinostat and panobinostat on SN-38 glucuronidation.
RESULTS
LC-MS/MS method validation
The chromatographic data were acquired and analysed using the Analyst v1.4.2 software package (Applied Biosystems/MDS SCIEX). The LC-MS/MS analysis was highly specific and selective as the peaks have a symmetrical resolution, with no interference around the retention time (tR) of the analytes. The retention times were as follows: SN38 and SN-38-d3 eluting at tR = 4.24 min, and SN-38G and SN-38G-d3 eluting at tR = 3.97 min (Figure 1). Accuracy was expressed as the percentage of the mean value measured over the true value of QC samples, whereas precision at each QC concentration was expressed as the coefficient of variation by calculating the standard deviation as a percentage of the mean calculated concentration. Briefly, excellent linearity of SN-38 and SN-38G was demonstrated to be within the range of 10 to 500 nmol/L. A weighting factor of 1/x was used to minimise deviations from the slope of the curve at lower concentrations resulting from bigger variance at higher concentrations. Good linearity was achieved, with a coefficient of determination (r2) of > 0.996 for both SN-38 and SN-38G. This implied a strong linear relationship between the peak area ratio and concentration. The intra-day and inter-day precisions and accuracies for QC samples were summarised in Table 1. The intra-day precision was between 1.26-2.41% for SN-38 and 1.62-6.24% for SN-38G and the inter-day precision was 1.67-3.38% for SN-38 and 4.22-8.30% for SN-38G. The accuracy was between 91.14%-100.78% for SN-38 and 91.45%-105.60% for SN-38G. All these data met the requirement of the FDA guidelines [33]. Therefore, this method was considered sufficiently accurate, sensitive and reproducible for the simultaneous determination of SN-38 and SN-38G throughout a wide dynamic range.
Figure 1. Representative chromatograms of SN-38, SN-38-d3, SN-38G and SN-38G-d3.
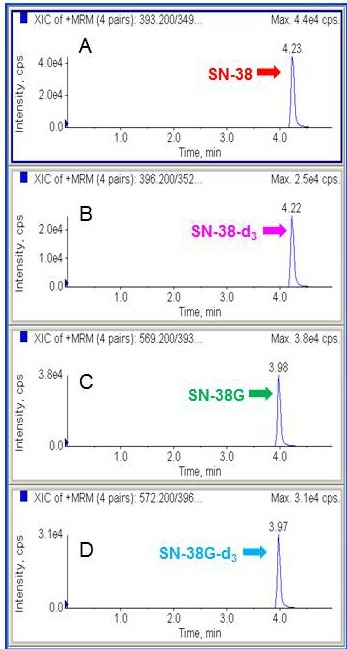
Peak of SN-38 A. Peak of SN-38 internal standard using isotopic sister of the analyte, SN-38-d3 B. Peak of SN-38G C. Peak of SN-38G internal standard, SN-38G-d3 D.
Table 1. Intra-day and inter-day precision and accuracy of SN-38 (A) and SN-38G (B).
| (A) | QC samples | |||||
|---|---|---|---|---|---|---|
| Intra-day (n = 3) | Inter-day (n = 3) | |||||
| Nominal conc. (nmol/L) |
Quantified conc. (Mean ± S.D, nmol/L) |
Accuracy (%) | CV (%) | Quantified conc. (Mean ± S.D, nmol/L) |
Accuracy (%) | CV (%) |
| 30 | 27.33 ± 0.44 | 91.14 | 1.59 | 29.15 ± 1.80 | 97.28 | 6.24 |
| 150 | 147.00 ± 3.54 | 97.94 | 2.41 | 144.50 ± 2.33 | 96.20 | 1.62 |
| 350 | 352.67 ±4.46 | 100.78 | 1.26 | 346.00 ± 9.26 | 98.80 | 2.69 |
| (B) | QC samples | |||||
|---|---|---|---|---|---|---|
| Intra-day (n = 3) | Inter-day (n = 3) | |||||
| Nominal conc. (nmol/L) |
Quantified conc. (Mean ± S.D, nmol/L) |
Accuracy (%) | CV (%) | Quantified conc. (Mean ± S.D, nmol/L) |
Accuracy (%) | CV (%) |
| 30 | 27.43 ± 0.46 | 91.45 | 1.67 | 27.93 ± 1.93 | 93.18 | 6.92 |
| 150 | 143.67 ± 4.02 | 95.71 | 2.80 | 145.00 ± 6.10 | 96.60 | 4.22 |
| 350 | 369.00 ± 12.46 | 105.36 | 3.38 | 369.70 ± 30.68 | 105.60 | 8.30 |
Effects of belinostat on SN-38 glucuronidation by UGT1A1
As the concentration of belinostat increased, the formation rate of SN-38G decreased in a dose-dependent manner, demonstrating an increase in the inhibition of SN-38 glucuronidation (Figure 2A), thus suggesting that the inhibition type was non-competitive. Upon addition of belinostat at 100 and 200 μmol/L, intrinsic clearance (CLint) was markedly decreased by ~ 62% (from 21.59 to 8.28 μL/min/mg protein) and 81% (from 21.59 to 4.20 μL/min/mg protein) respectively (p < 0.05) (Figure 2B).
Figure 2. Enzyme kinetics of glucuronidation of SN-38 by UGT1A1.
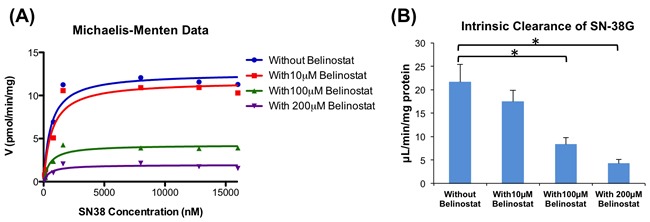
Michaelis-Menten graph of SN-38G formation when different concentrations of SN-38 were incubated with belinostat (0, 10, 100 and 200 μmol/L) A. Apparent intrinsic clearance of SN-38G B. By using a paired t-test, the incubations with 100 and 200 μmol/L belinostat were significantly different (p < 0.05) from the incubation without belinostat (*).
Effects of belinostat on SN-38 glucuronidation by pooled HLMs
Belinostat was also found to strongly inhibit SN-38 glucuronidation by the 3 types of pooled HLMs (50 donor pool, UGT1A1*1*28 and UGT1A1*28*28 allelic variants) in a dose-dependent manner (Figure 3). The 50 donor pool HLMs, were used to represent the wild-type homozygote for (TA)6TAA allele (6/6), while the UGT1A1*1*28 and UGT1A1*28*28 allelic variant HLMs are heterozygote (6/7) and homozygote (7/7) for (TA)7TAA allele, respectively. Based on the analysis of the Michaelis-Menten graphs (Figure 3A, 3C and 3E), belinostat also showed non-competitive inhibition against SN-38 glucuronidation by HLMs. In the 50 donor pool HLMs (6/6) (Figure 3B), belinostat showed the highest inhibitory effect at 200 μmol/L, significantly decreasing the CLint by 68%, from 81.25 to 26.11 μL/min/mg protein (p < 0.05). At 100 μmol/L of belinostat, CLint was also significantly decreased by 56%, from 81.25 to 36.01 μL/min/mg protein (p < 0.05). In the UGT1A1*1*28 (6/7) and UGT1A1*28*28 (7/7) allelic variant HLMs (Figure 3D and 3F), similar significant decreases were detected, where CLint dropped by 79% (from 29.22 to 6.10 µL/min/mg protein) and 75% (from 5.40 to 1.34 µL/min/mg protein), respectively at 200 μmol/L of belinostat (p < 0.05).
Figure 3. Enzyme kinetics of glucuronidation of SN-38 by human liver microsomes (HLM).
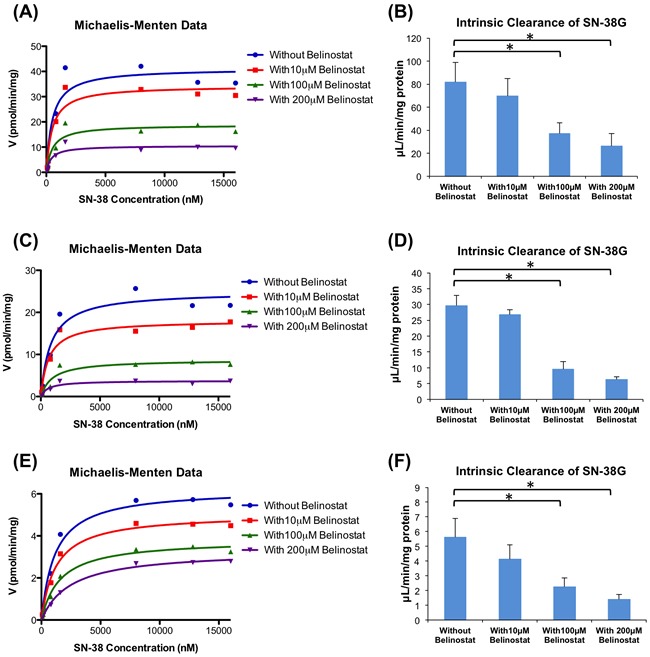
Michaelis-Menten graphs of SN-38G formation when different concentrations of SN-38 were incubated with belinostat (0, 10, 100 and 200 μmol/L) and 6/6 genotype A., 6/7 genotype C. and 7/7 genotype E.; Apparent intrinsic clearance of SN-38G in 6/6 genotype B., 6/7 genotype D. and 7/7 genotype F. By using a paired t-test, the incubations with 100 and 200 μmol/L belinostat were significantly different (p < 0.05) from the incubation without belinostat (*).
Our results also suggested the association of UGT1A1 genetic polymorphism and the inhibitory effect of belinostat on SN-38G formation. As illustrated in Figure 4A, SN-38 glucuronidation activities (Vmax) decreased significantly (p < 0.05) in the samples using variant 6/7 and 7/7 genotypes (24.96 pmol/min/mg and 6.23 pmol/min/mg, respectively), as compared to the samples with wild type 6/6 genotypes (41.13 pmol/min/mg). Moreover, co-administration of 10, 100, or 200 μmol/L belinostat and SN-38 led to further reduction in the SN-38G formation rates and this reduction occurred in a dose-dependent manner (Figure 4B-4D). In particular for 200 μmol/L of belinostat (Figure 4D), the rates of SN-38G formation were nearly similar for both variant 6/7 heterozygous and 7/7 homozygous genotypes, at 3.77 pmol/min/mg (decreased by ~ 64% compared to wild type) and 3.30 pmol/min/mg (decreased by ~ 68% compared to wild type), respectively, suggesting that belinostat, at high concentrations, is able to inhibit both heterozygous and homozygous genotypes to a similar extent. This implied that patients with heterozygous genotype may develop severe toxicity similar to that of the homozygous genotype, if a high dose of belinostat is prescribed. Since large populations worldwide are found to have heterozygous UGT1A1*28 genotype and it is thought to be at a lower risk of developing irrinotecan-related toxicity as compared to the homozygous genotype [14, 36], our finding can be clinically relevant as it may prompt extra caution when the combination of irinotecan and belinostat is given to patients with heterozygous genotype. Further investigation is warranted to confirm this finding in vivo.
Figure 4.
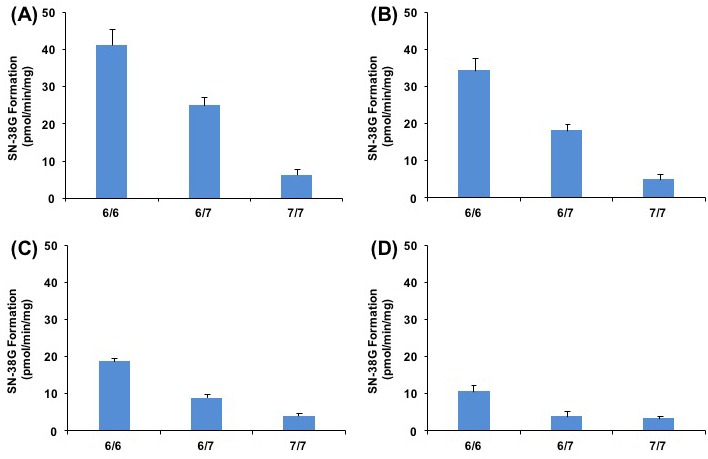
Relationship of UGT1A1*28 genotype with SN-38G formation rates (Vmax) at different concentration of belinostat in presence of 0 A., 10 B., 100 C., or 200 μmol/L D., on SN-38 glucuronidation.
Effects of vorinostat on SN-38 glucuronidation by UGT1A9
At a therapeutic concentration of vorinostat (2 µmol/L), no significant inhibition of UGT1A9 was observed. CLint of SN-38G increased from 7.48 to 12.12 μL/min/mg protein when incubated without and with vorinostat respectively, but the increase was found to be insignificant (p > 0.05).
Effects of panobinostat on SN-38 glucuronidation by UGT1A isoforms
Panobinostat, at both concentrations (20 and 200 nmol/L), was not found to have any inhibitory effect on all 3 UGT1A isoforms tested. Comparing 20 nmol/L and 200 nmol/L panobinostat to the incubations without panobinostat, the CLint increases were all insignificant (p > 0.05).
DISCUSSION
To our knowledge, this is the first report describing glucuronidation-mediated drug interaction of irinotecan with hydroxamate HDACis. In order to determine the concentration of SN38 and SN38G, a sensitive and robust LC-MS/MS method has been successfully developed and validated according to the FDA guidelines [33]. Excellent linearity was achieved with good precisions and accuracies for SN38 and SN38G. We have demonstrated that belinostat inhibits UGT1A1 non-competitively in a dose-dependent manner by decreasing the glucuronidation of SN-38, the active metabolite of irinotecan. However, this was not the case for vorinostat and panobinostat. This is consistent with other studies, which showed that SN-38 and belinostat are primarily deactivated by UGT1A1 [15, 28]. UGT1A1 has also been reported to have more than 1 binding site, and there are several known non-competitive inhibitors of UGTs enzymes, such as phenylbutazone and quinine [34]. However, the molecular basis of belinostat's non-competitive inhibition is unknown.
There has been interest in developing combination therapy with irinotecan and HDACis like vorinostat [35]. Notably, our results suggested that taking belinostat orally (simulated by its concentration at 10 μmol/L) with irinotecan would not have significant interactions, whereas using a high-dose intravenous belinostat infusion (simulated by its concentration at 200 μmol/L) with irinotecan could pose severe DDI problems. The data suggested that both drugs should not be simultaneously administered together via intravenous infusion. If this combination is needed, human interaction studies should be performed to establish the safety of doses administered. Furthermore, our novel findings can be useful for formulation scientists as they may design combination of anti-cancer products in the future to combat drug-resistant cancers. Our results suggest that irinotecan and belinostat should not be combined and formulated together for use due to their potential DDI, which may cause SN-38 driven toxicity. This finding would be of importance, especially since belinostat was FDA approved in 2014 [31].
Using recombinant UGT1A9, we observed that SN-38G formation was not reduced by vorinostat at its therapeutic concentration of 2 μmol/L, when given orally [32]. Clinically, our results suggested that there should be no drug interaction problem when administering oral vorinostat with intravenous irinotecan. In the future, formulation scientists could combine these 2 drugs in a formulation if it is shown that they have increased efficacy in treating cancer.
Currently, there have not been any studies demonstrating the UGT isoforms that are responsible for the glucuronidation of panobinostat. As such, we decided to study the isoforms (UGT1A1, UGT1A6 and UGT1A7) involved in SN-38 glucuronidation and investigate whether panobinostat inhibits any of these isoforms. In our present work, the results obtained suggested that there is no glucuronidation-mediated DDI between panobinostat and irinotecan via UGT1A1, UGT1A6 and UGT1A7. Hence, there should be no drug interaction problems when administering intravenous irinotecan with oral or intravenous panobinostat. Therefore, irinotecan and panobinostat can be used safely as a combination therapy in a clinical setting, and formulation scientists can formulate such combination products in the future.
Most importantly, it was stated and shown earlier in our findings that genetic polymorphic expression of UGT1A1 can cause inter-individual variability in irinotecan pharmacokinetics and pharmacodynamics [3]. Critically, the DDI between irinotecan and belinostat was found to be worsened to the same extent for both the UGT1A1*28 heterozygous and homozygous genotypes, highlighting the potential clinical significance as a larger proportion of patients with heterozygous genotype may have the same risk of developing severe toxicity as those of the homozygous genotype. It is noteworthy that the Caucasian population is particularly at risk, as 11.5% are UGT1A1*28 homozygotes and 51.4% are heterozygotes, increasing the total population at risk of severe toxicity to 66.9% [36]. Also in Asians, the Indian population is most at risk, as 1.7% are UGT1A1*28 homozygotes and 25% are heterozygotes, bringing more than a quarter of the population (26.7%) to a risk of severe toxicity [37].
Currently, individualised treatment of irinotecan can be tailored for patients who have been genotyped with the UGT1A1*28 allele, which predisposes them to higher risks of neutropenia [36]. However, it has also been shown that UGT1A1*28 is associated with reduced belinostat glucuronidation, which suggests that patients with this genotype may potentially have higher exposure to active belinostat resulting from impaired clearance of belinostat [17, 18, 28]. This has serious implications, as a higher concentration of belinostat due to the UGT1A1 mutation will result in a much stronger inhibition of SN-38 glucuronidation, thereby causing severe toxicity accordingly. Internationally, this polymorphism is also more common in Caucasians and Africans than Asians, with an allelic frequency of about 3-fold or more in Caucasian and African counterparts [38]. Thus, this suggests a greater risk of severe toxicity if combination therapy containing irinotecan and belinostat is given to patients harbouring the commonest genetic variant of UGT1A1.
Therefore, irinotecan and belinostat should be administered together with caution, and our results need careful evaluation in human interaction studies. As the results of our study could be clinical significant due to the high frequency of genetic variants of UGT1A1, we strongly believe that further investigations into the potential inhibition of other drugs including new substrates and inhibitors of UGT1A1 on irinotecan metabolism are warranted due to their severe adverse effects in combination therapies.
MATERIALS AND METHODS
Chemicals and reagents
SN-38, SN-38G and their internal standards, SN-38-d3 and SN-38G-d3 respectively, were obtained from Toronto Research Chemicals (Toronto, ON, Canada). Belinostat, vorinostat and panobinostat, were purchased from Selleck Chemicals (Houston, TX, USA). Human recombinant UDP-glucuronosyltransferases (UGT1A1, UGT1A6, UGT1A7, UGT1A9), UGT control, UGT reaction mix - solutions A and B, HLMs (50 donor pool, UGT1A1*1*28 and UGT1A1*28*28 allelic variants) were purchased from BD Gentest (San Jose, CA, USA). All other reagents were of HPLC grade or of the highest grade commercially available.
Liquid chromatography-tandem mass spectrometry analysis of SN-38 and SN-38G
The protein precipitation with acetonitrile was adopted for sample preparation for determination of SN-38 and SN-38G, similar toa previously described method in which methanol was used as a a protein precipitating agent [39]. Ten microliter aliquots were injected into a high-performance liquid chromatograph assay system (Agilent 1100, Germany) with tandem mass spectrometric detection (API 4000 triple-quadrupole mass spectrometer; AB Sciex, Concord, Canada). Baseline separation of the analytes was achieved on the Alltima C18 (150 mm x 2.1 mm, 5 μm) column. The mobile phase A and B consisted of 100% acetonitrile and 0.1% formic acid, respectively. Gradient elution was done for 5 minutes at the flow rate of 0.4 mL/min using the following stepwise gradient: 0 to 0.1 minutes, 84% A and 16% B; 0.1 to 1.6 minutes, 16% A and 64% B; and 1.6 to 5 minutes, back to 84% A and 16% B.
Incubations of SN-38 with belinostat
The incubation mixture (50 µL) consisted of SN-38 (80 - 16000 nmol/L), belinostat at different concentrations (0, 10, 100 and 200 µmol/L), MgCl2 (8 mmol/L), Tris-HCl buffer at pH 7.4 (50 mmol/L), alamethicin in dH2O (0.025 mg/mL) and UDPGA (5 mmol/L) in Eppendorf tubes. After pre-incubation, the reaction was started with the addition of UGT1A1 or pooled HLMs (0.5 mg/mL). Reaction mixtures were incubated at 37°C for 30 minutes. To ensure clinical relevance, the Cmax of SN-38 in clinical trials falls within the range being studied [40]. Also, belinostat at 10 µmol/L, 100 µmol/L and 200 µmol/L were chosen as they represented the Cmax of oral, low-dose and high-dose intravenous administration respectively [41–43]. Appropriate control reactions were also conducted with UGT control as the microsomal protein in a similar fashion.
Incubations of SN-38 with vorinostat
SN-38 was incubated in the presence of vorinostat (0 and 2 µmol/L) and UGT1A9 (0.5 mg/mL). Vorinostat 2 µmol/L was chosen as it represented the Cmax following oral administration. UGT1A9 was chosen since vorinostat and SN-38 share this similar minor metabolic pathway [29].
Incubations of SN-38 with panobinostat
SN-38 was incubated in the presence of panobinostat (0, 20 and 200 nmol/L) and various enzymes in the UGT1A family (UGT1A1, UGT1A6, UGT1A7 at 0.5 mg/mL). Panobinostat at 20 nmol/L and 200 nmol/L were chosen as they represented the Cmax following oral and intravenous administration respectively [44]. UGT1A1, UGT1A6 and UGT1A7 were chosen since these UGTs could be responsible for glucuronidating panobinostat [45].
Data analysis
All experiments were carried out in triplicate. SN-38G formation velocity (ν) was calculated as Cm, 30 min/incubation time/microsomal protein concentration, where Cm, 30 min was the metabolite concentration after 30 min incubation. Plots of substrate concentration, S (X axis) versus ν (Y axis) were then constructed. Km and Vmax were calculated by the Michaelis-Menten Equation 1 below using GraphPad software v5.01 (GraphPad Software, Inc., San Diego, CA, USA). The kinetic parameter Vmax/Km, which is equivalent to intrinsic clearance (CLint) (Equation 2), was derived from the slope of the plot of the rate of product formation versus substrate concentration. Comparisons of CLint between two groups were performed by a paired t-test. Data were considered statistically significant when p < 0.05.
ν = [S] x Vmax/([S] + Km) Equation 1
CLint = Vmax/Km Equation 2
Acknowledgments
This research is supported by the National Research Foundation Singapore and the Singapore Ministry of Education under their Research Centres of Excellence (RCE) Initiative; and Clinician Scientist Award (Senior Investigator Category) By the NMRC for Translational Pipeline: Developing novel therapeutics for cancer treatment, including the role of histone deacetylase inhibitor. The authors extend their appreciation to the International Scientific Partnership Program ISPP at King Saud University for funding this research work through ISPP#0091.
Footnotes
CONFLICTS OF INTEREST
The authors declare that no competing interests exist.
REFERENCES
- 1.Beijnen JH, Schellens JH. Drug interactions in oncology. The Lancet Oncology. 2004;8:489–496. doi: 10.1016/S1470-2045(04)01528-1. [DOI] [PubMed] [Google Scholar]
- 2.Hirose K, Kozu C, Yamashita K, Maruo E, Kitamura M, Hasegawa J, Omoda K, Murakami T, Maeda Y. Correlation between plasma concentration ratios of SN-38 glucuronide and SN-38 and neutropenia induction in patients with colorectal cancer and wild-type UGT1A1 gene. Oncology Letters. 2012;3:694–698. doi: 10.3892/ol.2011.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith NF, Figg WD, Sparreboom A. Pharmacogenetics of irinotecan metabolism and transport: an update. Toxicology In Vitro. 2006;2:163–175. doi: 10.1016/j.tiv.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 4.Creemers GJ, Lund B, Verweij J. Topoisomerase I inhibitors: topotecan and irenotecan. Cancer Treatment Reviews. 1994;1:73–96. doi: 10.1016/0305-7372(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 5.Douillard J, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, Gruia G, Awad L, Rougier P. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. The Lancet. 2000;9209:1041–1047. doi: 10.1016/s0140-6736(00)02034-1. [DOI] [PubMed] [Google Scholar]
- 6.Humerickhouse R, Lohrbach K, Li L, Bosron WF, Dolan ME. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Research. 2000;5:1189–1192. [PubMed] [Google Scholar]
- 7.Rivory LP, Bowles MR, Robert J, Pond SM. Conversion of irinotecan (CPT-11) to its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), by human liver carboxylesterase. Biochemical Pharmacology. 1996;7:1103–1111. doi: 10.1016/0006-2952(96)00457-1. [DOI] [PubMed] [Google Scholar]
- 8.Guichard S, Terret C, Hennebelle I, Lochon I, Chevreau P, Frétigny E, Selves J, Chatelut E, Bugat R, Canal P. CPT-11 converting carboxylesterase and topoisomerase I activities in tumour and normal colon and liver tissues. British Journal of Cancer. 1999;80:364–370. doi: 10.1038/sj.bjc.6690364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senter PD, Beam KS, Mixan B, Wahl AF. Identification and activities of human carboxylesterases for the activation of CPT-11, a clinically approved anticancer drug. Bioconjugate Chemistry. 2001;6:1074–1080. doi: 10.1021/bc0155420. [DOI] [PubMed] [Google Scholar]
- 10.Rivory LP, Robert J. Identification and kinetics of a -glucuronide metabolite of SN-38 in human plasma after administration of the camptothecin derivative irinotecan. Cancer Chemotherapy and Pharmacology. 1995;2:176–179. doi: 10.1007/BF00689205. [DOI] [PubMed] [Google Scholar]
- 11.Iyer L, King CD, Whitington PF, Green MD, Roy SK, Tephly TR, Coffman BL, Ratain MJ. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. Journal of Clinical Investigation. 1998;4:847–854. doi: 10.1172/JCI915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagné JF, Montminy V, Belanger P, Journault K, Gaucher G, Guillemette C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38) Molecular Pharmacology. 2002;3:608–617. doi: 10.1124/mol.62.3.608. [DOI] [PubMed] [Google Scholar]
- 13.de Jong FA, de Jonge MJ, Verweij J, Mathijssen RH. Role of pharmacogenetics in irinotecan therapy. Cancer Letters. 2006;1:90–106. doi: 10.1016/j.canlet.2005.04.040. [DOI] [PubMed] [Google Scholar]
- 14.Hanioka N, Ozawa S, Jinno H, Ando M, Saito Y, Sawada J. Human liver UDP-glucuronosyltransferase isoforms involved in the glucuronidation of 7-ethyl-10-hydroxycamptothecin. Xenobiotica. 2001;10:687–699. doi: 10.1080/00498250110057341. [DOI] [PubMed] [Google Scholar]
- 15.Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11) Clinical Cancer Research. 2001;8:2182–2194. [PubMed] [Google Scholar]
- 16.Iyer L, Das S, Janisch L, Wen M, Ramírez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. The Pharmacogenomics Journal. 2002;1:43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 17.Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL. UGT1A1* 28 genotype and irinotecan-induced neutropenia: dose matters. Journal of the National Cancer Institute. 2007;17:1290–1295. doi: 10.1093/jnci/djm115. [DOI] [PubMed] [Google Scholar]
- 18.Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, Yokoyama A, Saitoh S, Shimokata K, Hasegawa Y. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Research. 2000;24:6921–6926. [PubMed] [Google Scholar]
- 19.Kehrer DF, Mathijssen RH, Verweij J, de Bruijn P, Sparreboom A. Modulation of irinotecan metabolism by ketoconazole. J Clin Oncol. 2002;14:3122–3129. doi: 10.1200/JCO.2002.08.177. [DOI] [PubMed] [Google Scholar]
- 20.Yong WP, Ramirez J, Innocenti F, Ratain MJ. Effects of ketoconazole on glucuronidation by UDP-glucuronosyltransferase enzymes. Clin Cancer Res. 2005;18:6699–6704. doi: 10.1158/1078-0432.CCR-05-0703. [DOI] [PubMed] [Google Scholar]
- 21.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nature Reviews Drug Discovery. 2002;4:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 22.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. Journal of Cellular Biochemistry. 2009;4:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanji N, Ozawa A, Kikugawa T, Miura N, Sasaki T, Azuma K, Yokoyama M. Potential of histone deacetylase inhibitors for bladder cancer treatment. Expert Review of Anticancer Therapy. 2011;6:959–965. doi: 10.1586/era.10.230. [DOI] [PubMed] [Google Scholar]
- 24.Bapat SA. Modulation of gene expression in ovarian cancer by active and repressive histone marks. Epigenomics. 2010;1:39–51. doi: 10.2217/epi.09.38. [DOI] [PubMed] [Google Scholar]
- 25.Piekarz RL, Bates SE. Epigenetic modifiers: basic understanding and clinical development. Clinical Cancer Research. 2009;12:3918–3926. doi: 10.1158/1078-0432.CCR-08-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fazzone W, Wilson PM, Labonte MJ, Lenz HJ, Ladner RD. Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. International Journal of Cancer. 2009;2:463–473. doi: 10.1002/ijc.24403. [DOI] [PubMed] [Google Scholar]
- 27.Kim JC, Shin ES, Kim CW, Roh SA, Cho DH, Na YS, Kim TW, Kim MB, Hyun YL, Ro S, Kim SY, Kim YS. In vitro evaluation of histone deacetylase inhibitors as combination agents for colorectal cancer. Anticancer Research. 2009;8:3027–3034. [PubMed] [Google Scholar]
- 28.Wang LZ, Ramírez J, Yeo W, Chan MY, Thuya WL, Lau JY, Wan SC, Wong AL, Zee YK, Lim R, Lee SC, Ho PC, Lee HS, et al. Glucuronidation by UGT1A1 Is the Dominant Pathway of the Metabolic Disposition of Belinostat in Liver Cancer Patients. PloS One. 2013;1:e54522. doi: 10.1371/journal.pone.0054522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balliet RM, Chen G, Gallagher CJ, Dellinger RW, Sun D, Lazarus P. Characterization of UGTs active against SAHA and association between SAHA glucuronidation activity phenotype with UGT genotype. Cancer Research. 2009;7:2981–2989. doi: 10.1158/0008-5472.CAN-08-4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clive S, Woo MM, Nydam T, Kelly L, Squier M, Kagan M. Characterizing the disposition, metabolism, and excretion of an orally active pan-deacetylase inhibitor, panobinostat, via trace radiolabeled 14C material in advanced cancer patients. Cancer Chemotherapy and Pharmacology. 2012;4:513–522. doi: 10.1007/s00280-012-1940-9. [DOI] [PubMed] [Google Scholar]
- 31.Beleodaq, in [package insert] 2014, Spectrum Pharmaceuticals, Inc. Irvine, CA: [Google Scholar]
- 32.Zolinza, in [package insert] 2013, Merck & Co., Inc. Whitehouse Station, NJ: [Google Scholar]
- 33.US Food and . Drug Administration, Guidance for Industry: Bioanalytical Method Validation, 2001, US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Rockville, MD: [Google Scholar]
- 34.Liu Y, Ramírez J, House L, Ratain MJ. The UGT1A1*28 Polymorphism Correlates with Erlotinib's Effect on SN-38 Glucuronidation. Eur J Cancer. 2010;46:2097–2103. doi: 10.1016/j.ejca.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chinnaiyan P, Chowdhary S, Potthast L, Prabhu A, Tsai YY, Sarcar B, Kahali S, Brem S, Yu HM, Rojiani A, Murtagh R, Pan E. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro Oncol. 2012;1:93–100. doi: 10.1093/neuonc/nor187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sciences Health Authority Association between UGT1A1 variant alleles and irinotecan-induced severe neutropenia. Adverse Drug Reaction News. 2010;12:1. [Google Scholar]
- 37.Stingl JC, Bartels H, Viviani R, Lehmann ML, Brockmöller J. Relevance of UDP-glucuronosyltransferase polymorphisms for drug dosing: A quantitative systematic review. Pharmacol Ther. 2014;1:92–116. doi: 10.1016/j.pharmthera.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 38.Beutler E, Gelbart T, Demina A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA. 1998;14:8170-–8174. doi: 10.1073/pnas.95.14.8170. 0-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Peer CJ, Alfaro R, Tian T, Spencer SD, Figg WD. Quantification of irinotecan, SN38, and SN38G in human and porcine plasma by ultra high-performance liquid chromatography-tandem mass spectrometry and its application to hepatic chemoembolization. J Pharm Biomed Anal. 2012;62:140–148. doi: 10.1016/j.jpba.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camptosar, in [package insert] 2012, Pfizer Inc. New York, NY: [Google Scholar]
- 41.Steele N, Plumb JA, Vidal L, Tjørnelund J, Knoblauch P, Buhl-Jensen P, Molife R, Brown R, de Bono JS, Evans TR. Pharmacokinetic and pharmacodynamic properties of an oral formulation of the histone deacetylase inhibitor Belinostat (PXD101) Cancer Chemotherapy and Pharmacology. 2011;6:1273–1279. doi: 10.1007/s00280-010-1419-5. [DOI] [PubMed] [Google Scholar]
- 42.Steele NL, Plumb JA, Vidal L, Tjørnelund J, Knoblauch P, Rasmussen A, Ooi CE, Buhl-Jensen P, Brown R, Evans TR, DeBono JS. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clinical Cancer Research. 2008;3:804–810. doi: 10.1158/1078-0432.CCR-07-1786. [DOI] [PubMed] [Google Scholar]
- 43.Yeo W, Chung HC, Chan SL, Wang LZ, Lim R, Picus J, Boyer M, Mo FK, Koh J, Rha SY, Hui EP, Jeung HC, Roh JK, et al. Epigenetic Therapy Using Belinostat for Patients With Unresectable Hepatocellular Carcinoma: A Multicenter Phase I/II Study With Biomarker and Pharmacokinetic Analysis of Tumors From Patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. Journal of Clinical Oncology. 2012;27:3361–3367. doi: 10.1200/JCO.2011.41.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catalano MG, Pugliese M, Gargantini E, Grange C, Bussolati B, Asioli S, Bosco O, Poli R, Compagnone A, Bandino A, Mainini F, Fortunati N, Boccuzzi G. Cytotoxic activity of the histone deacetylase inhibitor panobinostat (LBH589) in anaplastic thyroid cancer in vitro and in vivo. International Journal of Cancer. 2012;3:694–704. doi: 10.1002/ijc.26057. [DOI] [PubMed] [Google Scholar]
- 45.Rios GR, Tephly TR. Inhibition and active sites of UDP-glucuronosyltransferases 2B7 and 1A1. Drug Metabolism and Disposition. 2002;12:1364–1367. doi: 10.1124/dmd.30.12.1364. [DOI] [PubMed] [Google Scholar]