

Dear Editor
The ichthyoses are rare skin disorders linked by the common finding of scale and concomitant barrier function abnormalities. Recently, mutations in PNPLA1 which encodes patatin-like phopholipase domain containing 1, and plays a role in the formation of the epidermal lipid barrier, have been identified as rare cause of non-syndromic ARCI1–6.
We identified subjects with PNPLA1 mutations within our registry of 732 ichthyosis kindreds. DNA was isolated from blood, and either multiplex targeted next generation sequencing (NGS) of 42 genes known to cause disorders of keratinization or WES (Supplementary Table 1) was performed. Fourteen unrelated ARCI probands were found to be compound heterozygous or homozygous for mutations in PNPLA1 which were confirmed with Sanger sequencing. PNPLA1 mutations segregated with disease in all kindreds, five of which were consanguineous (Supplementary Figures 1–14).
A total of 16 different PNPLA1 mutations were observed, including two which result in early termination, a splice site mutation, and 13 missense substitutions at conserved residues (Figure 1, Supplementary Figure 15). All missense mutations are within the more highly conserved N-terminal half of the protein, and all but two are clustered within the patatin domain. Two subjects are homozygous for missense mutations at S53, the nucleophilic serine in the putative lipid hydrolase site, and one is homozygous for a missense mutation at D172, the other critical residue in the catalytic dyad.
Figure 1. Spectrum of cutaneous phenotypes and PNPLA1 mutation sites in subjects with ichthyosis.
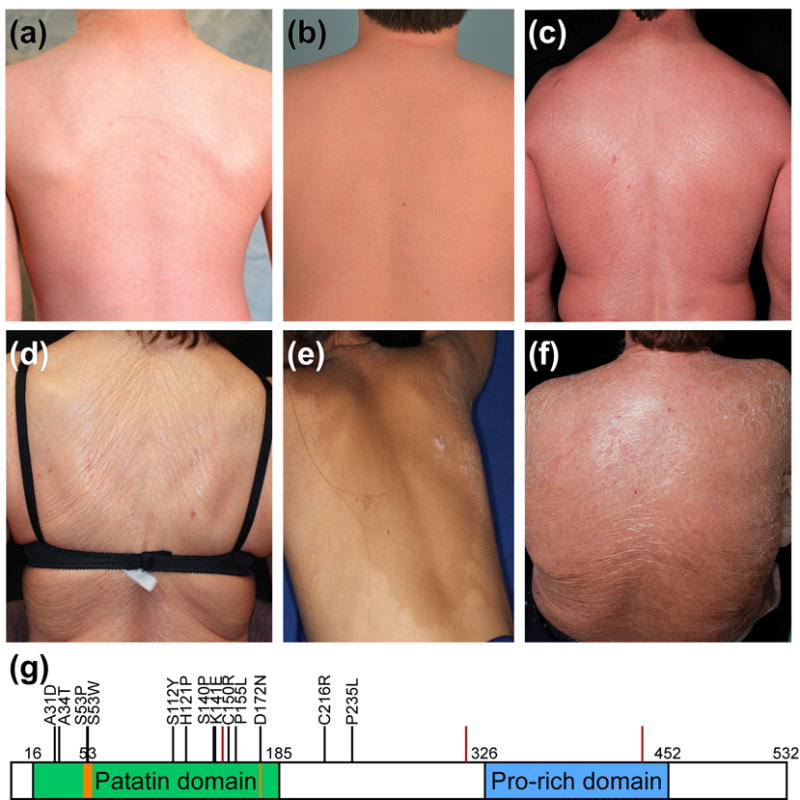
Extent and severity of erythema and scale vary significantly and include: (a) ICH136-1 and (b) ICH431-1, mild erythema and fine white scale; (c) ICH201-4, moderate-severe erythema and fine white scale; (d) ICH162-2 and (e) ICH561-1, minimal erythema and plate-like scale; and (f) ICH201-1, moderate-to-severe erythema with plate-like scale. (g) PNPLA1 protein domains: patatin domain (green), lipid hydrolase catalytic dyad (orange), and proline-rich domain (blue) are indicated; numbers specify amino acid position. Locations of mutations reported herein are shown with black bars and the amino acid change (missense mutations) or red bars (splice site and frameshift mutations).
The phenotypes of all subjects with PNPLA1 mutations are described in Table 1, with representative clinical photos provided in Supplementary Figures 1–14. At birth, seven subjects presented with a collodion membrane (one with vernix-like hyperkeratosis), and eight showed generalized erythema and/or scaling. Mature phenotypes include scale that may be fine or plate-like, and erythema ranging from minimal to severe (Figure 2). The presence and degree of ectropion and palmoplantar keratoderma are variable, although they are either absent or mild in the majority of subjects. Only seven of 19 subjects were born with a collodion membrane.
Table 1.
Characteristics of subjects with PNPLA1 mutations.
| Subject | Mutation(s) | Neonatal Presentation | Scale | Erythema | Ectropion | PPK | Consanguinity |
|---|---|---|---|---|---|---|---|
| ICH136-1 | c.335C>A; p.S112Y c.464C>T; p.P155L |
collodion1 | fine white | mild | none | none | no |
| ICH162-1 | c.92C>A; p.A31D c.464C>T; p.P155L |
collodion | plate-like | minimal | severe | mild | no |
| ICH162-2 | c.92C>A; p.A31D c.464C>T; p.P155L |
Non-collodion, presented in infancy with scale and erythema | plate-like | minimal | mild2 | mild | no |
| ICH201-1 | c.100G>A; p.A34T c.418T>C; p.S140P |
generalized scale & erythema | plate-like | moderate | mild | mild | no |
| ICH201-3 | c.418T>C; p.S140P homozygous | generalized scale & erythema | fine white | moderate-severe | none | mild | no |
| ICH201-4 | c.418T>C; p.S140P homozygous | generalized scale & erythema | fine white | moderate-severe | none | none | no |
| ICH215-1 | c.514G>A; p.D172N homozygous | collodion | unknown3 | no | |||
| ICH422-1 | c.418T>C; p.S140P c.448T>C; p.C150R |
collodion | fine white | mild | none | none | no |
| ICH431-1 | c.1300del.G; p.A434fs homozygous | Non-collodion, presented at 1 month with scale and erythema | fine white | mild | none | none | no |
| ICH454-1 | c.646T>C; p.C216R homozygous | generalized scale | plate-like | minimal | none | mild | yes |
| ICH454-3 | c.646T>C; p.C216R homozygous | generalized scale | plate-like | minimal | none | mild | yes |
| ICH454-4 | c.646T>C; p.C216R homozygous | generalized scale | plate-like | minimal | none | mild | yes |
| ICH459-1 | c.362A>C; p.H121P c.438+2C>G |
collodion | fine white | minimal | none | none | no |
| ICH561-1 | c.1300del.G; p.A434fs homozygous | unknown | extremities plate-like; trunk has fine white scale | minimal | none | mild | unknown4 |
| ICH590-1 | c.939G>T, c.940-952del.TGGGTTCCCAAAG; p.E313Dfs homozygous | unknown | lower extremities plate-like; trunk fine white | mild-moderate | mild | none | yes |
| ICH592-1 | c.704C>T; p.P235L homozygous | collodion | fine white | minimal | none | none | yes |
| ICH600-1 | c.421A>G; p.K141E homozygous | generalized scale & erythema | Plate-like | minimal | none | mild | yes |
| ICH650-1 | c.157T>C; p.S53P homozygous | generalized scale | plate-like | moderate | mild | mild | no |
| ICH658-1 | c.158C>G; p.S53W homozygous | collodion | fine white on trunk; plate-like on lower extremities | mild-moderate | mild | none | yes |
PPK: palmoplantar keratoderma; hom.: homozygous;
excessive vernix;
ectropion did not present until late adulthood;
subject moved out of the country at age 6 weeks and was lost to follow-up;
subject is adopted.
Generally, the spectrum of phenotypic severity appears difficult to correlate with specific PNPLA1 genotypes. While some subjects with the same genotype exhibit consistent clinical features, there are other subjects with identical or similar mutations who show notable variation in phenotype. For instance, the affected siblings of kindred ICH162 (Supplementary Figure 2), both of whom are compound heterozygous for the same missense mutations, vary significantly in the severity of their presentations. ICH162-1 was born collodion and developed plate-like scale, palmoplantar keratoderma, and severe ectropion, whereas her sister had generalized scale and erythema at birth with no collodion membrane, and now has fine white scale; mild ectropion presented only within the past two years at age 80.
Interestingly, we report two families that appear to display dominant inheritance, but in which sequencing revealed PNPLA1 mutations consistent with recessive inheritance. In kindred ICH201 (Supplementary Figure 3), which was previously published as autosomal dominant ichthyosis 7, there are actually two different recessive PNPLA1 genotypes in affected individuals. The proband (ICH201-1) is compound heterozygous for A34T and S140P, while her two affected children (ICH201-3 and ICH201-4) are both homozygous for S140P, having presumably inherited a second copy of this mutation from their unaffected father. Mutation A34T has been previously been observed with homozygous inheritance in a prior report of a kindred from Galicia, Spain 8. Kindred ICH201 is also from Galicia, and collection of the extended family history revealed additional family members with ichthyosis (the proband’s deceased sister and a deceased nephew from another sister), despite no known history of consanguinity. These observations suggest that A34T and S140P may be founder mutations present at low frequency in Galicia, a region in which founder mutations in TGM1 have also been reported 9. Kindred ICH454 (Supplementary Figure 7) also is notable for an affected parent with two affected children and the resulting appearance of dominant inheritance. In this family, all three are homozygous for the same PNPLA1 mutation as a result of the consanguineous union of the proband and a first cousin who is a heterozygous carrier.
Two of the missense mutations we report are distal to the patatin domain (aa16-185, Figure 1). Mutation C216R is homozygous in the three affected members of kindred ICH454 (described above), and mutation P235L is homozygous in subject ICH592-1, a child of first cousins of Turkish descent born with a collodion membrane and now exhibiting fine white scale and minimal erythema (Supplementary Figure 11). These mutations suggest that residues outside the canonical ezymatic region may nevertheless be critical to protein function.
In a cohort of 450 ichthyosis subjects in whom we have made a genetic diagnosis employing targeted NGS of 43 genes or WES, the 14 unrelated probands with pathogenic PNPLA1 mutations we report here show PNPLA1 to be an important, if relatively rare, cause of ARCI.
Supplementary Material
Acknowledgments
We thank the study subjects and their families for their invaluable contribution to this work; Carol Nelson-Williams, Charlie Tian, Corey Saraceni, and Soheila Sotoudeh for technical assistance.
Funding Sources: The work was supported by R01 AR068392 and the Yale Center for Mendelian Genomics (U54 HG006504).
Abbreviations
- ARCI
autosomal recessive congenital ichthyosis
- LI
lamellar ichthyosis
- CIE
congenital ichthyosiform erythroderma
Footnotes
Conflict of interest: The authors declare no conflict of interest relevant to this work.
References
- 1.Grall A, Guaguere E, Planchais S, et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat Genet. 2012;44:140–7. doi: 10.1038/ng.1056. [DOI] [PubMed] [Google Scholar]
- 2.Lee E, Rahman OU, Khan MT, et al. Whole exome analysis reveals a novel missense PNPLA1 variant that causes autosomal recessive congenital ichthyosis in a Pakistani family. Journal of dermatological science. 2016;82:46–8. doi: 10.1016/j.jdermsci.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad F, Ansar M, Mehmood S, et al. A novel missense variant in the PNPLA1 gene underlies congenital ichthyosis in three consanguineous families. J Eur Acad Dermatol Venereol. 2015 doi: 10.1111/jdv.13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellström Pigg M, Bygum A, Gånemo A, et al. Spectrum of Autosomal Recessive Congenital Ichthyosis in Scandinavia: Clinical Characteristics and Novel and Recurrent Mutations in 132 Patients. Acta Derm Venereol. 2016 doi: 10.2340/00015555-2418. [DOI] [PubMed] [Google Scholar]
- 5.Vahidnezhad H, Youssefian L, Saeidian AH, et al. Gene Targeted Next Generation Sequencing Identifies PNPLA1 Mutations in Patients with a Phenotypic Spectrum of Autosomal Recessive Congenital Ichthyosis: The Impact of Consanguinity. The Journal of investigative dermatology. 2016 doi: 10.1016/j.jid.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Wilson PA, Gardner SD, Lambie NM, et al. Characterization of the human patatin-like phospholipase family. J Lipid Res. 2006;47:1940–9. doi: 10.1194/jlr.M600185-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Toribio J, Fernandez Redondo V, Peteiro C, et al. Autosomal dominant lamellar ichthyosis. Clin Genet. 1986;30:122–6. doi: 10.1111/j.1399-0004.1986.tb00580.x. [DOI] [PubMed] [Google Scholar]
- 8.Fachal L, Rodriguez-Pazos L, Ginarte M, et al. Identification of a novel PNPLA1 mutation in a Spanish family with autosomal recessive congenital ichthyosis. The British journal of dermatology. 2014;170:980–2. doi: 10.1111/bjd.12757. [DOI] [PubMed] [Google Scholar]
- 9.Fachal L, Rodríguez-Pazos L, Ginarte M, et al. Multiple local and recent founder effects of TGM1 in Spanish families. PLoS One. 2012;7:e33580. doi: 10.1371/journal.pone.0033580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.