

Abstract
The nanoscale engineering of nucleic acids has led to exciting molecular technologies for high-end biological imaging. The predictable base pairing, high programmability, and superior new chemical and biological methods used to access nucleic acids with diverse lengths and in high purity, coupled with computational tools for their design, have allowed the creation of a stunning diversity of nucleic acid--based nanodevices. Given their biological origin, such synthetic devices have a tremendous capacity to interface with the biological world, and this capacity lies at the heart of several nucleic acid--based technologies that are finding applications in biological systems. We discuss these diverse applications and emphasize the advantage, in terms of physicochemical properties, that the nucleic acid scaffold brings to these contexts. As our ability to engineer this versatile scaffold increases, its applications in structural, cellular, and organismal biology are clearly poised to massively expand.
Keywords: AFM, aptamers, FISH, functional bioimaging, live imaging, super-resolution microscopy, nucleic acid nanodevices, Spinach, structural DNA nanotechnology
INTRODUCTION
DNA and RNA are highly programmable molecular scaffolds, and this property enables the integration of molecular signals as well as the fabrications of molecular assemblies with nanoscale precision in three dimensions. In turn, this development has yielded a class of nucleic acid--based nanodevices showing increasing promise as reporters of biological phenomena. In this review, we consider key molecular technologies based on nucleic acids for imaging biological processes, ranging from in vitro to in vivo applications. Each application outlined herein leverages a certain physical, chemical, or biological property inherent in the nucleic acid scaffold. We discuss this fundamental property and how it is exploited to achieve functionality of the nucleic acid nanodevice.
The nucleic acid scaffold has a fairly conserved dimension (approximately 2 nm wide), as seen in double-stranded DNA (dsDNA) or double-stranded RNA (dsRNA), regardless of sequence (1). Using the specificity of Watson--Crick base pairing and the uniformity of the pitch of B-DNA, one can molecularly glue rod-like helices together in virtually any desirable orientation in space to realize a variety of shapes (2, 3). One may also realize structural motifs, termed tiles, by displaying sticky ends or complementary single-stranded regions in specific orientations. These tiles may be glued together in different orientations on the basis of molecular logic governing which orientations turn sticky. These tiling approaches yield a variety of two-dimensional (2D) architectures (4, 5). Another way to achieve micrometer-sized superstructures with nanoscale precision, as well as molecular addressability, is DNA origami (6). In this method, a long single-stranded piece of DNA, typically that of the M13 phage, is folded into precise shapes using a large collection of shorter, single strands of DNA termed staple strands or staples. Each staple hybridizes to spatially remote regions on the M13 phage DNA, bringing these regions into close spatial proximity; the net result is the folding of the longer DNA into a specific 2D or three-dimensional (3D) shape with nanoscale addressability. With computer-aided design principles, M13-phage DNA can be folded into virtually any shape (7). Such computer-aided design incorporates hierarchy in interaction strengths of different domains, the tuning of which is possible owing to the high tunability of Watson--Crick base pairing.
The high uniformity of the 2-nm DNA helix, its easy identification, and its ability to yield molecularly addressable nanostructures allow the correlation of computer-predicted device geometries with nanoscale atomic force microscopy (AFM) topography images of DNA origami structures. This combination has yielded a new development in which DNA origami templates, due to their high periodicity and uniform structure, serve as locators of precisely positioned protein molecules. These locators enable the imaging of protein activity or chemical reactivity in vitro at the single-molecule level.
The specificity of Watson--Crick base pairing underlies the antisense recognition property associated with nucleic acids. Fluorescence in situ hybridization (FISH) and molecular beacon technologies, which exploit antisense recognition, have been absorbed readily into mainstream biology to visualize RNA and DNA molecules in fixed cells and represent the simplest form of this class of nucleic acid nanodevices. These nanodevices function as high visibility probes of the RNA layer of gene regulation, in which the essential antisense recognition of a desired RNA is transduced to a structural change in the nucleic acid device. The conformationally altered nanodevice can be subjected to different kinds of amplification strategies to achieve enhanced visualization of target RNAs. Given the tremendous tunability in Watson--Crick base pairing affinities, one can manipulate the complementary oligonucleotide sequences to precisely control their binding and unbinding rates. This property has resulted in an exciting new technology termed DNA-PAINT (point accumulation for imaging in nanoscale topography) that couples super-resolution imaging with the high rates of blinking due to transient binding and dissociation of short, fluorescent oligonucleotides to image fixed cells below the diffraction limit (8–10).
Single-stranded DNA or RNA can fold into 3D motifs with specific, synthetic functionality, termed functional nucleic acids (11). RNA has the additional property of genetic encodability. Thus, a new molecular technology has emerged that uses genetically encodable, functional, synthetic RNA motifs to image RNA as well as small molecules in the cytosol of living cells (12, 13). Such RNA nanodevices also use the modular property of the RNA scaffold. Thus, one can judiciously combine functional modules onto a single transcript of RNA such that the functions of each of these modules can be either independent or interdependent (14).
Like the RNA scaffold, DNA is also modular and has the added advantage of hydrolytic stability. Thus, a new DNA-based, quantitative, functional imaging technology has emerged that can report on small-molecule concentrations within compartments of living cells. Complementary to RNA-based imaging technology, these DNA nanodevices exploit the modularity of the DNA scaffold to encode on a single nanodevice (a) a sensing module, (b) a reporting module, and (c) a targeting module that localize the nanodevice in a cellular compartment (15–17). The capacity to combine bright, synthetic organic fluorophores with the biological interfacability of nucleic acid devices allows quantitative functional imaging in live model organisms.
We therefore discuss bioimaging applications of nucleic acid nanodevices in three broad categories. First, we consider a class of DNA nanodevices used to explore the structure and function of proteins by in vitro imaging. Second, we discuss the deployment of nucleic acid nanodevices to image proteins, DNA, and RNA in fixed cells. Third, we examine a range of nucleic acid nanodevices fabricated for various applications in live imaging.
IN VITRO IMAGING OF BIOMOLECULES
Imaging with Atomic Force Microscopy
The uniform dimensions, the relatively nonconductive nature of dsDNA and dsRNA, and the easy physical identification of the approximately 2-nm wide filamentous feature predispose DNA and RNA to being probed by AFM. Using a sharp probe tip that is raster scanned over a surface, AFM detects surface topography by measuring the distances between the probe and surface. Currently, most AFM studies use the tapping mode under physiological conditions (18, 19). In this mode, the tip contacts the surface with high periodicity, which provides a contour map of the surface with accuracies of approximately 1 nm in the Z-axis. AFM has been widely deployed to image, below the diffraction limit, biological phenomena associated with nucleic acids, such as complex formation, enzymatic reactions, and conformational changes of biomacromolecular assemblies (20). In this section, we discuss a selection of DNA nanodevices that leverage the one-dimensional (1D), 2D, or 3D periodic structure of DNA to reveal molecular insight into biological phenomena and chemical reactivity. One of the early examples of AFM imaging of DNA investigated dsDNA in complex with Escherichia coli RNA polymerase (RNAP) (21). This study enabled the visualization of RNAP binding and bending of DNA, confirming the tension requirement within dsDNA for RNAP binding.
AFM has played a central role in validating the premise that DNA could be used to rationally build 1D, 2D, and 3D superstructures with nanoscale precision, in turn validating the self-assembly and design principles of structural DNA nanotechnology (2, 22). DNA architectures have been used as molecular breadboards to position proteins at designated locations on a periodic DNA lattice assembled either using tile-based assembly (23) or DNA origami (24). Such protein positioning is frequently mediated by biotin--streptavidin interactions or aptamer--protein interactions by displaying biotin or aptamer domains, respectively, at defined locations on the DNA architecture. For example, Chhabra et al. (25) simultaneously immobilized platelet-derived growth factor (PDGF) as well as thrombin on a 2D DNA origami surface by displaying their cognate aptamers. Such precise modulation of distance between protein-binding motifs has been applied to study distance dependencies associated with fundamental molecular phenomena, such as cooperativity in protein binding (24), as well as to multienzyme catalysis (26). Cooperativity was studied by positioning individual bivalent aptamer ligands excessively far apart, so that the former cannot participate in cooperative binding with the latter, thus preventing thrombin binding. Distance dependencies on catalysis were elegantly demonstrated in a study by Wilner et al. (26), in which the enzymes glucose oxidase and horseradish peroxidase were precisely positioned to vary the distance between the two enzymes and the product formation was measured by spectrophotometry. The study demonstrated that the net catalytic activity of the enzyme cascade could be rationally tuned. This concept was later translated inside a living system in which Delebecque et al. (27) expressed a 2D RNA array in bacteria that effectively surface immobilized fusion proteins of (Fe-Fe) hydrogenase and ferredoxin. Modulation of the interenzyme distance resulted in a 48-fold greater efficiency of hydrogen production.
High-speed AFM (HS-AFM) permits the direct visualization of dynamic processes at subsecond temporal resolution at the level of single molecules (28). HS-AFM has captured dynamics of nucleic acid assemblies and their complexes with diverse proteins such as DNA-modifying or repairing enzymes (29–31). Interrogating DNA origami architectures using HS-AFM has also been applied to study bond-breaking reactions and diffusion kinetics. For example, chemical reactions such as thiol--disulphide exchange or singlet oxygen-mediated bond rupture could be imaged at single-molecule resolution on a 2D DNA origami nanodevice (32). Essentially, the chemically reactive centers in question, such as disulphide linkages or double bonds, are first positioned on a DNA origami breadboard by incorporating them in biotin-labeled staple strands. Addition of streptavidin to the solution leads to its immobilization on the DNA origami surface, which serves as a marker in AFM imaging owing to its height difference. Addition of a reactant such as dithiothreitol (for disulphide exchange) or eosin (as a singlet oxygen generator) results in bond rupture, as revealed by loss of the surface feature corresponding to the immobilized streptavidin. This method is potentially generalizable to the single-molecule study of any bond-making or bond-breaking reaction and the associated diffusion kinetics in macromolecules in real time.
DNA origami frameworks thus function as excellent locators for displaying a dsDNA template for AFM imaging. DNA origami AFM locators have been used to image enzymatic reactions on dsDNA templates at single-molecule resolution. Precise positioning of the attachment sites of the dsDNA template on the origami framework can modulate the tension and bending of the displayed dsDNA substrate. Many DNA-modifying enzymes require that their dsDNA substrate possesses the capacity to bend (33). For example, methylation on adenine by EcoRI methyltransferase (M.EcoRI) occurs upon a 55–59° bending of dsDNA. Endo et al. (34) used 2D DNA origami to investigate the feasibility of DNA methylation by M.EcoRI on 64-bp and 74-bp dsDNA fixed to impart higher tension and lower conformational flexibility of the former compared with the latter (Figure 1a). Addition of M.EcoRI to the origami surface showed complexation and methylation of the 74 bp-dsDNA template and no reduced reactivity on the 64-bp dsDNA template. Such imaging approaches can be used to assay the role of conformational flexibility of dsDNA toward diverse enzymatic reactions.
Figure 1.
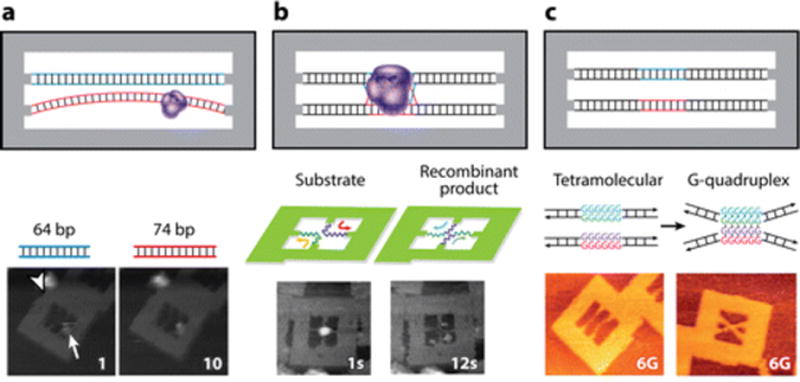
DNA origami as atomic force microscopy (AFM) locators. (a) Visualization of DNA methylation by high-speed AFM (HS-AFM) via EcoRI methyltransferase on a 74-bp double-stranded DNA (dsDNA) substrate. Panelamodified from Reference 34 with permission. (b) Visualization of DNA recombination by Cre recombinase showing only the starting material and end product. Panelbmodified from Reference 31 with permission. (c) Visualization of G-quadruplex formation by two G-rich sequences positioned proximally. Panelcmodified from Reference 37 with permission.
DNA origami has been used to directly visualize various stages of transcription, such as RNAP binding, sliding along a DNA template, RNA synthesis, and RNAP dissociation (30). A strip of DNA origami acts as an AFM observation scaffold onto which a 1-kb template dsDNA is attached at fixed positions. The addition of RNAP to the system before HS-AFM imaging of the RNAP--dsDNA complexes enables the visualization of RNAP sliding on a template DNA that is easily identified by its incorporation in the DNA origami frame. Further, the newly transcribed RNA could also be imaged by AFM by incorporating biotinylated UTP in the transcription mix before addition of streptavidin.
More recently Suzuki et al. (31) have used origami locators to image DNA recombination using HS-AFM (Figure 1b). They have employed Cre recombinase, which recognizes a 34-bp LoxP domain incorporated in two segments of dsDNA. Cre forms a synaptic complex with two such adjacent dsDNA LoxP domains, leading to the recombination products via a characteristic Holliday junction intermediate. Substrate dsDNA strands are positioned in a predesigned cavity on an origami AFM locator either in parallel or in antiparallel orientations incorporating specific bending angles. Synaptic complex formation and recombination events could be imaged in real time using HS-AFM. Holliday junction intermediates crossing at right angles on the origami locator afford the recombination products, in contrast to the intermediates crossing at 60°, which give the product in different proportions (Figure 1b). HS-AFM imaging revealing dsDNA bending requirements for protein activity has been applied to transcription factors, such as SRY (sex determining region Y)-box 2 (Sox2) and paired box 6 (Pax6) (35), and base excision repair enzymes, such as 8-oxoguanine glycosylase (hOgg1) and T4-pyramidine dimer glycosylase (PDG) (36).
Another interesting application of HS-AFM is in the imaging of conformational changes within a dsDNA template displayed on a DNA origami observation platform. A good example is the molecular mechanism of G-quadruplex formation induced by HIV-1 nucleocapsid proteins. Nucleocapsid proteins NCp15 and NCp7 are known to stabilize G quadruplexes owing to their chaperone activity (29). HS-AFM imaging of DNA origami locators presenting G-quadruplex--forming domains captured molecular phenomena associated with this process, such as protein sliding, 3D hopping between DNA strands, G-quadruplex formation, and protein dissociation. In fact, the power of DNA origami locators as imaging probes for AFM is best illustrated by the observation of key in-pathway intermediates in G-quadruplex formation, such as G hairpins and G triplexes (Figure 1c) (37). The direct observation of these intermediates had, until then, proved elusive by any known experimental technique.
APPLICATION IN CELLULAR IMAGING OF FIXED SYSTEMS
Visualization of Endogenous Nucleic Acids
Antisense recognition is a property unique to the nucleic acid scaffold. Antisense recognition of endogenous nucleic acid targets by synthetic fluorescent oligonucleotides to pinpoint localization or abundance of these targets within the cell is widely deployed in biology. Commonly termed fluorescence in situ hybridization (FISH), this method is used routinely to visualize genomic DNA loci and single RNA transcripts within their cellular environment in fixed systems. Numerous fluorescent oligonucleotide probes hybridize along the target sequence and yield an enhanced fluorescent signal. The first report of in situ hybridization was made by Pardue & Gall (38) in 1969, when the authors looked at Drosophila salivary gland chromosomes using radiolabeled probes. Radiolabels were soon replaced by fluorescent labels for visualization of 5S rRNA in polytene chromosomes of Drosophila (39). This early research used an indirect method of visualization, compared with present-day FISH methodology in which the oligonucleotide, carrying a hapten label, is detected immunohistochemically by a fluorophore-tagged antibody against the hapten (Figure 2a). FISH with fluorescently labeled oligonucleotides was subsequently realized in 1980 (40). The improvement of DNA synthesis and the use of branched DNA probes, coupled with robotics and image analysis, have made assays of transcriptome heterogeneity at the single-cell level possible. For example, using 50-nucleotide long fluorescent oligonucleotides, tandemly hybridized to their target mRNA, has allowed the visualization of individual β-actin mRNA molecules within mammalian cells (41). Importantly, FISH probe sizes can be dramatically reduced from approximately 50 nucleotide to 17—22 nucleotide by using a large probe set of singly labeled fluorescent oligonucleotides (typically a set of 48 probes). By using Laplacian of Gaussian (LoG) filters and deconvolution, one can simultaneously detect three transcripts in chinese hamster ovary (CHO) cells and in a carcinoma cell line A549; this method can also be applied to other systems such as yeast, Drosophila, and Caenorhabditis elegans (42). In an effort to further shorten the probe length yet preserve specificity, nucleic acid analogs such as locked or peptide nucleic acids have been used (43–46) owing to their higher melting temperatures following hybridization to their target RNAs. Signal amplification is necessary when detecting short RNA sequences as seen in microRNAs, which can accommodate only a single antisense probe (47). Furthermore, different endogenous targets can be labeled with FISH probes bearing spectrally different fluorophores. Such multivalent FISH techniques have also been applied to live-cell imaging with single-transcript sensitivity (48, 49).
Figure 2.
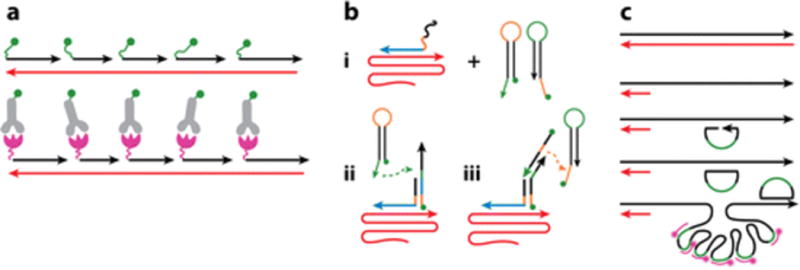
Applications in visualizing endogenous nucleic acids in fixed systems. (a) Fluorescence in situ hybridization (FISH) detects an endogenous RNA/DNA target (red) using antisense oligonucleotides (black) labeled with a fluorophore (green) or antibody (gray) against a hapten (magenta). Panelamodified from Reference 49a with permission. (b) Site-specific amplification of FISH signaling using hybridization chain reaction (HCR) technology. (i) An initiator DNA device (blue) binds to a specific sequence on the target mRNA (red) and (ii) initiates hybridization to its complementary sequence, present on the first fluorescently labeled hairpin. (iii) The hairpin thus opens up, leading to hybridization of the second fluorescent hairpin. Complementary sequences are shown in orange and green. This process results in a cascade of alternating hybridization events between two distinct fluorescent, metastable DNA hairpins. Panelbmodified from Reference 51 with permission. (c) Use of padlock probes (greenandblacksemicircles) against a target mRNA (red) location specifically generates a rolling circle amplicon detected by fluorescent oligonucleotides (pink) against the padlock sequence (green).Panelcmodified from Reference 55 with permission.
However, FISH still suffers from low signal-to-noise ratio owing to the limited number of fluorophores that can be tiled to the target transcript without amplification, as well as inadequate probe specificity for short sequences and poor uptake of longer length probe sequences. To overcome these limitations and to enable single-step, multiplexed RNA imaging, a triggered hybridization chain reaction (HCR) has been devised (50). The HCR technique uses two fluorescently labeled nucleic acid devices that are complementary to each other but coexist as metastable hairpin structures (Figure 2b). A nucleic acid initiator device is required to hybridize to the input domain of one of the hairpin sequences, leading to its opening and hybridization to the complementary sequence in the second hairpin device. The initiator device thus elicits a chain reaction of hybridization events that leads to a nicked dsDNA polymer. Thus, this reaction is amplified locally at the site of hybridization within the cell, resulting in approximately 200-fold brighter labeling of an endogenous DNA or RNA target than observed with conventional FISH methods. Such an approach is extendable to multiplexed RNA-FISH detection in whole-mount embryos with impressive clarity (50, 51).
Another method with intrinsic signal amplification potential is in situ polymerase chain reaction (PCR), first described in 1990, which uses PCR or reverse transcriptase PCR to amplify a gene of interest in a fixed cell. Initially, fixed cells suspended in a PCR reaction mixture were thermally cycled in microtubes using a conventional block cycler, centrifuged onto glass slides, and intracellular PCR products were visualized by in situ hybridization or immunohistochemistry. However, the use of this method has been limited owing to low amplification efficiency and low reproducibility due to the diffusion of PCR products from the site of synthesis within the cells as well as extracellular stochastic generation of amplicons (52, 53). To overcome this hurdle, padlock probes have been used to detect mRNAs. Padlock probes are devices that hybridize onto cDNA generated (cDNA is generated) by reversely transcribed primers from the endogenous target. This hybridization results in the formation of a nicked single-stranded circular DNA hybridized to the cDNA. The nick is sealed by a DNA ligase, and rolling circle amplification is initiated by the phi29 phage polymerase (54). The DNA nanostructure or the rolling chain product that is formed is detected by a fluorescent oligonucleotide complementary to the padlock sequence (Figure 2c). Because DNA ligases are sensitive to mismatches flanking the nick, this method is capable of distinguishing single-nucleotide polymorphisms with single-molecule sensitivity. This technology has been used for multiplexed detection of three genes in mouse tissue (55). A similar concept has been used to visualize endogenous RNAs by fluorescence in situ RNA sequencing developed by the Church laboratory (56).
Super-Resolution Imaging Using Nucleic Acids
Fluorescence based imaging has improved our understanding of endogenous nucleic acid localisation within cells. On one hand, diffraction limited microcopy has dramatically improved signal to noise ratios which are criticial for sensitive detection. On the other hand, super-resolution microscopy has grown tremendously, bridging the gap between conventional optical microscopy and EM in many ways. DNA architectures have found a niche in super-resolution microscopy as powerful probes for calibration. Here, hybridization is the key property of nucleic acids that underlies the utlility of these molecular probes, as one can leverage sequence specificity and tunability of association strengths to enable the detection of multiple cellular targets.
Calibration standards for super-resolution microscopy
An important property of DNA origami is the combination of nanoscale-to-mesoscale architectures that possess nucleotide-level addressability. The capacity to computationally model these architectures allows one to obtain internucleotide spacings with 6-nm resolution (57, 58). As a result, various DNA origami architectures have been fabricated as imaging standards to calibrate super-resolution microscopes, such as direct stochastic optical reconstruction microscopy (dSTORM), structured illumination microscopy (SIM), or stimulated emission depletion (STED) (59). This concept was first outlined by Simmel & Tinnefeld in 2009 (10), when they used super-resolution fluorescence microscopy to optically image single-layer DNA origami structures. In their study, two fluorophores were positioned at a distance of approximately 90 nm on a rectangular DNA origami structure by simply modifying two staple strands at the 5′end of the upper surface. The origami structures simultaneously displayed biotin groups protruding on the opposite face for immobilization on a streptavidin-modified glass surface and were subsequently imaged using total internal reflection fluorescence microscopy (TIRFM) and AFM. This structure was then imaged using three different kinds of super-resolution methods, namely (a) single-molecule high-resolution imaging with photobleaching, (b) dSTORM, and (c) blink microscopy (60–62), and, from the observed distance in each technique, the associated imaging resolution could be determined.
Many super-resolution methods such as STED and its variants, SIM, ground state depletion microscopy followed by individual molecule return (GSDIM), super-resolution optical fluctuation imaging (SOFI), and others, require diverse parameters such as laser intensity, acquisition rate, activation laser settings, image analysis, and image rendering to be optimized in order to realize the manufacturer specifications on image resolution. DNA origami can be used to simultaneously calibrate distances as well as brightness by displaying specific numbers of fluorophores at well-defined distances. Schmied et.al. (63) used a rectangular DNA origami that functioned as an imaging standard covering a full range of distances by positioning ATTO647N fluorophores at distances of 6, 12, or 18 nm (Figure 3a). The ability to position a stoichiometrically precise number of dyes in programmable geometries and in exactly the same molecular environment on DNA origami yields uniform photophysical characteristics with near-negligible sample variation. Such DNA origami architectures function as a brightness standard (Figure 3b). DNA origami nanopillars bearing fluorophores positioned at specific distances from the surface enable calibration of the Z-axis resolution in super-resolution microscopy (64). SIM has a typical intermark distance of 120 to 180 nm and requires a high number of dyes, as does STED microscopy. Localization-based super-resolution techniques such as stochastic optical reconstruction microscopy (STORM) and photoactivated localization microscopy (PALM), which are based on single-blinking or switchable molecules, require fewer dyes with tunable switching kinetics. With DNA origami--based nanorulers, defined standards for each of these fluorescence techniques have been created (65).
Figure 3.
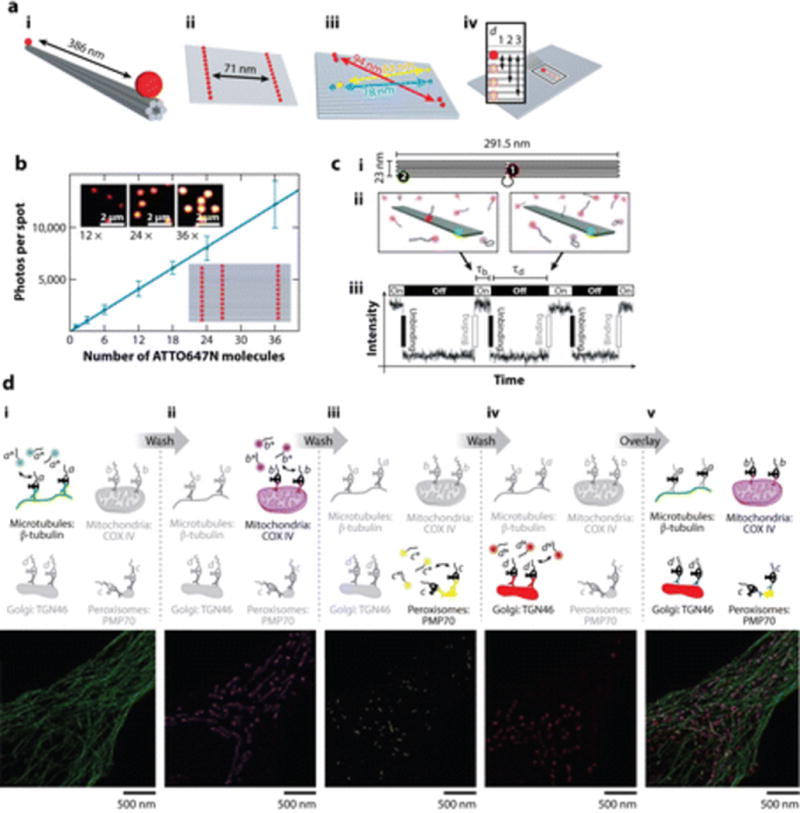
DNA-guided super-resolution microscopy. (a) DNA origami distance calibration standards for (i) diffraction-limited microscopy using a DNA N-helix bundle, (ii) STED microscopy using a 2D DNA origami nanoruler, (iii) STORM technique using a 2D DNA origami for different wavelengths, (iv) 2D DNA origami bearing multiple interdye distances. Panelamodified from Reference 63 with permission. (b) DNA origami brightness calibration standards. The lower inset shows various numbers of dyes, and the upper inset shows fluorescence images of brightness standards. Scale bar is 2 µm. Panelbmodified from Reference 63 with permission. (c) DNA-PAINT imaging using (i) an origami breadboard incorporating a fluorescent staple strand (green) and a docking site for an imager strand (red). (ii) Hybridization of the imager strand to the docking site results in bright fluorescence in TIRFM. (iii) A typical intensity-versus-time trace shows low and high fluorescence in the dissociated (τd) and bound states (τb), respectively, of the imager strand. Panel cmodified from Reference 9 with permission. (d) Exchange-PAINT imaging in fixed cells. (i--iv) Four cellular targets are labeled with an antibody carrying a unique DNA-PAINT docking sequence. An imager strand (a,b,c or d) specific to a docking sequence is added sequentially to visualize each cellular target, leading to multiplexing. Scale bar is 500 nm. (v) Overlay of all four targets. Paneldmodified from Reference 8 with permission. Abbreviations: PAINT, point accumulation for imaging in nanoscale topography; STED, stimulated emission depletion; STORM, stochastic optical reconstruction microscopy; TIRFM, total internal reflection fluorescence microscopy.
DNA-guided super-resolution imaging of cellular architectures
The PAINT (point accumulation for imaging in nanoscale topography) method, developed by Sharanov & Hochstrasser (66),was used to image membranes to a resolution of 25 nm. This method used a fluorescent dye, Nile Red, that fluoresced brightly upon membrane insertion and was weakly fluorescent when unbound. Universal PAINT (uPAINT) is a powerful adaptation of this method that uses (a) fluorescent antibodies against membrane proteins that gives molecular information and (b) a diverted TIRFM laser angled obliquely to yield a highly inclined laminated optical (HILO) sheet to abolish the fluorescence of unbound, labeled antibodies. uPAINT has captured super-resolution images and real-time trajectories of membrane proteins in live cells (67). DNA-PAINT, developed by Jungmann et al. (9), can now image cytosolic targets at super resolution, albeit on fixed cells. It exploits the fundamental property that hybridization and dissociation rates of antisense oligonucleotides are tunable by either changing the length, sequence composition, or concentration of antisense oligonucleotide probes. Thus, an immobilized DNA origami architecture bearing a docking strand (i.e., a single-stranded segment 9–10 nucleotides long projecting out at a defined position) can be imaged by imager strands (Figure 3c). Imager strands are fluorescently labeled oligonucleotides that are 9–10 nucleotides long, have high KD, have high specificity due to antisense recognition, and are designed to have low melting temperatures to enable repeated binding and unbinding events. Each binding event brings the imager strands into the evanescent regime and can be monitored by TIRFM. The fact that binding is based on DNA hybridization allows for the precise control over fluorescence ON and OFF times ranging from milliseconds to seconds. The fluorescence ON time can be adjusted by changing the concentration of imager strands in solution, and the fluorescence OFF time can be controlled by the length of the imager-docking strand duplex. After stochastic reconstruction of diffraction-limited TIRFM images, the binding sites on a DNA origami are clearly resolved (Figure 3c) (9). This technology has been used to determine the number of available docking sites on functional DNA origami nanostructures (68). A critical advantage of DNA-PAINT is its bleaching independence, which enables imaging over long durations and the use of diverse fluorophores. Building on uPAINT, Jungmann et al. treated fixed and permeabilized cells with antitubulin and anti-COX IV antibodies, each linked to a docking strand with a specific sequence, and then used two complementary imager strands to obtain super-resolution images of microtubules and mitochondria using HILO microscopy (8). The use of DNA sequences allows one to have several pairs of imager-docker duplexes with similar binding energetics yet show adequate sequence specificity to enable multiplexed imaging. Exchange-PAINT uses sequential multiplexing in which imager strands to different cellular targets can be applied to the same biological sample by simply washing out and exchanging the solution with an imaging strand for a different target (Figure 3d). Exchange-PAINT has been used to achieve four-color super-resolution images of intracellular architectures, namely microtubules, mitochondria, the Golgi complex, and peroxisomes (8).
APPLICATIONS IN LIVE CELL IMAGING
Visualization of Endogenous Nucleic Acids
Fixation of cells precludes the study of dynamic, live processes. The need to image the dynamic movements of intracellular RNAs in living cells has stimulated the development of many diverse RNA devices and new concepts in the design of synthetic functional RNAs.
Antisense-based recognition
Among the most straightforward approaches for imaging RNAs in cells is to transfect or inject fluorescently labeled antisense probes (69, 70) and molecular beacons (71–74). The probe hybridizes to its target RNA, enabling imaging of the RNA by fluorescence microscopy.
Molecular beacons enable real-time tracking of nucleic acids in live cells and can be considered one of the earliest forms of functional nucleic acid nanodevices (75). Molecular beacons are DNA sequences comprising a target-recognition region of approximately 15–30 bases flanked by two short complementary domains at the 5′ and 3′ termini. This combination forces the entire sequence into a stem--loop structure in the absence of the endogenous target. In this hairpin form, a quencher and a fluorophore, which are each located at a different terminus of the molecular beacon, are brought into close proximity, such that fluorescence is quenched effectively. In the presence of a target DNA or RNA molecule, hybridization between the target and the loop sequence of the beacon occurs, and the stronger intermolecular hybridization breaks the weaker stem helix. The spatial separation of the fluorophore and quencher results in the restoration of fluorescence (76, 77). Molecular beacons are generally microinjected into cells to study the transport and localization of endogenous RNAs. Alternatively, one can exploit reversible permeabilization of the plasma membrane using pore-forming toxins or cell-penetrating peptides (73, 78). Molecular beacons have been used to track cytosolic localization of RNAs such as oskar mRNA in Drosophila melanogaster oocytes during development (72). Dual fluorescence (Förster) resonance energy transfer (FRET) molecular beacons have been used to study K-ras mRNA localization in living cells; the study used probe hybridization to affect intermolecular FRET (74). This approach can perturb cellular function, and reliable delivery of these probes to the cytosol, rather than endosomes, is challenging (79).
Protein-based RNA recognition
Genetic encoding of reporters allows cells to express them, thus bypassing perturbative methods of introduction within cells (described above), which can affect diverse signaling pathways and/or cell viability. One can image RNAs by repurposing naturally occurring RNA--protein interactions. This approach relies on proteins that bind to specific RNA elements with high affinity and specificity. To image RNAs, the RNA-binding proteins are heterologously expressed as green fluorescent protein (GFP)--tagged proteins and their cognate RNA-binding elements are incorporated into heterologously expressed RNA sequences. Thus, the RNA-binding protein is converted into a tool to deliver GFP to an RNA. This approach has been used in the bacteriophage MS2 coat protein MS2-GFP and Pumilio--split fluorescent protein imaging systems.
MS2-GFP is the first example of a genetically encoded system for RNA imaging (80). This system consists of the bacterial MS2 phage coat protein, which binds to the MS2 RNA hairpin with high affinity. The MS2 coat protein is expressed as a GFP fusion protein that binds RNAs expressed to contain an RNA tag comprising up to 48 copies of the MS2 hairpin (81, 82). This technique was recently used to image heterologously expressed individual β-actin mRNAs in neurons in a live mouse (83). An analogous system is a protein-RNA pair comprising the λN peptide and the 24-nucleotide boxB RNA motif that binds with 22-nM affinity. The boxB motif efficiently folds modularly into a hairpin and can therefore be judiciously incorporated into longer target RNA sequences without disrupting the target RNA structure. The λN peptide is a 22--amino acid segment obtained from the transcriptional antitermination protein N of phage λ that is fused to an enhanced green fluorescent protein (EGFP) and an M9 nuclear localization signal. When expressed in cells, λN-EGFP binds to a target RNA of interest that incorporates four boxB sequences. This technique has been used to image the localization of RNA molecules in the cytosol of eukaryotic cells (84).
In contrast to the MS2-GFP system, which allows imaging of heterologously expressed RNAs, the Pumilio--split fluorescent protein imaging system allows imaging of endogenous RNAs. This approach uses Pumilio homology domains (PUM-HDs), which can be engineered to interact with nearly any 8-nucleotide RNA site (85). Imaging RNAs with PUM-HDs uses a split fluorescent protein imaging approach to achieve highly specific detection of endogenous RNAs. Two halves of a fluorescent protein are fused to two different PUM-HDs that bind to adjacent 8-nucleotide sites within an endogenous transcript. When each PUM-HD binds to adjacent sites in an RNA, the two halves of the fluorescent protein come together, which reconstitutes fluorescence (85–87). This approach benefits from low background, because the initial fluorescent protein halves are dark, as well as from the ability to target endogenous RNAs. However, only one GFP is tethered to the RNA of interest. Conversely, the MS2 approach tethers dozens of GFPs, resulting in sensitivity down to single RNA transcripts.
Aptamer-based recognition
Because of its genetically encodable nature, RNA itself presents an exciting and more direct route to imaging cellular RNA molecules (14). The Spinach complex is an RNA mimic of GFP (13) and an RNA aptamer that binds fluorophores resembling hydroxybenzylidene imidazolinone (HBI), the fluorophore in GFP. Spinach binds the otherwise nonfluorescent 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), resulting in highly enhanced fluorescence (13, 88). Similar to the mechanism by which GFP induces fluorescence in HBI, Spinach binds to DFHBI and switches it to a highly fluorescent state. As a result, DFHBI exhibits nearly undetectable fluorescence in cells except when bound to a Spinach aptamer (13). This property allows Spinach to be used as a tag to fluorescently mark and image specific RNAs in living cells (13).
Visualization of Small Molecules
Small molecules inside the cell, whether second messengers or metabolites, are crucial for cell--cell communication (89). Thus, imaging spatiotemporal fluctuations of these small molecule concentrations is essential to understanding cell signaling and is termed functional imaging. Owing to their molecular recognition capabilities, DNA and RNA can be used as functional imaging probes. As a molecular scaffold, DNA as well as RNA are highly modular: Each module can execute functions that can be either dependent or independent of each other (90, 91). For example, an mRNA molecule may be envisaged as an endogenous, nucleic acid device comprising modules such as the 5′ cap, the 5′ untranslated region (UTR), the coding region, the 3′UTR, and the polyA tail, each of which have a specific function. Here the 5′ cap and the polyA tail have interdependent functions to enhance translation (92), whereas the coding region has an independent function, as seen when encoding fusion proteins. In fact, as organisms increase in complexity, the sizes of DNA molecules composing the genome increase enormously, whereas the sizes of proteins or lipid molecules change very little. Thus, modularity is an extremely important property to achieve complex functionality, to achieve multifunctionality, or to encode a hierarchy in functionality. Probes for functional imaging therefore need to possess more than one functionality.
In the cytosol
Typically, genetically encoded, protein-based reporters for small molecules or ions in the cytosol comprise two fluorescent proteins flanking a central target-binding domain (93, 94). When the target molecule binds, it induces a conformational change that repositions the fluorescent proteins, which leads to changes in FRET that can be imaged by fluorescence microscopy in live cells (95). Protein-based sensors using this strategy have been developed to detect dynamic changes in calcium, glucose, cyclic nucleotides, and other molecules (96). A limitation of these protein-based sensors is their requirement of a protein that binds the target molecule of interest accompanied by a conformational change sufficient to induce a large change in FRET. For many metabolites, proteins with these properties are not readily available. Thus, fluorescent reporters for many important metabolites and signaling molecules have not been generated.
Allosteric Spinach-based sensors
Given RNA’s modular nature, it is a particularly powerful tool for obtaining fluorescent scaffolds that are responsive to small molecules. Spinach can be designed so that the constitutively fluorescent Spinach--DFHBI complex can be converted to make its fluorescence conditional to the binding of a specific metabolite (14). These sensors comprise two modules with interdependent functions: Spinach and a target-binding aptamer. The target-binding aptamer is integrated into Spinach using a structurally critical stem as an entry point (14). Because most aptamers are unstructured without their cognate ligand, their insertion into the Spinach module causes the disruption of a critical stem in Spinach. As a result, Spinach cannot fold, bind DFHBI, or yield a fluorescent signal. However, when the cognate ligand does bind its aptamer module, the aptamer folds, resulting in the folding of the critical stem in Spinach (Figure 4a). This folding leads to Spinach fluorescence that can be detected in vitro or in cells (14). One study used this approach to fuse Spinach to specific aptamers, thereby generating a range of fluorescent reporters for small molecules such as adenosine diphosphate (ADP), S-adenosylmethionine (SAM), and other metabolites (14). These reporters were used to image the dynamics and turnover of SAM and ADP in live bacteria (14). This approach was subsequently applied to detect and image cyclic di-GMP, cyclic di-AMP, cyclic adenosine monophosphate -guanosine monophosphate (AMP-GMP), in addition to various proteins in living cells (97–100). These allosteric Spinach-based sensors demonstrate the versatility of RNA as a platform for creating genetically encoded sensors for imaging cellular metabolites. A circularly permuted version of Spinach can also be used to image metabolites whose aptamers are sensitive to modifications at their 3′ or 5′ termini (101).
Figure 4.
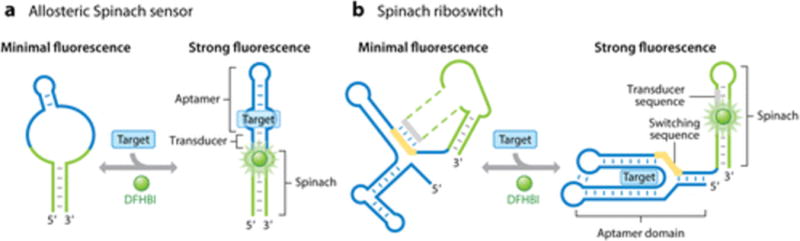
Sensing mechanisms of Spinach riboswitches and allosteric Spinach sensors. (a) Secondary structure and mechanism of allosteric Spinach sensors comprising a target-binding aptamer (blue) and Spinach (green) and responsive to a target molecule (blue rectangle). (b) Secondary structure and mechanism of Spinach riboswitches. Spinach is integrated into a riboswitch sequence (blue) and becomes responsive to a metabolite (blue rectangle). A transducer sequence (gray) is released from a metabolite-sensitive switching sequence (yellow) in the aptamer domain of the riboswitch and folds Spinach and binds DFHBI (green ball). Abbreviation: DFHBI, 3,5-difluoro-4-hydroxybenzylidene imidazolinone.
Spinach riboswitches for metabolite detection
One can also convert metabolite-induced conformational switches found in riboswitches into fluorescence readouts using Spinach technology. Riboswitches are RNA regulatory elements found primarily in bacterial mRNAs (102). Riboswitches contain binding domains that recognize specific intracellular metabolites and that undergo a conformational switch upon binding their cognate ligand. These conformational switches typically activate or inhibit translation of the mRNA but can regulate other aspects of mRNA metabolism (103). Spinach riboswitches are synthetic, RNA-based, genetically encoded metabolite reporters that leverage natural riboswitch sensing mechanisms (104). Typically, when the metabolite binds a riboswitch, portions of the riboswitch are structurally remodeled, resulting in the formation of a new helix. In a Spinach riboswitch, Spinach is inserted into the riboswitch so that the newly formed helix enables the folding of Spinach. Using this approach, naturally occurring riboswitches that bind thiamine pyrophosphate, adenine, and guanine were converted into Spinach riboswitches that fluoresce upon binding these metabolites in a concentration-dependent manner (104).
Spinach riboswitches employ a different mechanism than that used in allosteric Spinach reporters (14, 97, 98, 105) (Figure 4b). Spinach riboswitches switch between two folded conformations, in which the switching module is associated either with the ligand-binding pocket of the riboswitch or with Spinach. In contrast, allosteric Spinach sensors switch from an unfolded to a folded state. Notably, the sensors do not appear to differ in terms of the signal-to-noise ratio and kinetics of activation/deactivation (14). Spinach riboswitches score higher than allosteric Spinach sensors, in that they retain the high analyte specificity and physiological binding affinity of the riboswitches from which they were derived. Conversely, allosteric Spinach sensors rely on aptamers that are typically generated using in vitro aptamer-selection approaches such as SELEX (106, 107). Such in vitro--generated aptamers may not exhibit the requisite selectivity to discriminate against diverse metabolites with structures similar to the desired target molecule. In principle, negative selection can be used to obtain metabolite-selective aptamers during SELEX (108). However, negative selection cannot be performed against all the diverse structurally related metabolites in the cell, especially if some of them are unknown. Aptamer affinity is also a critical parameter needed for proper sensor function (109). In most cases, aptamers should exhibit binding affinities comparable with the cellular metabolite concentration for fluorescence levels to directly correspond to the metabolite concentration (109). However, obtaining aptamers with a predetermined binding affinity using SELEX is typically challenging (110). Naturally occurring riboswitches have evolved to exhibit in vivo binding affinities that correlate to the intracellular concentrations of their targets. Thus, sensors based on riboswitches rather than SELEX-derived aptamers are more likely to display affinity and selectivity tuned for live systems.
In subcellular organelles
Complementary to Spinach-based technologies, which find applicability in probing the cytoplasmic or nucleoplasmic environment of live cells, is a new DNA-based technology that probes the lumenal environment of subcellular organelles. This technology combines (a) the brightness of organic fluorophores, (b) the biological interfacability of the nucleic acid scaffold, and (c) the capacity to encode synthetic function in these devices to achieve quantitative imaging of membrane-bound organelles in living cells.
DNA nanodevices as probes for quantitative imaging
The Krishnan group has exploited the modularity inherent in the DNA scaffold to create a variety of DNA-based fluorescent analyte-measuring devices (91, 111). In this class of nanodevices, a single DNA nanodevice comprises three modules that function independently of each other: (a) a sensing module that consists of a functional nucleic acid or ion-sensitive dye that is sensitive to the analyte, (b) a reporting module comprising a pair of organic fluorophores to measure the concentration of the analyte, and (c) a targeting module that localizes the nanodevice to a specific subcellular compartment within a live cell in which the analyte concentration is to be measured.
The sensing and reporting modules together form a measuring module. The reporter module incorporates a pair of fluorophores that, after binding or recognition of the analyte by the sensing module, undergoes a change in photophysical properties. Given that the analyte recognition domain has a single binding event for the analyte, the measuring module can measure 1.5 to 2 orders of magnitude of concentration of the analyte centered on the dissociation constant (KD). The ability to achieve large conformational changes upon association with an analyte is an important property of the nucleic acid scaffold that allows for the realization of quantitative sensors. Functional nucleic acids are folded in space into relatively compressed, globular shapes that enclose a cavity, within which the analyte binds. The same functional nucleic acid motif can also exist in an extended state when base paired with a complementary strand in the absence of the analyte. If the KD of analyte binding of the functional nucleic acid motif is well matched against an available complementary strand, one can realize nanodevices that show large conformational changes, from linear to folded, resulting in dynamic ranges that simply dwarf those observed in other biomolecular scaffolds (112). By integrating these large conformational changes with the attractive fluorescent properties of small-molecule organic fluorophores, such DNA reporters show large dynamic ranges with high signal-to-noise ratios, thus enabling the measurement of analyte concentrations with high accuracy inside living cells (16).
A few design principles by which this tuning may be achieved are discussed below. For example, consider the case of pH sensing with the i-motif, in which the i-motif can be considered as an aptamer for protons. An i-motif is a four-stranded DNA structure that is formed from four stretches of cytosine-rich segments held together by hemiprotonated cytosines, which form noncanonical C:C+ base pairs, and that exists only at acidic pH (113). This property has been used to engineer DNA nanodevices termed I-switches that undergo a pH-induced conformational switch in which the sensing module comprises an i-motif and the reporting module comprises a pair of Alexa dyes that undergoes FRET under acidic conditions (114). pH reporters based on the i-motif illustrate how one can tune reporter characteristics of the measuring module. By increasing the number of cytosines, one can shift the KD to higher pH (115). However, this shift increases the cooperativity of the pH-induced structural transition and simultaneously narrows the overall pH regime that the device reports on. An alternative to changing the KD without affecting the cooperativity is the use of chemically modified cytosines, such as methylcytosine or bromocytosine, that have higher or lower pKa (acid dissociation constant) levels and can accordingly shift the pH sensitivity of the measuring module to higher or lower pH regimes (112). An interesting trick to create a broad pH-sensitive regime is to employ a mixture of pH reporters, each responding to different pH regimes. The mixture then has an overall broader pH sensitivity (116, 117). However, with such a mixture of reporters the response characteristics change along with the proportion of devices in the mixture. Although an apparent disadvantage for batch-to-batch reproducibility, it presents an effective way to achieve tunable pH sensitivity at desired pH regimes. In a similar vein, tuning the KD of binding leading to quantitative reporters at different concentration of the analyte has also been achieved using a cyclic AMP (cAMP)--sensitive nucleic acid device termed cAMPhor. cAMPhor uses a cAMP aptamer developed by the Breaker group (118) and has been shown to fluorescently map cAMP levels within membrane-bound compartments (101).
A whole range of ion-measuring devices can be similarly realized by using ion-sensitive dyes that undergo a change in photophysical properties upon interaction with a specific ion. Ion-sensitive dyes can substitute the functional nucleic acid motif of the sensing module. The function of the nucleic acid scaffold in such a context is to yield a molecularly homogenous system that couples an ion-sensitive dye to an ion-insensitive dye in a specific stoichiometry by simple hybridization. This technique has been demonstrated in the case of a DNA-based chloride sensor, termed Clensor (16). The sensing module of Clensor is a chloride-sensitive fluorophore, 10,10′-bis[3-carboxypropyl]-9,9′-biacridinium dinitrate termed BAC, which undergoes chloride-specific collisional quenching in cells (119). The reporter module of Clensor comprises BAC and a pH-insensitive fluorophore such as Alexa 647 that allows ratiometric quantification of chloride. The ability to quantify chloride has been used to fluorescently assay the activity of the relevant chloride ion channels within living cells. For example, Clensor was used to pinpoint the localization and function of the ClC (ClC stands for Chloride channel) family of chloride channels inside Drosophila hemocytes (16).
Targeting DNA nanodevices
To quantify analytes in subcellular organelles, one needs bifunctional molecular devices. The first function can be obtained using a measuring module, as described above. The second function can be obtained by encoding the device with targeting information that can localize the nucleic acid device to a specific subcellular organelle. Diverse pathogenic cargo (such as viruses or bacteria that infect the cell) or molecular cargo that the cell needs for its survival (such as nutrients, hormones, signaling peptides, and proteins) enter the cell via specific endocytic pathways (120). Each pathway can be accessed by encoding the molecular programs required to engage a specific receptor or trafficking pathway (Figure 5). Thus, the targeting module of this class of nucleic acid nanodevices can incorporate these specific molecular signals for targeting to intracellular locations of choice. A selection of these molecular programs for targeting are discussed below, using ion-sensing DNA nanodevices as a focal point.
Figure 5.
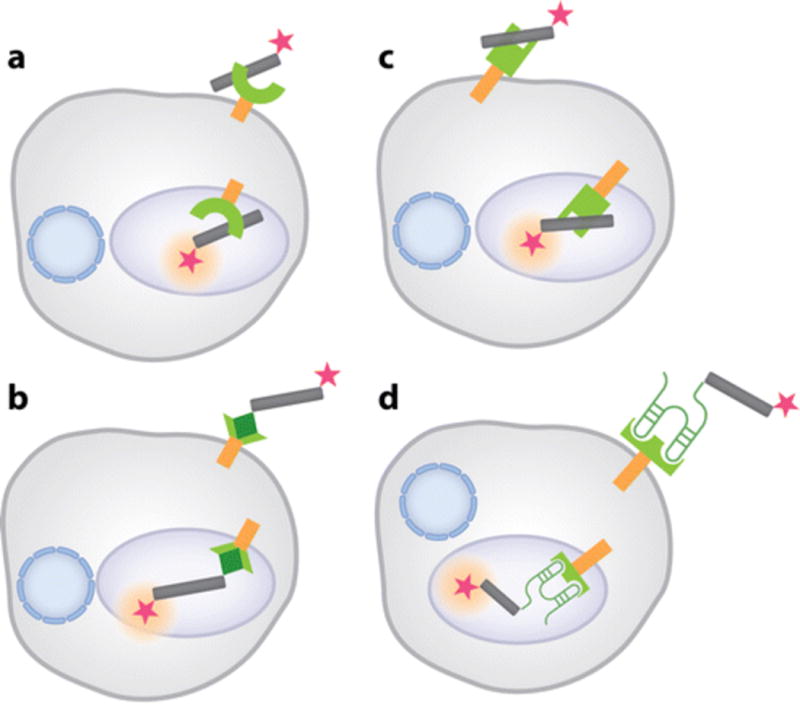
Molecular programs that target DNA nanodevices to intracellular compartments. (a) anionic ligand binding receptors ( ALBRs) or scavenger receptors bind a polyanionic DNA nanodevice (gray cylinders) and target it to endolysosomal organelles. (b) An endocytic ligand (dark green diamond) conjugated to a DNA nanodevice localizes it in organelles along the relevant endocytic receptor (green) pathway. (c) A sequence-specific DNA-binding protein (green) functions as a synthetic receptor to localize a DNA nanodevice within a target organelle by fusion to a shuttle protein (orange). (d) An aptamer (green hairpins) to the extracellular domain (green) of a trafficking protein is integrated to a DNA nanodevice for localization to a target organelle. All devices carry fluorescent tags (magenta stars)
Endocytic ligand and cognate receptor
Endocytic ligands, such as proteins like epidermal growth factor (EGF) or small molecules like folic acid, bind to their cognate receptors. In other words, the EGF receptor or the folate receptor resident at the plasma membrane and are subsequently trafficked inside the cell to specific endocytic organelles (121, 122). DNA reporters are most frequently targeted by simply chemically conjugating the relevant endocytic ligand to the nucleic acid assembly, while ensuring that ligand function is not compromised. The DNA reporter engages the relevant receptor through the chemically conjugated ligand and is then internalized by cells (Figure 5b). Thus, the lumenal ionic environment of various endocytic organelles, such as early, late, and recycling endosomes as well as lysosomes, have been mapped by DNA reporters (15, 16). For example, transferrin binds to the transferrin receptor and is subsequently trafficked to the recycling endosomes before shuttling back to the plasma membrane (123). Modi et al. (15) exploited this pathway to probe pH gradients by using a fluorescent DNA-based pH reporter termed the I-switch in Drosophila SR2+ cells. Similarly, a DNA tetrahedron bearing folic acid was targeted to KB cells expressing folate receptors (124) in a mouse model.
-
▪
Anionic Ligand Binding Receptor (ALBR) and the negatively charged DNA backbone. DNA’s phosphate backbone makes it polyanionic. This inherent negative charge has been exploited to target DNA devices to cells that express the ALBRs (125) or scavenger receptors (53) in macrophages (126). The Krishnan group used the ALBR pathway in Drosophila haemocytes to deliver the I-switch and Clensor to organelles along the endocytic pathway (Figure 5a). The precise measurement of pH and chloride in organelles along the endolysosomal pathway were thus enabled. This targeting mechanism proved robust enough to target other DNA nanodevices, such as DNA polyhedra (127), to coelomocytes in a live multicellular organism like C. elegans (17).
-
▪
Aptamers against cell surface proteins. In another variation, targeting may be achieved by molecularly programming nucleic acid assemblies with aptamers to cell surface receptors. Aptamers are known to bind a variety of chemicals like ions, small molecules, and even proteins (11). By integrating an aptamer to the extracellular domain of a trafficking protein onto a nanodevice, one can traffic a DNA nanodevice from the extracellular milieu to a subcellular compartment (Figure 5d). Recently, Saha et al. (16) integrated a well-known RNA aptamer against the transferrin receptor with a DNA nanodevice, termed Clensor-Tf, to localize Clensor-Tf within recycling endosomes. The aptamer module in Clensor-Tf tightly binds the extracellular domain of the transferrin receptor population on the plasma membrane of cells, leading to its uptake and eventual enrichment in recycling endosomes. Similarly, Chang et al. (128) targeted a DNA icosahedron by integrating an aptamer to the MUC1 protein to deliver doxorubicin to epithelial cancer cells. Another example is the delivery of a DNA pyramid to HeLa cells using an aptamer to nucleolin present on the plasma membrane (129). Aptamer-based targeting can also be used to deliver functional enzymes to a designated subcellular organelle. Chen et al. (130) conjugated a lysosomal enzyme, L-iduronidase to a DNA aptamer, which binds to the transferrin receptor and successfully delivered L-iduronidase to lysosomes in cultured mouse fibroblasts. Similarly, Chu et al. (131) delivered gelonin, a plant toxin that inhibits protein synthesis to cancer cells, that overexpressed prostate-specific membrane antigen and induced selective cell death.
-
▪
Chimera of synthetic DNA receptors. A generalizable synthetic, molecular programming strategy was recently outlined by Modi et al. (114). The advantages of this strategy are that it circumvents both the need for an aptamer to a cell-surface protein as well as the need to chemically conjugate endocytic ligands to DNA nanodevices. The strategy uses a synthetic, sequence-specific DNA binding protein that is expressed as a chimera with a suitable shuttle protein for DNA nanodevice localization within a given target organelle. A shuttle protein traffics between the plasma membrane and the target organelle. A chimera of the DNA binding protein fused to the extracellular domain of the trafficking protein results in the display of a synthetic receptor for DNA by the trafficking protein on the cell surface. The DNA device binds its synthetic receptor and is trafficked intracellularly to its subcellular destination (Figure 5c). Modi et al. (114) thus successfully targeted the I-switch to the trans-Golgi network (TGN) by expressing a chimera of a sequence-specific DNA-binding protein with a proprotein convertase, furin, that shuttles between the plasma membrane and the TGN (132).
OUTLOOK
The nucleic acid scaffold has a number of attractive physicochemical properties as well as biological advantages, many of which have been harnessed to create nanodevices for several imaging applications outlined above. However, a few properties of the scaffold have not yet been harnessed, and these could open up new vistas for biological imaging. For example, electron-dense DNA and RNA can diffract electrons efficiently (133, 134). Thus, reminiscent of AFM-based locator DNA devices, one could envisage DNA origami locator devices designed for cryo--electron microscopy to derive structures for biomolecules that are challenging to observe owing to their small size (H. Dietz and S. Scheres, unpublished data, personal communication. Further, the periodicity of DNA and its well-defined structure might aid refinement methods substantially. Another property of DNA that has not yet been exploited for imaging is its immunogenicity (135). One can use DNA nanodevices to simultaneously stimulate and image the activated immune system. A third property is the embryonic field of RNA localization, in which RNA sequence information also encodes localization within the cell (136). This property could be used to understand biological phenomena associated with spatial positioning of RNA, perhaps at synapses (137), in exosomes (138), or in the cytosol during development (139).
Although the first study of Exchange-PAINT provides a glimpse into the capacity to multiplex with the nucleic acid scaffold (8), parallelization has remained underutilized in nucleic acid--guided biological imaging. One can obtain several sequence combinations of sufficient diversity and identical melting temperatures to perform highly parallelized imaging operations on the same biological sample. Functional imaging probes based on DNA/RNA nanodevices can be bar-coded to protein targets with spectrally distinct fluorophores. Although pH has been measured in two pathways at the same time inside the same cell (114), multiplexed functional imaging of several organelles at a time is yet to be achieved.
Live imaging with nucleic acids is rife with possibilities. Live imaging with super resolution microscopy also remains an outstanding challenge for nucleic acid nanodevices. A plethora of metabolites remain to be imaged and can be detected by using Spinach riboswitches. Currently, at least 100 riboswitches have been computationally identified by comparative analysis of bacterial genomes but are designated as orphans because their target is not known (140–145). Because many riboswitches can be converted into Spinach riboswitches, it is important to deorphanize these riboswitches to expand the repertoire of metabolites that can be imaged.
Finally, the true potential of such novel nucleic acid--based molecular technologies lies in successfully interfacing them with other chemical technologies. For example, consider the use of oligodeoxyfluorosides, which are oligonucleotides composed primarily of fluorophores rather than standard nucleobases. The phosphodiester backbone in the oligodeoxyfluoroside confers aqueous solubility (146–149), and the combination of fluorophores produces a palette of distinct emission wavelengths in response to a single excitation wavelength (147). Coupled with their interfacability to automated synthesis (149) and their ability to sense various metabolites and small molecules (150, 151), they present great potential for live imaging. Other high-end imaging probes such as quantum dots (152, 153) or fluorescent nucleic acid--metal nanoclusters (154) are of particular interest owing to their brightness and potential targetability, which may make them suitable for long-duration in vivo imaging and correlative microscopy. Developments toward multicolor RNA detection probes similar to Spinach technology, but with improved brightness and higher emission maxima, are highly desirable for multiplexed imaging of RNA molecules in living systems (155).
Several cellular compartments still remain to be targeted by externally introducible DNA nanodevices, and the applications of these devices to a variety of model organisms still remain unexplored. Combining these devices with protein-based localization technologies, such as SNAP-tag (156), nanobodies (157), or carbohydrate--based (158) or lipid--based (159) click chemistries, will allow new regions of the cell to be probed with unprecedented clarity.
Acknowledgments
YK acknowledges financial support from the Human Frontier Science Program, DBT-Wellcome Trust India Alliance, the Brain Research Foundation and the University of Chicago. Our sincere apologies to those colleagues whose work could not be included due to space limitations.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review. Y.K. and S.R.J. are authors of patents related to I-switch/Clensor technology and Spinach technology, respectively.
Literature Cited
- 1.Saenger W. Principles of Nucleic Acid Structure. New York: Springer-Verlag; 1984. [Google Scholar]
- 2.Jones MR, Seeman NC, Mirkin CA. Programmable materials and the nature of the DNA bond. Science. 2015;347:1260901. doi: 10.1126/science.1260901. [DOI] [PubMed] [Google Scholar]
- 3.Seeman NC. Nanomaterials based on DNA. Annu. Rev. Biochem. 2010;79:65–87. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winfree E, Liu F, Wenzler LA, Seeman NC. Design and self-assembly of two-dimensional DNA crystals. Nature. 1998;394:539–44. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 5.Rothemund PW, Papadakis N, Winfree E. Algorithmic self-assembly of DNA Sierpinski triangles. PLOS Biol. 2004;2:424. doi: 10.1371/journal.pbio.0020424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothemund PWK. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 7.Douglas SM, Marblestone AH, Teerapittayanon S, Vazquez A, Church GM, Shih WM. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 2009;37:5001–6. doi: 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jungmann R, Avendaño MS, Woehrstein JB, Dai M, Shih WM, Yin P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods. 2014;11:313–18. doi: 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jungmann R, Steinhauer C, Scheible M, Kuzyk A, Tinnefeld P, Simmel FC. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 2010;10:4756–61. doi: 10.1021/nl103427w. [DOI] [PubMed] [Google Scholar]
- 10.Steinhauer C, Jungmann R, Sobey TL, Simmel FC, Tinnefeld P. DNA origami as a nanoscopic ruler for super-resolution microscopy. Angew. Chem. Int. Ed. Engl. 2009;48:8870–73. doi: 10.1002/anie.200903308. [DOI] [PubMed] [Google Scholar]
- 11.Cho EJ, Lee J-W, Ellington AD. Applications of aptamers as sensors. Annu. Rev. Anal. Chem. 2009;2:241–64. doi: 10.1146/annurev.anchem.1.031207.112851. [DOI] [PubMed] [Google Scholar]
- 12.Filonov GS, Moon JD, Svensen N, Jaffrey SR. Broccoli: Rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 2014;136:16299–308. doi: 10.1021/ja508478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paige JS, Wu KY, Jaffrey SR. RNA mimics of green fluorescent protein. Science. 2011;333:642–46. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paige JS, Nguyen-Duc T, Song W, Jaffrey SR. Fluorescence imaging of cellular metabolites with RNA. Science. 2012;335:1194. doi: 10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Modi S, MG S, Goswami D, Gupta GD, Mayor S, Krishnan Y. A DNA nanomachine that maps spatial and temporal pH changes inside living cells. Nat. Nanotechnol. 2009;4:325–30. doi: 10.1038/nnano.2009.83. [DOI] [PubMed] [Google Scholar]
- 16.Saha S, Prakash V, Halder S, Chakraborty K, Krishnan Y. A pH-independent DNA nanodevice for quantifying chloride transport in organelles of living cells. Nat. Nanotechnol. 2015;10:645–51. doi: 10.1038/nnano.2015.130. [DOI] [PubMed] [Google Scholar]
- 17.Surana S, Bhat JM, Koushika SP, Krishnan Y. An autonomous DNA nanomachine maps spatiotemporal pH changes in a multicellular living organism. Nat. Commun. 2011;2:340. doi: 10.1038/ncomms1340. [DOI] [PubMed] [Google Scholar]
- 18.Morris V, Kirby A, Gunning A. Atomic Force Microscopy for Biologists. Imperial College Press; 2010. [Google Scholar]
- 19.Lyubchenko YL, Shlyakhtenko LS, Ando T. Imaging of nucleic acids with atomic force microscopy. Methods. 2011;54:274–83. doi: 10.1016/j.ymeth.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajendran A, Endo M, Sugiyama H. Single-molecule analysis using DNA origami. Angew. Chem. Int. Ed. Engl. 2012;51:874–90. doi: 10.1002/anie.201102113. [DOI] [PubMed] [Google Scholar]
- 21.Rivetti C, Guthold M, Bustamante C. Wrapping of DNA around the E. coli RNA polymerase open promoter complex. EMBO J. 1999;18:4464–75. doi: 10.1093/emboj/18.16.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seeman NC. Nucleic acid junctions and lattices. J. Theor. Biol. 1982;99:237–47. doi: 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 23.Yan H, Park SH, Finkelstein G, Reif JH, LaBean TH. DNA-templated self-assembly of protein arrays and highly conductive nanowires. Science. 2003;301:1882–84. doi: 10.1126/science.1089389. [DOI] [PubMed] [Google Scholar]
- 24.Rinker S, Ke Y, Liu Y, Chhabra R, Yan H. Self-assembled DNA nanostructures for distance-dependent multivalent ligand-protein binding. Nat. Nanotechnol. 2008;3:418–22. doi: 10.1038/nnano.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chhabra R, Sharma J, Ke Y, Liu Y, Rinker S, et al. Spatially addressable multiprotein nanoarrays templated by aptamer-tagged DNA nanoarchitectures. J. Am. Chem. Soc. 2007;129:10304–5. doi: 10.1021/ja072410u. [DOI] [PubMed] [Google Scholar]
- 26.Wilner OI, Weizmann Y, Gill R, Lioubashevski O, Freeman R, Willner I. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nanotechnol. 2009;4:249–54. doi: 10.1038/nnano.2009.50. [DOI] [PubMed] [Google Scholar]
- 27.Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. Organization of intracellular reactions with rationally designed RNA assemblies. Science. 2011;333:470–74. doi: 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- 28.Ando T, Uchihashi T, Kodera N. High-speed AFM and applications to biomolecular systems. Annu. Rev. Biophys. 2013;42:393–414. doi: 10.1146/annurev-biophys-083012-130324. [DOI] [PubMed] [Google Scholar]
- 29.Rajendran A, Endo M, Hidaka K, Tran PLT, Mergny JL, et al. HIV-1 nucleocapsid proteins as molecular chaperones for tetramolecular antiparallel g-quadruplex formation. J. Am. Chem. Soc. 2013;135:18575–85. doi: 10.1021/ja409085j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Endo M, Tatsumi K, Terushima K, Katsuda Y, Hidaka K, et al. Direct visualization of the movement of a single T7 RNA polymerase and transcription on a DNA nanostructure. Angew. Chem. Int. Ed. Engl. 2012;51:8778–82. doi: 10.1002/anie.201201890. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki Y, Endo M, Katsuda Y, Ou K, Hidaka K, Sugiyama H. DNA origami based visualization system for studying site-specific recombination events. J. Am. Chem. Soc. 2014;136:211–18. doi: 10.1021/ja408656y. [DOI] [PubMed] [Google Scholar]
- 32.Voigt NV, Tørring T, Rotaru A, Jacobsen MF, Ravnsbaek JB, et al. Single-molecule chemical reactions on DNA origami. Nat. Nanotechnol. 2010;5:200–3. doi: 10.1038/nnano.2010.5. [DOI] [PubMed] [Google Scholar]
- 33.Youngblood B, Reich NO. Conformational transitions as determinants of specificity for the DNA methyltransferase EcoRI. J. Biol. Chem. 2006;281:26821–31. doi: 10.1074/jbc.M603388200. [DOI] [PubMed] [Google Scholar]
- 34.Endo M, Katsuda Y, Hidaka K, Sugiyama H. Regulation of DNA methylation using different tensions of double strands constructed in a defined DNA nanostructure. J. Am. Chem. Soc. 2010;132:1592–97. doi: 10.1021/ja907649w. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto S, De D, Hidaka K, Kim KK, Endo M, Sugiyama H. Single molecule visualization and characterization of Sox2-Pax6 complex formation on a regulatory DNA element using a DNA origami frame. Nano Lett. 2014;14:2286–92. doi: 10.1021/nl4044949. [DOI] [PubMed] [Google Scholar]
- 36.Endo M, Katsuda Y, Hidaka K, Sugiyama H. A versatile DNA nanochip for direct analysis of DNA base-excision repair. Angew. Chem. Int. Ed. Engl. 2010;49:9412–16. doi: 10.1002/anie.201003604. [DOI] [PubMed] [Google Scholar]
- 37.Rajendran A, Endo M, Hidaka K, Sugiyama H. Direct and single-molecule visualization of the solution-state structures of G-hairpin and G-triplex intermediates. Angew. Chem. Int. Ed. Engl. 2014;53:4107–12. doi: 10.1002/anie.201308903. [DOI] [PubMed] [Google Scholar]
- 38.Pardue ML, Gall JG. Molecular hybridization of radioactive DNA to the DNA of cytological preparations. PNAS. 1969;64:600–4. doi: 10.1073/pnas.64.2.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rudkin GT, Stollar BD. High resolution detection of DNA-RNA hybrids in situ by indirect immunofluorescence. Nature. 1977;265:472–73. doi: 10.1038/265472a0. [DOI] [PubMed] [Google Scholar]
- 40.Bauman JG, Wiegant J, Borst P, van Duijn P. A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochromelabelled RNA. Exp. Cell Res. 1980;128:485–90. doi: 10.1016/0014-4827(80)90087-7. [DOI] [PubMed] [Google Scholar]
- 41.Femino AM, Fay FS, Fogarty K, Singer RH. Visualization of single RNA transcripts in situ. Science. 1998;280:585–90. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- 42.Raj A, van den Bogaard P, Rifkin Sa, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008;5:877–79. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martens UM, Zijlmans JM, Poon SS, Dragowska W, Yui J, et al. Short telomeres on human chromosome 17p. Nat. Genet. 1998;18:76–80. doi: 10.1038/ng0198-018. [DOI] [PubMed] [Google Scholar]
- 44.Wienholds E, Kloosterman WP, Miska E, Alvarez-saavedra E, Berezikov E, et al. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–11. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- 45.Kloosterman WP, Wienholds E, de Bruijn E, Kauppinen S, Plasterk RHA. In situ detection of miRNAs in animal embryos using LNA-modified oligonucleotide probes. Nat. Methods. 2006;3:27–29. doi: 10.1038/nmeth843. [DOI] [PubMed] [Google Scholar]
- 46.Lansdorp PM, Verwoerd NP, Van De Rijke FM, Dragowska V, Little MT, et al. Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet. 1996;5:685–91. doi: 10.1093/hmg/5.5.685. [DOI] [PubMed] [Google Scholar]
- 47.Lu J, Tsourkas A. Imaging individual microRNAs in single mammalian cells in situ. Nucleic Acids Res. 2009;37:1–10. doi: 10.1093/nar/gkp482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown J, Saracoglu K, Uhrig S, Speicher MR, Eils R, Kearney L. Subtelomeric chromosome rearrangements are detected using an innovative 12-color FISH assay (M-TEL) Nat. Med. 2001;7:497–501. doi: 10.1038/86581. [DOI] [PubMed] [Google Scholar]
- 49.Speicher MR, Gwyn Ballard S, Ward DC. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat. Genet. 1996;12:368–75. doi: 10.1038/ng0496-368. [DOI] [PubMed] [Google Scholar]
- 49a.Speicher MR, Carter NP. The new cytogenetics: blurring the boundaries with molecular biology. Nat. Rev. Genet. 2005;6:782–92. doi: 10.1038/nrg1692. [DOI] [PubMed] [Google Scholar]
- 50.Evanko D. Hybridization chain reaction. Nat. Methods. 2004;1:186–87. [Google Scholar]
- 51.Choi HMT, Chang JY, Trinh La, Padilla JE, Fraser SE, Pierce Na. Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat. Biotechnol. 2010;28:1208–12. doi: 10.1038/nbt.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian X, Lloyd RV. Recent developments in signal amplification methods for in situ hybridization. Diagn. Mol. Pathol. 2003;12:1–13. doi: 10.1097/00019606-200303000-00001. [DOI] [PubMed] [Google Scholar]
- 53.Bagasra O. Protocols for the in situ PCR-amplification and detection of mRNA and DNA sequences. Nat. Protoc. 2007;2:2782–95. doi: 10.1038/nprot.2007.395. [DOI] [PubMed] [Google Scholar]
- 54.Larsson C, Koch J, Nygren A, Janssen G, Raap AK, et al. In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes. Nat. Methods. 2004;1:227–32. doi: 10.1038/nmeth723. [DOI] [PubMed] [Google Scholar]
- 55.Larsson C, Grundberg I, Söderberg O, Nilsson M. In situ detection and genotyping of individual mRNA molecules. Nat. Methods. 2010;7:395–97. doi: 10.1038/nmeth.1448. [DOI] [PubMed] [Google Scholar]
- 56.Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 2015;10:442–58. doi: 10.1038/nprot.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim DN, Kilchherr F, Dietz H, Bathe M. Quantitative prediction of 3D solution shape and flexibility of nucleic acid nanostructures. Nucleic Acids Res. 2012;40:2862–68. doi: 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Castro CE, Kilchherr F, Kim DN, Shiao EL, Wauer T, et al. A primer to scaffolded DNA origami. Nat. Methods. 2011;8:221–29. doi: 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- 59.Huang B, Bates M, Zhuang X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gordon MP, Ha T, Selvin PR. Single-molecule high-resolution imaging with photobleaching. PNAS. 2004;101:6462–65. doi: 10.1073/pnas.0401638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steinhauer C, Forthmann C, Vogelsang J, Tinnefeld P. Superresolution microscopy on the basis of engineered dark states. J. Am. Chem. Soc. 2008;130:16840–41. doi: 10.1021/ja806590m. [DOI] [PubMed] [Google Scholar]
- 62.Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010;190:165–75. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmied JJ, Gietl A, Holzmeister P, Forthmann C, Steinhauer C, et al. Fluorescence and super-resolution standards based on DNA origami. Nat. Methods. 2012;9:1133–34. doi: 10.1038/nmeth.2254. [DOI] [PubMed] [Google Scholar]
- 64.Schmied JJ, Raab M, Forthmann C, Pibiri E, Wünsch B, et al. DNA origami-based standards for quantitative fluorescence microscopy. Nat. Protoc. 2014;9:1367–91. doi: 10.1038/nprot.2014.079. [DOI] [PubMed] [Google Scholar]
- 65.Schmied JJ, Forthmann C, Pibiri E, Lalkens B, Nickels P, et al. DNA origami nanopillars as standards for three-dimensional superresolution microscopy. Nano Lett. 2013;13:781–85. doi: 10.1021/nl304492y. [DOI] [PubMed] [Google Scholar]
- 66.Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. PNAS. 2006;103:18911–16. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giannone G, Hosy E, Levet F, Constals A, Schulze K, et al. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J. 2010;99:1303–10. doi: 10.1016/j.bpj.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Derr ND, Goodman BS, Jungmann R, Leschziner AE, Shih WM, Reck-Peterson SL. Tug-of-war in motor protein ensembles revealed with a programmable DNA origami scaffold. Science. 2012;338:662–65. doi: 10.1126/science.1226734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Molenaar C, Abdulle A, Gena A, Tanke HJ, Dirks RW. Poly(A)+ RNAs roam the cell nucleus and pass through speckle domains in transcriptionally active and inactive cells. J. Cell Biol. 2004;165:191–202. doi: 10.1083/jcb.200310139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Molenaar C, Marras SA, Slats JCM, Truffert J-C, Lemaitre M, et al. Linear 2′ O-Methyl RNA probes for the visualization of RNA in living cells. Nucleic Acids Res. 2001;29:e89. doi: 10.1093/nar/29.17.e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bao G, Tsourkas A, Santangelo PJ. Engineering nanostructured probes for sensitive intracellular gene detection. Mech. Chem. Biosyst. 2004;1:23–36. [PubMed] [Google Scholar]
- 72.Bratu DP, Cha BJ, Mhlanga MM, Kramer FR, Tyagi S. Visualizing the distribution and transport of mRNAs in living cells. PNAS. 2003;100:13308–13. doi: 10.1073/pnas.2233244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nitin N, Santangelo PJ, Kim G, Nie S, Bao G. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic Acids Res. 2004;32:e58. doi: 10.1093/nar/gnh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tyagi S, Alsmadi O. Imaging native β-actin mRNA in motile fibroblasts. Biophys. J. 2004;87:4153–62. doi: 10.1529/biophysj.104.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 1996;14:303–8. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 76.Monroy-Contreras R, Vaca L. Molecular beacons: powerful tools for imaging RNA in living cells. J. Nucleic Acids. 2011;2011:741723. doi: 10.4061/2011/741723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bao G, Rhee WJ, Tsourkas A. Fluorescent probes for live-cell RNA detection. Annu. Rev. Biomed. Eng. 2009;11:25–47. doi: 10.1146/annurev-bioeng-061008-124920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giles RV, Spiller DG, Grzybowski J, Clark RE, Nicklin P, Tidd DM. Selecting optimal oligonucleotide composition for maximal antisense effect following streptolysin O-mediated delivery into human leukaemia cells. Nucleic Acids Res. 1998;26:1567–75. doi: 10.1093/nar/26.7.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Santangelo PJ, Alonas E, Jung J, Lifland AW, Zurla C. Probes for intracellular RNA imaging in live cells. Methods Enzymol. 2012;505:383–99. doi: 10.1016/B978-0-12-388448-0.00028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 1998;2:437–45. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- 81.Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, et al. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr. Biol. 2003;13:161–67. doi: 10.1016/s0960-9822(02)01436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shav-Tal Y, Darzacq X, Shenoy SM, Fusco D, Janicki SM, et al. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Park HY, Lim H, Yoon YJ, Follenzi A, Nwokafor C, et al. Visualization of dynamics of single endogenous mRNA labeled in live mouse. Science. 2014;343:422–24. doi: 10.1126/science.1239200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Daigle N, Ellenberg J. λN-GFP: an RNA reporter system for live-cell imaging. Nat. Methods. 2007;4:633–36. doi: 10.1038/nmeth1065. [DOI] [PubMed] [Google Scholar]
- 85.Ozawa T, Natori Y, Sato M, Umezawa Y. Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nat. Methods. 2007;4:413–19. doi: 10.1038/nmeth1030. [DOI] [PubMed] [Google Scholar]
- 86.Tilsner J. Pumilio-based RNA in vivo imaging. Methods Mol. Biol. 2015;1217:295–328. doi: 10.1007/978-1-4939-1523-1_20. [DOI] [PubMed] [Google Scholar]
- 87.Tilsner J, Linnik O, Christensen NM, Bell K, Roberts IM, et al. Live-cell imaging of viral RNA genomes using a Pumilio-based reporter. Plant J. 2009;57:758–70. doi: 10.1111/j.1365-313X.2008.03720.x. [DOI] [PubMed] [Google Scholar]
- 88.Song W, Strack RL, Svensen N, Jaffrey SR. Plug-and-play fluorophores extend the spectral properties of Spinach. J. Am. Chem. Soc. 2014;136:1198–201. doi: 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bradshaw RA, Dennis EA. Handbook of Cell Signaling. New York: Academic; 2004. [Google Scholar]
- 90.Krishnan Y, Simmel FC. Nucleic acid based molecular devices. Angew. Chem. Int. Ed. Engl. 2011;50:3124–56. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]
- 91.Krishnan Y, Bathe M. Designer nucleic acids to probe and program the cell. Trends Cell Biol. 2012;22:624–33. doi: 10.1016/j.tcb.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 92.Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991;5:2108–16. doi: 10.1101/gad.5.11.2108. [DOI] [PubMed] [Google Scholar]
- 93.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002;3:906–18. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 94.Frommer WB, Davidson MW, Campbell RE. Genetically encoded biosensors based on engineered fluorescent proteins. Chem. Soc. Rev. 2009;38:2833–41. doi: 10.1039/b907749a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lakowicz JR. Principles of Fluorescence Spectroscopy. New York: Springer; 1999. [Google Scholar]
- 96.Lindenburg L, Merkx M. Engineering genetically encoded FRET sensors. Sensors. 2014;14:11691–713. doi: 10.3390/s140711691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song W, Strack RL, Jaffrey SR. Imaging bacterial protein expression using genetically encoded RNA sensors. Nat. Methods. 2013;10:873–75. doi: 10.1038/nmeth.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kellenberger CA, Wilson SC, Sales-Lee J, Hammond MC. RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. J. Am. Chem. Soc. 2013;135:4906–9. doi: 10.1021/ja311960g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kellenberger CA, Chen C, Whiteley AT, Portnoy Da, Hammond MC. RNA-based fluorescent biosensors for live cell imaging of second messenger cyclic di-AMP. J. Am. Chem. Soc. 2015;137:6432–35. doi: 10.1021/jacs.5b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kellenberger CA, Wilson SC, Hickey SF, Gonzalez TL, Su Y, et al. GEMM-I riboswitches from Geobacter sense the bacterial second messenger cyclic AMP-GMP. PNAS. 2015;112:5383–88. doi: 10.1073/pnas.1419328112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sharma S, Zaveri A, Visweswariah SS, Krishnan Y. A fluorescent nucleic acid nanodevice quantitatively images elevated cyclic adenosine monophosphate in membrane-bound compartments. Small. 2014;10:1–5. doi: 10.1002/smll.201400833. [DOI] [PubMed] [Google Scholar]
- 102.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chem. Biol. 2002;9:1043–49. doi: 10.1016/s1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 103.Serganov A, Nudler E. A decade of riboswitches. Cell. 2013;152:17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.You M, Litke JL, Jaffrey SR. Imaging metabolite dynamics in living cells using a Spinach-based riboswitch. PNAS. 2015;112:E2756–65. doi: 10.1073/pnas.1504354112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kellenberger CA, Hammond MC. In vitro analysis of riboswitch-Spinach aptamer fusions as metabolite-sensing fluorescent biosensors. Methods Enzymol. 2015;550:147–72. doi: 10.1016/bs.mie.2014.10.045. [DOI] [PubMed] [Google Scholar]
- 106.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–10. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 107.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–22. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 108.Stoltenburg R, Reinemann C, Strehlitz B. SELEX---a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007;24:381–403. doi: 10.1016/j.bioeng.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 109.Strack RL, Jaffrey SR. New approaches for sensing metabolites and proteins in live cells using RNA. Curr. Opin. Chem. Biol. 2013;17:651–55. doi: 10.1016/j.cbpa.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Klug SJ, Famulok M. All you wanted to know about SELEX. Mol. Biol. Rep. 1994;20:97–107. doi: 10.1007/BF00996358. [DOI] [PubMed] [Google Scholar]
- 111.Bhatia D, Sharma S, Krishnan Y. Synthetic, biofunctional nucleic acid-based molecular devices. Curr. Opin. Biotechnol. 2011;22:475–84. doi: 10.1016/j.copbio.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 112.Halder S, Krishnan Y. Design of ultrasensitive DNA-based fluorescent pH sensitive nanodevices. Nanoscale. 2015;7:10008–12. doi: 10.1039/c5nr01158b. [DOI] [PubMed] [Google Scholar]
- 113.Liu D, Balasubramanian S. A proton-fuelled DNA nanomachine. Angew. Chem. Int. Ed. Engl. 2003;42:5734–36. doi: 10.1002/anie.200352402. [DOI] [PubMed] [Google Scholar]
- 114.Modi S, Nizak C, Surana S, Halder S, Krishnan Y. Two DNA nanomachines map pH changes along intersecting endocytic pathways inside the same cell. Nat. Nanotechnol. 2013;8:459–67. doi: 10.1038/nnano.2013.92. [DOI] [PubMed] [Google Scholar]
- 115.Modi S, Halder S, Nizak C, Krishnan Y. Recombinant antibody mediated delivery of organelle-specific DNA pH sensors along endocytic pathways. Nanoscale. 2014;6:1144–52. doi: 10.1039/c3nr03769j. [DOI] [PubMed] [Google Scholar]
- 116.Idili A, Vallée-Bélisle A, Ricci F. Programmable pH-triggered DNA nanoswitches. J. Am. Chem. Soc. 2014;136:5836–39. doi: 10.1021/ja500619w. [DOI] [PubMed] [Google Scholar]
- 117.Lannes L, Halder S, Krishnan Y, Schwalbe H. Tuning the pH Response of i-Motif DNA Oligonucleotides. ChemBioChem. 2015;16:1647–56. doi: 10.1002/cbic.201500182. [DOI] [PubMed] [Google Scholar]
- 118.Koizumi M, Breaker RR. Molecular recognition of cAMP by an RNA aptamer. Biochemistry. 2000;39:8983–92. doi: 10.1021/bi000149n. [DOI] [PubMed] [Google Scholar]
- 119.Sonawane ND, Thiagarajah JR, Verkman AS. Chloride concentration in endosomes measured using a ratioable fluorescent Cl indicator: evidence for chloride accumulation during acidification. J. Biol. Chem. 2002;277:5506–13. doi: 10.1074/jbc.M110818200. [DOI] [PubMed] [Google Scholar]
- 120.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 121.Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014;24:26–34. doi: 10.1016/j.tcb.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sabharanjak S, Mayor S. Folate receptor endocytosis and trafficking. Adv. Drug Deliv. Rev. 2004;56:1099–109. doi: 10.1016/j.addr.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 123.Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002;54:561–87. doi: 10.1124/pr.54.4.561. [DOI] [PubMed] [Google Scholar]
- 124.Lee H, Lytton-Jean AKR, Chen Y, Love KT, Park AI, et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 2012;7:389–93. doi: 10.1038/nnano.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Albrecht C. Review of Joseph R. Lakowicz: Principles of Fluorescence Spectroscopy, 3rd edition. Anal. Bioanal. Chem. 2008;390:1223–24. [Google Scholar]
- 126.Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013;13:621–34. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- 127.Bhatia D, Surana S, Chakraborty S, Koushika SP, Krishnan Y. A synthetic icosahedral DNA-based host-cargo complex for functional in vivo imaging. Nat. Commun. 2011;2:339. doi: 10.1038/ncomms1337. [DOI] [PubMed] [Google Scholar]
- 128.Chang M, Yang CS, Huang DM. Aptamer-conjugated DNA icosahedral nanoparticles as a carrier of doxorubicin for cancer therapy. ACS Nano. 2011;5:6156–63. doi: 10.1021/nn200693a. [DOI] [PubMed] [Google Scholar]
- 129.Charoenphol P, Bermudez H. Aptamer-targeted DNA nanostructures for therapeutic delivery. Mol. Pharm. 2014;11:1721–25. doi: 10.1021/mp500047b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen C-hB, Dellamaggiore KR, Ouellette CP, Sedano CD, Lizadjohry M, et al. Aptamer-based endocytosis of a lysosomal enzyme. PNAS. 2008;105:15908–13. doi: 10.1073/pnas.0808360105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chu TC, Marks JW, Lavery LA, Faulkner S, Rosenblum MG, et al. Aptamer:toxin conjugates that specifically target prostate tumor cells. Cancer Res. 2006;66:5989–92. doi: 10.1158/0008-5472.CAN-05-4583. [DOI] [PubMed] [Google Scholar]
- 132.Molloy SS, Thomas L, VanSlyke JK, Stenberg PE, Thomas G. Intracellular trafficking and activation of the furin proprotein convertase: localization to the TGN and recycling from the cell surface. EMBO J. 1994;13:18–33. doi: 10.1002/j.1460-2075.1994.tb06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Selmi DN, Adamson RJ, Attrill H, Goddard AD, Gilbert RJC, et al. DNA-templated protein arrays for single-molecule imaging. Nano Lett. 2011;11:657–60. doi: 10.1021/nl1037769. [DOI] [PubMed] [Google Scholar]
- 134.Bai X-C, Martin TG, Scheres SHW, Dietz H. Cryo-EM structure of a 3D DNA-origami object. PNAS. 2012;109:20012–17. doi: 10.1073/pnas.1215713109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 136.Hwang H-W, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- 137.Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, et al. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol. Cell. 2011;42:673–88. doi: 10.1016/j.molcel.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011;2:282. doi: 10.1038/ncomms1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Martin KC, Ephrussi A. mRNA localization: gene expression in the spatial dimension. Cell. 2009;136:719–30. doi: 10.1016/j.cell.2009.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Weinberg Z, Wang JX, Bogue J, Yang J, Corbino K, et al. Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol. 2010;11:R31. doi: 10.1186/gb-2010-11-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, et al. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. PNAS. 2004;101:6421–26. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baker JL, Sudarsan N, Weinberg Z, Roth A, Stockbridge RB, Breaker RR. Widespread genetic switches and toxicity resistance proteins for fluoride. Science. 2012;335:233–35. doi: 10.1126/science.1215063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, et al. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 2007;35:4809–19. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, et al. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–79. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 145.Nelson JW, Sudarsan N, Furukawa K, Weinberg Z, Wang JX, Breaker RR. Riboswitches in eubacteria sense the second messenger c-di-AMP. Nat. Chem. Biol. 2013;9:834–39. doi: 10.1038/nchembio.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Teo YN, Kool ET. Polyfluorophore excimers and exciplexes as FRET donors in DNA. Bioconjug. Chem. 2009;20:2371–80. doi: 10.1021/bc9003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Teo YN, Wilson JN, Kool ET. Polyfluorophores on a DNA backbone: a multicolor set of labels excited at one wavelength. J. Am. Chem. Soc. 2009;131:3923–33. doi: 10.1021/ja805502k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wilson JN, Gao J, Kool ET. Oligodeoxyfluorosides: strong sequence dependence of fluorescence emission. Tetrahedron. 2007;63:3427–33. doi: 10.1016/j.tet.2006.07.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wilson JN, Kool ET. Fluorescent DNA base replacements: reporters and sensors for biological systems. Org. Biomol. Chem. 2006;4:4265–74. doi: 10.1039/b612284c. [DOI] [PubMed] [Google Scholar]
- 150.Kwon H, Jiang W, Kool ET. Pattern-based detection of anion pollutants in water with DNA polyfluorophores. Chem. Sci. 2015;6:2575–83. doi: 10.1039/c4sc03992k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Samain F, Dai N, Kool ET. Differentiating a diverse range of volatile organic compounds with polyfluorophore sensors built on a DNA scaffold. Chemistry. 2011;17:174–83. doi: 10.1002/chem.201002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Farlow J, Seo D, Broaders KE, Taylor MJ, Gartner ZJ, Jun Y-W. Formation of targeted monovalent quantum dots by steric exclusion. Nat. Methods. 2013;10:1203–5. doi: 10.1038/nmeth.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Banerjee A, Grazon C, Nadal B, Pons T, Krishnan Y, Dubertret B. Fast, efficient, and stable conjugation of multiple DNA strands on colloidal quantum dots. Bioconjug. Chem. 2015;26:1582–89. doi: 10.1021/acs.bioconjchem.5b00221. [DOI] [PubMed] [Google Scholar]
- 154.Yuan Z, Chen Y-C, Li H-W, Chang H-T. Fluorescent silver nanoclusters stabilized by DNA scaffolds. Chem. Commun. 2014;50:9800–15. doi: 10.1039/c4cc02981j. [DOI] [PubMed] [Google Scholar]
- 155.Dolgosheina EV, Jeng SCY, Panchapakesan SSS, Cojocaru R, Chen PSK, et al. RNA Mango aptamer-fluorophore: a bright, high-affinity complex for RNA labeling and tracking. ACS Chem. Biol. 2014;9:2412–20. doi: 10.1021/cb500499x. [DOI] [PubMed] [Google Scholar]
- 156.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 157.Muyldermans S. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 2013;82:775–97. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 158.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, et al. Copper-free click chemistry for dynamic in vivo imaging. PNAS. 2007;104:16793–97. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, et al. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J. Am. Chem. Soc. 2009;131:4967–75. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]