

Abstract
Alcoholic cardiomyopathy (ACM) can develop after consumption of relatively large amounts of alcohol over time or from acute binge drinking. Of the many factors implicated in the etiology of ACM, chronic perturbation in protein balance has been strongly implicated. This review focuses on recent contributions (since 2010) in the area of protein metabolism and cardiac function related to ACM. Data reviewed include that from in vitro and preclinical in vivo animal studies where alcohol or an oxidative metabolite was studied and outcome measures in either cardiomyocytes or whole heart pertaining to protein synthesis or degradation were reported. Additionally, studies on the contractile properties of cardiomyocytes were also included to link signal transduction with function. Methodological differences including the potential impact of sex, dosing and duration/timing of alcohol administration are addressed. Acute and chronic alcohol consumption decreases cardiac protein synthesis and/or activation of proteins within the regulatory mammalian/mechanistic target of rapamycin complex (mTORC1) pathway. Albeit limited, evidence suggests that myocardial protein degradation via the ubiquitin pathway is not altered, while autophagy may be enhanced in ACM. Alcohol impairs ex vivo cardiomyocyte contractility in relation to its metabolism and expression of proteins within the growth factor pathway. Dysregulation of protein metabolism, including the rate of protein synthesis and autophagy, may contribute to contractile deficits and is a hallmark feature of ACM meriting additional sex-inclusive, methodologically consistent studies.
Keywords: protein synthesis, protein degradation, autophagy, heart function, myocardial contractility
Introduction
The detrimental impact of both acute and chronic consumption of excess alcohol (i.e., ethanol) is well-known in relation to several organ systems including the cardiovascular system. While lower levels of alcohol intake (1-2 standard drinks/day) are often cited as having a beneficial or protective effect, sustained higher levels of alcohol consumption can lead to alcoholic cardiomyopathy (ACM) (Krenz and Korthuis, 2012). Numerous pathological and molecular changes accompany the development and progression of ACM and as such, combinations of aberrant physiological processes likely contribute to ACM onset in a manner that is poorly defined. Among these mechanisms is the dysregulation of protein homeostasis often referred to as proteostasis, that includes protein imbalance (decreased synthesis, increased breakdown) as well as the improper folding, assembly and/or trafficking of intracellular proteins, all of which may ultimately impair organ function (Wiersma et al., 2016). A myriad of pathways and mechanisms contribute to the regulation of proteostasis and as a tissue composed of cells with limited proliferative capacity the importance of identifying alcohol-mediated defects cannot be underscored. This work will focus on two of the major cellular aspects of proteostasis, protein synthesis and protein degradation, following acute and chronic alcohol (Figure 1). The proteostatic response is activated by alcohol in an attempt to restore normal cellular, molecular and physiological function; however, this is rarely achieved especially under conditions where consumption of this toxic agent is excessive in amount and prolonged in duration.
Figure 1.
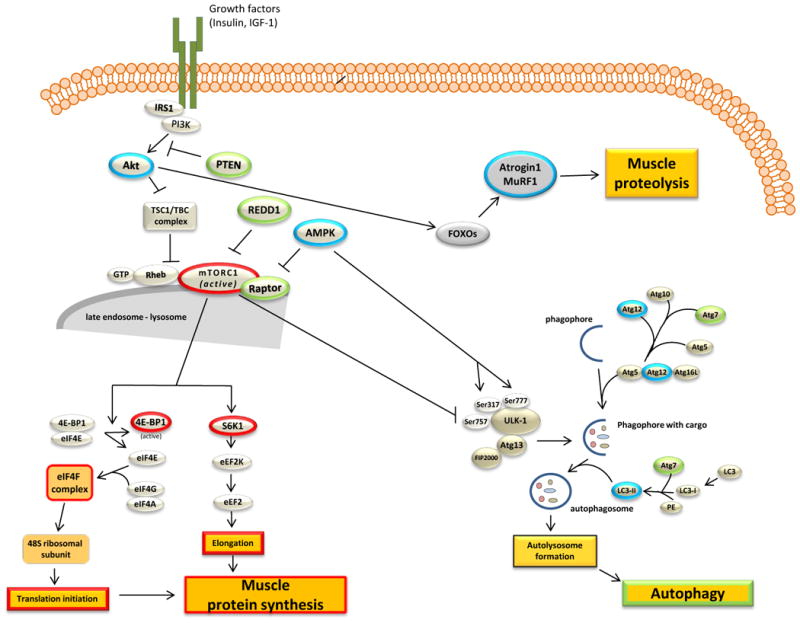
Overview of the signaling pathways involved in protein balance in relation to alcoholic cardiomyopathy. Proteins and processes that have been outlined in red are those that have be conclusively shown to be decreased by alcohol intoxication. Those proteins outlined in green are those that are increased, however in many cases an increase is indicative of an inhibitory action. Blue outlining represents those markers discussed but either no consensus or no change has been reported. Arrows typically represent activation and hashed lines indicate inhibition.
The purpose of this review is to highlight emerging data in relation to alcohol-induced changes in cardiac protein balance focusing primarily on research published since ∼ 2010 that included: (1) measurements made in vitro in cardiomyocytes or in vivo in heart tissue in response to physiological levels of alcohol or its oxidative metabolites encompassing endpoints related to protein synthesis or degradation; or (2) myocyte contractility or cardiac performance in relation to these conditions. Comprehensive searches using PubMed and GoogleScholar as well as previously cited references from related publications (primary articles and reviews) were performed. Throughout the review, the effects of acute versus chronic alcohol have been differentiated along with providing other protocol-related differences including alcohol dose and sex. Overall, current data corroborate past research showing alcohol decreases protein synthesis; however, our knowledge of alcohol-mediated changes in protein degradation has been greatly enhanced by recent work that has focused on delineating the effects of acute and chronic alcohol on autophagy.
Protein synthesis
Background
Seminal work related to ACM, protein synthesis and regulatory signaling pathways has been reviewed previously (Lang et al., 2005, Lang et al., 2001, Preedy et al., 1999), and therefore is only briefly highlighted below to provide context for the more recent findings presented herein.
The decrease in myocardial protein content is due in part to a suppression of global protein synthesis (Preedy et al., 1999, Preedy and Richardson, 1994, Preedy and Richardson, 1996, Siddiq et al., 1993) and the underlying mechanism may differ depending on the duration of alcohol exposure. For example, the decrement in cardiac protein synthesis is associated with impairment of both mRNA translation initiation and elongation after chronic alcohol (Lang et al., 1999, Vary et al., 2001a) while acute alcohol intoxication appears to selectively inhibit translation initiation (Lang et al., 2000a, Lang et al., 2000b). In regard to the expression of the regulatory proteins mediating these translational events, alcohol did not alter eukaryotic initiation factor (eIF)-2B signaling or the content/phosphorylation of eIF4E (Lang et al., 2003, Lang et al., 2000a, Lang et al., 2001, Lang et al., 1999, Vary et al., 2001a, Vary et al., 2001b), but instead exerts its inhibitory actions by decreasing the phosphorylation of eIF4E binding protein-1 (4E-BP1) (Lang et al., 2000a, Lang et al., 2001, Lang et al., 1999, Vary et al., 2001a, Vary et al., 2004) (Figure 1). The alcohol-induced dephosphorylation of 4E-BP1 increases the affinity of this protein for eIF4E and as a result the association of eIF4E with eIF4G is decreased leading to inhibition of cap-dependent mRNA translation and protein synthesis. Alcohol not only decreased the phosphorylation of 4E-BP1, but also decreased the phosphorylation of p70S6 Kinase 1 (S6K1) (Lang et al., 2000a, Vary and Lang, 2008, Vary et al., 2001b), another integral protein in the activation of translation initiation. The mammalian/mechanistic target of rapamycin complex (mTORC)-1, is a key signal integration site and regulator of protein synthesis in cardiac muscle via phosphorylation of its downstream substrates 4E-BP1 and S6K1 (Sciarretta et al., 2014). Both acute (male rats, 75 mmol/kg body weight (BW)) and chronic alcohol (male rats, 40% agar block and 10% in water, 20-26 wk) administration to rats decreased mTORC1 kinase activity in association with inhibition of protein synthesis (Vary et al., 2005, Vary et al., 2008). In recent years the response of these target proteins to alcohol has been confirmed and extended as discussed below.
Chronic alcohol, protein synthesis and signaling
Recent investigations continue to support the alcohol-induced inhibition of mTORC1 as evidenced by the decreased phosphorylation of S6K1 (Guo et al., 2012, Lang and Korzick, 2014) and 4E-BP1 (Lang and Korzick, 2014), as well as the autophosphorylation of mTOR (Elmadhun et al., 2014, Guo et al., 2012, Zhang et al., 2010) (Figure 1). However, the strain, age and sex of the animal may potentially represent confounding variables in response to alcohol. A recent systematic investigation has demonstrated that myocardial mTOR signaling is age-, sex-, strain- and substrate-dependent (Baar et al., 2016). For example, the ability of refeeding to increase the phosphorylation of ribosomal protein S6 (rpS6) – a downstream substrate of S6K1 – was blunted in old (26 month old) female mice [vs. young (6 month old) females], but not in old (30 month old) male mice (Baar et al., 2016). Further, when young male and female mice were directly compared, the extent of rpS6 and 4E-BP1 phosphorylation in heart was greater in females (Baar et al., 2016). Prior to this work, the effect of aging on mTOR signaling in heart was reported to be decreased in male rats and preserved by caloric restriction, thereby providing additional evidence of the potential modulatory effects of these intrinsic variables on mTOR signaling outcomes in the heart (Linford et al., 2007). Lastly, there is also consistent evidence for age-dependent alterations in proteostasis (Wiersma et al., 2016). For example, chronic alcohol consumption did not decrease S6K1 Thr389-phosphorylation in adult (8 month) male Sprague-Dawley, Long Evans (36% kcal liquid diet, 8 wks) and Fischer 344 (36% kcal liquid diet, 20 wks) rats, but did decrease S6K1 phosphorylation in older (22 month) female Fischer 344 rats (Lang et al., 2014, Lang and Korzick, 2014) as well as in male and female mice (liquid diet, 4% v/v, 8 wks) (Guo et al., 2012). Alternatively, chronic alcohol (female rats, 36% kcal liquid diet, 20 wks) decreased Ser65-phosphorylated 4E-BP1 to the same extent in both adult and aged rats (Lang and Korzick, 2014). Similar to reports of decreased phosphorylation of the primary substrates of mTOR (S6K1, 4E-BP1), phosphorylation of mTOR on Ser2448 was also reduced by chronic alcohol (male mice, 4% v/v liquid diet, 12 wk) and enhancing the metabolism of acetaldehyde via overexpression of mitochondrial aldehyde dehydrogenase (ALDH2) offset this decrease (Ge and Ren, 2012). The total amounts of various proteins within the mTOR complex 1 [regulatory associated protein of mTOR (raptor), DEP-domain containing mTOR-interacting protein (DEPTOR), Proline rich Akt substrate (PRAS40), G-beta like protein (GβL) and mTOR] were unchanged by chronic alcohol despite different feeding paradigms and animal models (Elmadhun et al., 2014, Guo et al., 2012, Lang and Korzick, 2014, Zhang et al., 2010); however, the binding of mTOR to raptor (a large scaffolding protein within mTORC1) was increased by chronic alcohol (36% kcal liquid diet, 8 wks) in female rats (Lang and Korzick, 2014) indicating a possible mechanism for the inhibition of this regulatory complex.
The ability of chronic alcohol to modulate various upstream effectors of mTORC1, including the stress activated proteins AMP-activated protein kinase (AMPK) and regulated in development and DNA damage responses-1 (REDD1), has also been investigated. In general, AMPK phosphorylation is increased by cellular stress leading to inhibition of mTORC1 and protein synthesis. In response to chronic alcohol consumption, AMPK Thr172-phosphorylation has been reported to be increased (female rats, 36% kcal liquid diet, 20 wks) (Lang and Korzick, 2014), decreased (male mice, 4% v/v liquid diet, 15 wks or 12 wks) (Ge et al., 2011b, Ge and Ren, 2012) and unchanged (male miniswine, 90 mL of 50% v/v alcohol, 1×/day, 7 wks) (Elmadhun et al., 2014). Further, in isolated cardiomyocytes AMPK phosphorylation was only decreased in those cardiomyocytes isolated from rats receiving a high dose (5 g/kg/d) of alcohol with no change noted following the low (0.5 g/kg/d) and medium dose (2.5 g/kg/d) (intragastric infusion, 22 wk) (Chen et al., 2010). The lack of a consistent alcohol-induced change in AMPK activity, despite suppression of mTOR and/or protein synthesis, suggests that increased AMPK phosphorylation is not necessary for the reduction in cardiac protein synthesis although direct mechanistic evaluation is lacking. REDD1, another stress-responsive mTORC1 repressor (Dennis et al., 2014, DeYoung et al., 2008, Vega-Rubin-de-Celis et al., 2010), was increased after chronic alcohol feeding in adult and older female rats (36% kcal liquid diet, 8 wks) (Lang and Korzick, 2014). Therefore, REDD1 represents a putative target for future investigations of the development of ACM considering its dual role in the regulation of protein synthesis and breakdown (i.e. autophagy).
The effects of chronic alcohol on cardiac Akt, which lies upstream of mTORC1 in the canonical pathway and which is activated by insulin, are equivocal. The phosphorylation of both Ser473 and Thr308 on Akt appears necessary for its complete activation; however, these sites are differentially regulated and infrequently measured in the same investigation. For example, Akt Thr308 phosphorylation was increased (male miniswine; 90 mL, 50%/vol daily, 7 wks) (Elmadhun et al., 2014), decreased (male mice, 4% v/v liquid diet, 16 wks) (Ge and Ren, 2012, Zhang et al., 2010) or unchanged (female rats, 36% kcal liquid diet, 20 wks) (Lang and Korzick, 2014) while Ser473 phosphorylation was unchanged (female rats, 36% kcal liquid diet, 20 wks) or decreased (male mice, 4% v/v liquid diet, 12 wks) (Ge and Ren, 2012) after chronic alcohol (Lang and Korzick, 2014). The discrepant findings may be related to differences in animal models (miniswine vs. mice or rats) and/or alcohol dose and duration with the lowest dose increasing its phosphorylation (Elmadhun et al., 2014). Alternatively, within study consistency was evident as the directionality of change in Akt was the same as in the phosphorylation of regulatory proteins either up or downstream of Akt. This included an increase in phosphatase and tensin homolog (PTEN), a negative regulator of the Akt pathway, when Akt was decreased in chronic alcohol-fed (4% v/v liquid diet, 16 wk) male mice (Zhang et al., 2010) and no change in PRAS40 phosphorylation when Akt was not altered by chronic alcohol (female rats, 36% kcal liquid diet, 20 wks) (Lang and Korzick, 2014). Lastly, Akt may also interact with the mTORC1 signaling pathway via inhibitory phosphorylation of tuberous sclerosis complex 2 (TSC2) (Inoki et al., 2002). To our knowledge the effect of chronic alcohol on TSC2 phosphorylation has not been reported in heart. As REDD1 may exert its effects via a TSC2-dependent mechanism, changes in this protein may be important to investigate in the future.
In line with the lack of consistent changes in Akt, chronic alcohol-induced alterations in indices of insulin action were also equivocal. First, in human donor hearts expression of insulin like growth factor I (IGF-I) was decreased although no correlation between mean daily alcohol consumption or cumulative lifetime dose was evident (Borrisser-Pairó et al., 2013). Next, in cardiomyocytes isolated from rats given alcohol for 22 wks glucose transporter type 4 (GLUT4) mRNA and protein was significantly decreased in medium (2.5 g/kg/d) and high (5 g/kg/d) dosed animals but not in those receiving a low dose (0.5 g/kg/d) (Chen et al., 2010). Contrary to this finding, moderate levels of daily alcohol consumption by male miniswine (90 mL, 50%/vol daily; 7 wks) increased insulin myocardial signaling as evidence by increased GLUT4 membrane translocation (Elmadhun et al., 2013, Elmadhun et al., 2014). However, numerous other indices (i.e., phosphorylation of insulin receptor substrate (IRS)-1 Ser612 and IRS2 Ser371, glycogen content and phosphorylation of glycogen synthase kinase (GSK)-3β Ser9) remained unchanged (Elmadhun et al., 2013) indicating a discrepancy between pathway activation and outcome. The authors did not address potential mechanisms leading to this inconsistency but instead focused on the congruent increases in PI3K, Akt and FOXO1. Perhaps future measurement of the binding of PI3K to IRS1 or assessment at different time points would provide a more sensitive measure to link signaling with GLUT4 translocation.
Conversely, higher alcohol doses decreased insulin-stimulated glucose uptake and signaling albeit in a strain dependent manner (Lang et al., 2014). For example, insulin-stimulated (but not basal) glucose uptake was attenuated in the right and left ventricles of alcohol-treated Sprague Dawley rats (male, 36% kcal liquid diet, 8 wks) (Lang et al., 2014); however, this effect was strain-dependent as the alcohol-induced decrease was not seen in Long Evans rats (36% kcal, liquid diet, 8 wks) (Lang et al., 2014). Likewise, only Sprague-Dawley rats showed a concomitant alcohol-induced decrease in insulin-stimulated Akt Ser473 and AS160 Thr642 phosphorylation (Lang et al., 2014). Further, IRS1 Ser307 phosphorylation in Sprague-Dawley, but not Long Evans rats, was increased by alcohol feeding, compared with controls, under both basal and insulin-stimulated conditions (Lang et al., 2014). Overall, these findings corroborate prior work showing insulin receptor, IRS-1 and Akt phosphorylation to be decreased in chronic alcohol-fed male mice (4% v/v liquid diet, 12 wks) (Li et al., 2009).
Acute alcohol and anabolic signaling
As stated above, early investigations revealed that acute alcohol decreased myocardial protein synthesis and mTORC1 signaling while recent work has continued to expand upon these observations to show a coordinated suppression of mTORC1 activity. Specifically, acute alcohol (3 g/kg BW, 3 days) in male mice increased phosphorylation of AMPK Thr172 (Ge et al., 2011a, Guo et al., 2010, Kandadi et al., 2013) and Raptor Ser792 (Ge et al., 2011a, Guo and Ren, 2012, Kandadi et al., 2013), and decreased mTOR Ser2448 phosphorylation (Ge et al., 2011a, Guo and Ren, 2012, Kandadi et al., 2013). A decrease in Akt Ser473 phosphorylation after acute alcohol intoxication was also reported (Ge et al., 2011a), although these data were in contrast with the increase in Akt Ser473 phosphorylation observed in female rats after a single dose of alcohol (1.5 g/kg BW) (El-Mas and Abdel-Rahman, 2014), again highlighting that a lower dose may increase Akt while higher levels tend to decrease its activation.
The mitogen activated protein kinase (MAPK) pathway is an alternative or complementary protein synthetic pathway that may also be negatively regulated by acute alcohol in a dose- and sex-specific manner. In response to repeated binge intake (male rats, 5 wk, 4 binges/wk, 5 g/kg BW) phosphorylation of p38 was increased, while extracellular signal related kinase (ERK)1/2 and Jun amino-terminal kinase (JNK) phosphorylation were unchanged (Gu et al., 2013). This pathway has also been assessed using an isolated perfused rat (Sprague-Dawley) heart model where hearts were acutely perfused with a low, moderate or high dose of alcohol prior to cardiomyocyte isolation (Umoh et al., 2014). Decreased phosphorylation of p38, ERK1 and ERK2 along with a decreased mRNA expression of ERK1 and p38 in cardiomyocytes after low-dose (5 mM) alcohol indicated pathway suppression. Moderate (25 mM) alcohol led to similar changes in protein levels of these markers but did not evoke significant changes in mRNA expression. Lastly, high-dose (100 mM) alcohol did not alter the mRNA expression of ERK1 and p38 nor the phosphorylation of ERK1 (Umoh et al., 2014). Many of these changes were Akt-dependent based on subsequent manipulation of this multifunctional protein. Dose-dependent regulation of ERK by acute alcohol was also confirmed in left ventricle tissue from female rats with a low dose (0.5 g/kg BW) evoking no change in phosphorylated Akt or ERK and a relatively higher dose (1.5 g/kg BW) resulting in a significant increase (El-Mas and Abdel-Rahman, 2014). This increase was prevented in ovariectomized rats indicating a dependence on estrogen availability at least in female animals. Nevertheless, concomitant changes in cardiac protein metabolism and/or function were not assessed and therefore the functional impact of these alcohol-induced changes in the MAPK pathway is currently unclear.
Despite continued progress in the general understanding of the regulatory and functional components of the mTORC1 pathway and subsequent protein synthesis under normal and pathological conditions, these advances have not been leveraged to provide a greater understanding of the underlying mechanisms of ACM. Therefore, future work should focus on elucidating the cellular mechanisms by which alcohol impairs cardiac protein synthesis and heart function by investigating newly identified proteins within the mTORC1 signaling cascade.
Protein degradation in alcoholic cardiomyopathy
Proteasome degradation
The other side of the protein balance equation includes degradative pathways that normally function to maintain heart health by removing damaged organelles and proteins. There are two primary degradative pathways, the ubiquitin proteasome pathway (UPP) and the autophagy pathway. Little work appears to have been performed investigating the effects of long-term alcohol consumption on myocardial UPP activity while, several recent studies have focused on the potential importance of autophagy.
The UPP is the primary catabolic pathway with ubiquitination marking proteins for degradation by the proteasome complex. Ubiquitination is regulated by a series of enzymatic reactions mediated by the E1, E2 and E3 ligases. Proteasome degradation occurs at the 26S proteasome which is composed of the 20S subunit and regulatory particles (19S or 11S proteasomes) (Figure 1). An early report noted that total myofibrillar and sarcoplasmic protein breakdown did not differ between chronic alcohol-fed (27% of kcal/day) and pair-fed control Wistar rats (Tiernan et al., 1985). Similarly, recent data indicated that cardiac 20S proteasome activity was not altered by chronic alcohol (36% kcal liquid diet, 20 wks) in adult female rats (Lang and Korzick, 2014). The mRNA expression of two commonly used markers of UPP activity, muscle atrophy F-box (MAFbx or atrogin-1) and muscle RING finger 1 (MuRF1) were also unchanged. In contrast, proteasome activity and mRNA expression of MuRF1 and atrogin-1 were increased in aged rats after chronic alcohol (Lang and Korzick, 2014). Therefore, from the available evidence, it has been posited that chronic alcohol does not alter the UPP in the absence of another superimposed catabolic condition, like aging.
Autophagy
Autophagy involves the degradation of cellular components under basal conditions to maintain cellular homeostasis, remove damaged organelles/proteins or provide energy during times of starvation (Gallagher et al., 2016). The study of alcohol-induced alterations in cardiac autophagy is relatively new and the data are somewhat ambiguous. However, the potential importance of this pathway is highlighted by work demonstrating that loss of autophagy related protein (Atg) 5 leads to cardiac hypertrophy, left ventricular dilation and systolic dysfunction in the absence of alcohol (Nakai et al., 2007).
During nutrient stress, AMPK is activated (phosphorylated) thereby increasing phosphorylation of Unc-51 like kinase (ULK) - 1 on Ser317 and Ser777 promoting autophagy. Conversely, nutrient abundance activates mTOR1 to inhibit autophagy by disrupting the interaction between AMPK and ULK1 via Ser757-phosphorylation of ULK1 (Kim et al., 2011). ULK1 is required for the initial formation of the Atg13 and FIP200 complex, and the development of the phagophore. Autophagosome formation is initiated by Atg7- and Atg10-mediated conjugation of Atg12 to Atg5, followed by the joining of Atg16L to Atg12-Atg5 to stimulate attachment to the phagophore. Microtubule associated protein 1A/1B-light chain 3B (LC3B) processing is also important for autophagosome formation in that the addition of phosphatidylethanolamine to LC3-I results in LC3-II formation and phagophore ring closure (i.e. autophagosome formation) (Figure 1). While the utility of measuring these proteins as surrogate markers of autophagy is limited (Mizushima, 2007), they are some of the only endpoints assessed thus far in relation to ACM.
Autophagy following acute alcohol
Each of the in vivo studies investigating the acute effects of alcohol on autophagy employed the same experimental design in which male mice received intraperitoneal injections of 3 g/kg BW alcohol for 3 days and hearts were excised 24 hours after the last dose (Ge et al., 2011a, Guo and Ren, 2012, Kandadi et al., 2013). Alcohol increased the protein content of autophagy markers including LC3B-II, LC3B-II/I and Atg7 (Ge et al., 2011a, Guo and Ren, 2012, Kandadi et al., 2013). Additionally, ULK1 Ser757 (normalized to total levels) was decreased suggesting autophagy activation (Guo and Ren, 2012, Kandadi et al., 2013). While the above mentioned results are convincing, their interpretation is less than definitive as the direct assessment of autophagic flux remains to be determined in hearts from alcohol-treated animals
Accordingly, autophagic flux has been assessed in vitro in isolated cardiomyocytes and cultured H9c2 cardiomyoblasts (Ge et al., 2011a, Guo et al., 2012, Kandadi et al., 2013). However, two of these studies showed no effect of adding lysosomal inhibitors during alcohol exposure (Ge et al., 2011a, Guo et al., 2012); protein expression of LC3 and p62 as well as LC3 positive autophagosome formation were not enhanced in the presence of lysosomal inhibitors as would be expected if alcohol increased autophagic flux. In contrast, the addition of lysosomal inhibitors prior to alcohol treatment in H9c2 cells accentuated the increase in LC3-II and LC3-II/I protein expression (Kandadi et al., 2013). The reason for these discordant findings is not immediately apparent especially as these studies were performed by the same research group. As the addition of lysosomal inhibitors did not alter LC3B protein or LC3B-positive cell accumulation it appears alcohol has little effect on autophagic flux despite increasing markers of autophagy (LC3B-II and Atg proteins). Future work might focus on clarifying these results and determining the precise in vivo relationship between alcohol and autophagy in the context of ACM.
Activation of AMPK has been posited as a mechanism linking acute alcohol with a possible increase in autophagy as knock down (whole-body) of AMPKα2 in mice normalized the majority of the alcohol-induced changes (Guo and Ren, 2012, Kandadi et al., 2013). Further, down-regulation of AMPK via the overexpression of alcohol dehydrogenase (ALDH2) which increases acetaldehyde removal, also decreased alcohol-induced autophagy by eliminating the increase in Beclin1, Atg7, LC3B-II and LC3B-II/I (Ge et al., 2011a). Moreover, incubation of freshly isolated cardiomyocytes with alcohol (240 mg/dL; 4-5 hr) in the presence and absence of an AMPK activator (AICAR) demonstrated no interaction of alcohol and AICAR on LC3B-II and LC3B-II/I thereby suggesting a common pathway may be effected by both drugs (Ge et al., 2011a). Conversely, inhibition of AMPK prevented the alcohol-induced increase in LC3B-II and LC3B-II/I highlighting the importance of this energy sensor in the alcohol-induced activation of autophagy (Ge et al., 2011a). Data from experiments using cultured H9c2 cells also suggest an alcohol-induced AMPK-dependent increase in autophagy. That is, alcohol (240 mg/dL, 4-5 hr or 160 mM, 6 hr) increased the number of LC3 positive autophagosomes, whereas pre-treatment with an autophagy inhibitor (3-MA), treatment with Compound C or knockout down of ULK1 via siRNA prevented this effect (Ge et al., 2011a, Guo and Ren, 2012, Kandadi et al., 2013). These autophagy inhibitors also reversed the acute alcohol-induced increase in LC3B-II and LC3B-II/I corroborating that short-term alcohol exposure stimulates autophagy via the AMPK-ULK1 axis in cardiomyocytes.
Autophagy following chronic alcohol
The effects of prolonged alcohol consumption on cardiac autophagy are less thoroughly studied and the available data are less definitive. For example, alcohol feeding (36% kcal, liquid diet, 20 wks) in adult female rats increased Atg7 protein expression and AMPK Thr172 phosphorylation; however, LC3B-II/I and Atg 12 protein remained unchanged (Lang and Korzick, 2014). Similar results were observed in older rats including an alcohol-induced increase in Atg7 and Atg12 protein, AMPK Thr172 phosphorylation and REDD1, but no change in LC3B-II/I (Lang and Korzick, 2014). In contrast, LC3-II, LC3-II/I, Atg7 and the number of LC3B positive cells in myocardial tissue were all increased by alcohol (4% v/v liquid diet, 8 wk) feeding to male and female mice (Guo et al., 2012) while in another study 12 wks of the same feeding regime increased LC3B-II/I without changing beclin1 (Ge and Ren, 2012). The alcohol-induced increase in autophagy in these latter two studies may be due to acetaldehyde as induction of LC3B-II, LC3B-II/I and Atg7, as well as the number of LC3B-positive cells were all increased in mice overexpressing alcohol dehydrogenase (ADH) compared to wild-type mice (Guo et al., 2012) and overexpression of ALDH2 to clear acetaldehyde prevented the increase in LC3B-II/I (Ge and Ren, 2012). Therefore, at this time, sufficient definitive evidence is lacking to conclude that chronic alcohol alters cardiac autophagy.
ACM and cardiac function
While alterations in cellular and molecular signaling are important to understanding the etiology and pathophysiology of ACM, it is also imperative to investigate associated functional changes produced by alcohol. There are two primary contexts where functional parameters have been measured. The first being in cardiomyocytes isolated from control animals and then treated with alcohol ex vivo (typically 240 mg/dl for several hours). The second experimental paradigm includes assessing cardiomyocytes isolated from animals treated with alcohol prior to sacrifice and cell isolation. As the results obtained from ex vivo alcohol treatment of cardiomyocytes are similar to those reported from animals acutely intoxicated with alcohol prior to cell isolation, they have been combined in the first section below.
Acute alcohol intoxication
Resting cell length of cardiomyocytes was generally unaltered after acute alcohol (Guo et al., 2012, Guo and Ren, 2012, Guo et al., 2010, Ma et al., 2010, Yuan et al., 2015); however, two investigations have reported an increase following 3 days of alcohol (3 g/kg BW) (Ge et al., 2011a, Kandadi et al., 2013). The preponderance of data indicate that peak shortening velocity is consistently decreased following acute alcohol (3 g/kg BW, 1 or 3 days) in male mice (Ge et al., 2011a, Guo et al., 2012, Guo and Ren, 2012, Guo et al., 2010, Kandadi et al., 2013, Ma et al., 2010). Likewise, these same studies demonstrated a consistent decrease in the maximal velocity of shortening/relengthening (±dL/dt), no change in time to peak shortening (TPS), and an increased time to 90% of relengthening (TR90) (Ge et al., 2011a, Guo et al., 2012, Guo and Ren, 2012, Guo et al., 2010, Kandadi et al., 2013, Ma et al., 2010). Such functional deficits appear dose-dependent as a lower alcohol dose (1 g/kg BW for 3 days) did not evoke commensurate changes in cardiac function (Yuan et al., 2015). While each of these markers is indicative of contractile dysfunction, the alcohol-induced increase in TR90 specifically suggests disrupted calcium resequestration or cycling when observed in conjunction with an increase in intracellular calcium decay time (Ge et al., 2011a, Guo et al., 2010, Kandadi et al., 2013). Collectively, these changes demonstrate significant diastolic dysfunction produced by acute alcohol (Ren and Wold, 2008).
Intracellular calcium handling in cardiomyocytes following acute alcohol has been measured by fluorescence imaging. The intracellular calcium concentrations are inferred from the ratio of fura-fluorescence intensity (FFI) at 360/380 nm and clearance of intracellular calcium was estimated from fluorescence decay time. Acute alcohol decreased the rise in intracellular calcium following an electrical stimulus (ΔFFI) (Ge et al., 2011a, Guo et al., 2010, Kandadi et al., 2013), increased the rate of decay of intracellular calcium (Ge et al., 2011a, Guo et al., 2010, Kandadi et al., 2013) and decreased calcium uptake in cardiomyocytes (Ge et al., 2011a). Further, acute alcohol decreased protein expression of sarcoplasmic reticulum calcium-ATPase (SERCA)-2a (an essential calcium transporter at the sarcoplasmic reticulum) and phospholamban phosphorylation which inhibits SERCA activity in the unphosphorylated state (Kandadi et al., 2013). These findings are consistent with an alcohol-induced impairment in calcium handling that would negatively impact myocardial contraction.
Chronic alcohol
Cardiomyocytes have also been isolated from chronic alcohol-fed animals for functional assessment. Chronic alcohol intake (16 wks) in male mice did not alter resting cell length, TPS, or protein levels of the sodium-calcium exchanger or phosphorylated phospholamban (Ge and Ren, 2012, Zhang et al., 2010). However, similar to acute alcohol, peak shortening and ±dL/dt were depressed while TR90 was increased (Ge and Ren, 2012, Zhang et al., 2010). These data are in agreement with earlier work from the same laboratory (Hintz et al., 2003). Additionally, peak shortening and ΔFFI were decreased, while intracellular calcium decay was increased in cardiomyocytes from alcohol-fed mice suggesting impaired calcium handling in response to contraction (Zhang et al., 2010). In cardiomyocytes from alcohol-fed (1 year) monkeys, resting cell length and the ratio of length-width in the cell was increased implying cardiac remodeling (Cheng et al., 2010). Cardiomyocyte contractile performance was also compromised as the velocity of shortening and relengthening was decreased along with several measurements of calcium regulation and intracellular movement (Cheng et al., 2010). Therefore, independent of the model, long-term alcohol intake leads to decrements in cardiac contractile function.
Alcohol metabolites
The majority of work investigating the relationship between alcohol metabolism and cardiac function has been performed by Ren and colleagues using male mice and consistent methodological approaches. Prior work had found that acetaldehyde negatively impacted cardiac structure and contractility, including perturbed calcium handling and myocardial excitation-contraction coupling (Ibrahim et al., 2014, Ren et al., 1997, Ren et al., 2002, Umoh et al., 2014, Zhang et al., 2004). Recently, defects in TPS, ±dL/dt, and TR90 were exaggerated by acute alcohol (3 g/kg BW, 3 days) in cardiomyocytes isolated from ADH overexpressing mice, compared to wild-type littermates (Guo et al., 2012, Guo et al., 2010). A similar impairment in each variable was noted in cardiomyocytes directly treated with acetaldehyde (i.e., no alcohol) (Guo et al., 2012). Calcium handling was also altered by ADH overexpression in response to alcohol as ΔFFI was decreased and intracellular calcium decay was increased to a greater extent than in alcohol-treated wild-type mice (Guo et al., 2010). Lastly, using the perfused heart model, alcohol along with ADH overexpression decreased ±dP/dt (Guo and Ren, 2010, Guo et al., 2010). Overall, these studies provide compelling evidence that acetaldehyde per se contributes to functional deficits in the heart produced by alcohol (Guo et al., 2012).
Acetaldehyde can also be produced via CYP2E1 which also appears to contribute to impaired contractility after chronic alcohol feeding (4% v/v liquid diet, 6 wk) (Zhang et al., 2013). When CYP2E1 was inhibited during the final 4 wks of alcohol feeding, the expected alcohol-induced decrements in resting cell length, peak shortening, ±dL/dt, and TR90 in cardiomyocytes were not observed (Zhang et al., 2013). Further, inhibition of CYP2E1 during alcohol feeding normalized calcium signaling, including intracellular calcium decay, ΔFFI and protein expression of the sodium-calcium exchanger and phospholamban (Zhang et al., 2013). Hence, these data substantiate the negative effects of excess acetaldehyde on cardiac function.
The other enzyme important in the metabolism of alcohol with a potential role in alcohol-induced cardiac dysfunction is aldehyde dehydrogenase (ALDH)-2. In cardiomyocytes derived from mice lacking ALDH2, alcohol-induced impairments in peak shortening, ±dL/dt, and TR90 were greater than changes seen in alcohol-treated wild-type mice (Ma et al., 2010). Similarly, inhibition of ALDH2 using cyanamide exaggerated the alcohol-induced changes in cardiomyocyte functional capacity (Ma et al., 2010). Conversely, when ALDH2 was overexpressed, thereby enhancing the metabolism of acetaldehyde to acetate, the alcohol-induced impairments in cardiomyocyte contraction were completely prevented (Ge et al., 2011a, Ge and Ren, 2012). These findings in combination with in vitro work using an ALDH2 activator in alcohol-treated cells provide compelling evidence of the negative impact the alcohol metabolites on cardiac function and the development of ACM.
Growth factors and autophagy related signaling
Insulin-like growth factor (IGF)-I lies upstream of a cascade of proteins implicated in anabolic signaling via mTORC1 and is decreased by alcohol; however, the role of IGF-I in alcohol-induced cardiac dysfunction is equivocal. For example, cardiac-specific overexpression of IGF-I ameliorated alcohol-induced decrements in peak shortening, ±dL/dt, TR90, and calcium signaling (ΔFFI, intracellular calcium decay, stimulus frequency induced peak shortening) in cardiomyocytes isolated from alcohol-fed mice (4% v/v liquid diet, 16 wk) (Zhang et al., 2010). In contrast, mice with liver deficiency of IGF-I (LID) and therefore minimal circulating IGF-I levels, had smaller chronic alcohol-induced defects in cardiomyocyte contractility including normalization of peak shortening, ±dL/dt, TR90, peak FFI, ΔFFI and intracellular calcium decay (Ge et al., 2011b). In this study, administration of exogenous IGF-I to LID mice reversed the protective effects of IGF-I deficiency in the context of chronic alcohol feeding (Ge et al., 2011b). As total and free/unbound IGF-I is reduced with chronic alcohol intake and is a contributing factor to ACM (Lang et al., 2005), the overexpression of IGF-I would be expected to be a beneficial treatment fulfilling the need for this growth factor to maintain protein synthesis and minimize loss of cardiac protein and contractility defects.
As highlighted above, AMPK is an mTORC1 inhibitor and autophagy activator. Through the use of transgenic mice as well as a pharmacological activator (AICAR) and inhibitor (Compound C), the role of AMPK in the modulation of contractility during ACM has been investigated. The preponderance of evidence indicates inhibition of AMPK activity can be beneficial for cardiac function in isolated cardiomyocytes following either in vitro (240 mg/dl, 4 hr) or in vivo acute alcohol intoxication (3 g/kg BW, 3 days) (Ge et al., 2011a, Guo and Ren, 2012, Guo et al., 2010, Kandadi et al., 2013). Moreover, AMPK deficiency prevented the alcohol-induced changes in cardiomyocytes contractility, ΔFFI and intracellular calcium decay (Kandadi et al., 2013), although other indices of calcium transport during contraction including peak FFI, resting FFI, SERCA2a and phospholamban phosphorylation remained altered from control values (Kandadi et al., 2013). However, contradictory findings were reported as AICAR-induced activation of AMPK prevented contractile dysfunction in alcohol-treated cardiomyocytes (Ge et al., 2011b). As identical concentrations, timing of drug administration and general protocols were followed the reason for this different response is unclear.
Inhibition of mTORC1 upon AMPK activation is one mechanism through which alcohol may perturb cardiomyocyte contractile function. In this regard, treatment of cardiomyocytes with the mTOR inhibitor rapamycin worsened the effects alcohol (240 mg/dl, 4-5 hr) on ±dL/dt and TR90, and did not improve the alcohol-induced decrease in peak shortening (Ge et al., 2011a). Thus, mTOR activation appears necessary for maintenance of contractile function following alcohol potentially due to its protein synthetic effects; however, this should be confirmed in models of chronic in vivo alcohol consumption.
Autophagy can be regulated by AMPK in both an mTORC1-dependent and -independent manner via site specific phosphorylation of ULK1 (as described above). Therefore, the previously discussed data pertaining to AMPK activation on cardiac function may also be related to the regulation of autophagy. Accordingly, inhibition of autophagy via 3-MA in either cardiomyocytes treated ex vivo with alcohol or from mice following acute alcohol intoxication (3 g/kg BW, 3 days) antagonized the alcohol-induced change in peak shortening, ±dL/dt and TR90, while resting cell length and TPS were unchanged (Ge et al., 2011a, Guo et al., 2012, Guo and Ren, 2012, Kandadi et al., 2013). Importantly, 3-MA also prevented the acetaldehyde-mediated decrements in contractile function (Guo et al., 2012). These data suggest that increased autophagy may contribute to the acute alcohol-induced impairment in contractile function and over time lead to the development of symptomatic cardiomyopathy.
Conclusion
This review has critically summarized recently published data, the majority of which substantiates and extends earlier evidence regarding the detrimental impact of acute and chronic alcohol on cardiac protein metabolism and contractility. Alcoholic cardiomyopathy is a multi-faceted disease whereby several factors contribute to its development and herein we present a case for the role of protein imbalance in that process. Ample evidence indicates that alcohol decreases protein synthesis via inhibition of translation initiation and/or elongation. Future work should focus on extending past findings to determine the role of newly identified protein targets and regulatory subunits within these pathways. Alternatively, while alcohol does not appear to alter the global UPP pathway, it is possible that alcohol modulates the degradation of select individual cardiac proteins and therefore represents another potential area of investigation. Our knowledge of the role of autophagy in ACM is continuing to expand and will be important to our further understanding of the disease. As the majority of work in this area has been contributed by one laboratory, independent confirmation of results and possible mechanisms is needed.
Throughout we have indicated specific pertinent methodological characteristics to show there is often a lack of consistency between the experimental models. Although the use of different models can represent a strength when comparable alcohol-induced effects are observed, it also represents a limitation when the results differ between models. Finally, a greater effort needs to be made to include both male and female animals within research protocols. While it is well-accepted that women are more susceptible to ACM and lower doses may be required for symptom development, the majority of research has been performed in male animals. Given the new NIH initiative to conduct research in males and females, more scientists should be encouraged to address potential sex differences in the etiology of ACM.
Acknowledgments
Support: This work was supported by a grant from NIAAA R37 AA011290 (C.H.L) and F32 AA023422 (J.L.S)
References
- Baar EL, Carbajal KA, Ong IM, Lamming DW. Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell. 2016;15:155–166. doi: 10.1111/acel.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrisser-Pairó F, Antúnez E, Tobías E, Fernández-Solà J. Insulin-like growth factor 1 myocardial expression decreases in chronic alcohol consumption. Regen Med Res. 2013;1:3. doi: 10.1186/2050-490X-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wang F, Sun X, Zhou J, Gao L, Jiao Y, Hou X, Qin CY, Zhao J. Chronic ethanol feeding impairs AMPK and MEF2 expression and is associated with GLUT4 decrease in rat myocardium. Exp Mol Med. 2010;42:205–215. doi: 10.3858/emm.2010.42.3.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HJ, Grant KA, Han QH, Daunais JB, Friedman DP, Masutani S, Little WC, Cheng CP. Up-regulation and functional effect of cardiac β3-adrenoreceptors in alcoholic monkeys. Alcohol Clin Exp Res. 2010;34:1171–1181. doi: 10.1111/j.1530-0277.2010.01194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MD, Coleman CS, Berg A, Jefferson LS, Kimball SR. REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci Signal. 2014;7 doi: 10.1126/scisignal.2005103. ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Nongenomic effects of estrogen mediate the dose-related myocardial oxidative stress and dysfunction caused by acute ethanol in female rats. Am J Physiol Endocrinol Metab. 2014;306:E740–747. doi: 10.1152/ajpendo.00465.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmadhun NY, Lassaletta AD, Burgess T, Sabe AA, Sellke FW. Alcohol consumption improves insulin signaling in the myocardium. Surgery. 2013;154:320–327. doi: 10.1016/j.surg.2013.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmadhun NY, Sabe AA, Lassaletta AD, Sellke FW. Alcohol consumption mitigates apoptosis and mammalian target of rapamycin signaling in myocardium. J Am Coll Surg. 2014;218:1175–1181. doi: 10.1016/j.jamcollsurg.2013.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher LE, Williamson LE, Chan EY. Advances in Autophagy Regulatory Mechanisms. Cells. 2016;5 doi: 10.3390/cells5020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W, Guo R, Ren J. AMP-dependent kinase and autophagic flux are involved in aldehyde dehydrogenase-2-induced protection against cardiac toxicity of ethanol. Free Radic Biol Med. 2011a;51:1736–1748. doi: 10.1016/j.freeradbiomed.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W, Li Q, Turdi S, Wang XM, Ren J. Deficiency of insulin-like growth factor 1 reduces vulnerability to chronic alcohol intake-induced cardiomyocyte mechanical dysfunction: role of AMPK. J Cell Mol Med. 2011b;15:1737–1746. doi: 10.1111/j.1582-4934.2010.01158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W, Ren J. mTOR-STAT3-notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. J Cell Mol Med. 2012;16:616–626. doi: 10.1111/j.1582-4934.2011.01347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Fink AM, Chowdhury SA, Geenen DL, Piano MR. Cardiovascular responses and differential changes in mitogen-activated protein kinases following repeated episodes of binge drinking. Alcohol Alcohol. 2013;48:131–137. doi: 10.1093/alcalc/ags090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Hu N, Kandadi MR, Ren J. Facilitated ethanol metabolism promotes cardiomyocyte contractile dysfunction through autophagy in murine hearts. Autophagy. 2012;8:593–608. doi: 10.4161/auto.18997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Ren J. Alcohol dehydrogenase accentuates ethanol-induced myocardial dysfunction and mitochondrial damage in mice: role of mitochondrial death pathway. PLoS One. 2010;5:e8757. doi: 10.1371/journal.pone.0008757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Ren J. Deficiency in AMPK attenuates ethanol-induced cardiac contractile dysfunction through inhibition of autophagosome formation. Cardiovasc Res. 2012;94:480–491. doi: 10.1093/cvr/cvs127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Scott GI, Ren J. Involvement of AMPK in alcohol dehydrogenase accentuated myocardial dysfunction following acute ethanol challenge in mice. PLoS One. 2010;5:e11268. doi: 10.1371/journal.pone.0011268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintz KK, Relling DP, Saari JT, Borgerding AJ, Duan J, Ren BH, Kato K, Epstein PN, Ren J. Cardiac overexpression of alcohol dehydrogenase exacerbates cardiac contractile dysfunction, lipid peroxidation, and protein damage after chronic ethanol ingestion. Alcohol Clin Exp Res. 2003;27:1090–1098. doi: 10.1097/01.ALC.0000075823.73536.DD. [DOI] [PubMed] [Google Scholar]
- Ibrahim BM, Fan M, Abdel-Rahman AA. Oxidative stress and autonomic dysregulation contribute to the acute time-dependent myocardial depressant effect of ethanol in conscious female rats. Alcohol Clin Exp Res. 2014;38:1205–1215. doi: 10.1111/acer.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Kandadi MR, Hu N, Ren J. ULK1 plays a critical role in AMPK-mediated myocardial autophagy and contractile dysfunction following acute alcohol challenge. Curr Pharm Des. 2013;19:4874–4887. doi: 10.2174/1381612811319270010. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenz M, Korthuis RJ. Moderate ethanol ingestion and cardiovascular protection: from epidemiologic associations to cellular mechanisms. J Mol Cell Cardiol. 2012;52:93–104. doi: 10.1016/j.yjmcc.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CH, Derdak Z, Wands JR. Strain-dependent differences for suppression of insulin-stimulated glucose uptake in skeletal and cardiac muscle by ethanol. Alcohol Clin Exp Res. 2014;38:897–910. doi: 10.1111/acer.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CH, Frost RA, Deshpande N, Kumar V, Vary TC, Jefferson LS, Kimball SR. Alcohol impairs leucine-mediated phosphorylation of 4E-BP1, S6K1, eIF4G, and mTOR in skeletal muscle. Am J Physiol Endocrinol Metab. 2003;285:E1205–1215. doi: 10.1152/ajpendo.00177.2003. [DOI] [PubMed] [Google Scholar]
- Lang CH, Frost RA, Kumar V, Vary TC. Impaired myocardial protein synthesis induced by acute alcohol intoxication is associated with changes in eIF4F. Am J Physiol Endocrinol Metab. 2000a;279:E1029–1038. doi: 10.1152/ajpendo.2000.279.5.E1029. [DOI] [PubMed] [Google Scholar]
- Lang CH, Frost RA, Kumar V, Wu D, Vary TC. Impaired protein synthesis induced by acute alcohol intoxication is associated with changes in eIF4E in muscle and eIF2B in liver. Alcohol Clin Exp Res. 2000b;24:322–331. [PubMed] [Google Scholar]
- Lang CH, Frost RA, Summer AD, Vary TC. Molecular mechanisms responsible for alcohol-induced myopathy in skeletal muscle and heart. Int J Biochem Cell Biol. 2005;37:2180–2195. doi: 10.1016/j.biocel.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Lang CH, Kimball SR, Frost RA, Vary TC. Alcohol myopathy: impairment of protein synthesis and translation initiation. Int J Biochem Cell Biol. 2001;33:457–473. doi: 10.1016/s1357-2725(00)00081-9. [DOI] [PubMed] [Google Scholar]
- Lang CH, Korzick DH. Chronic alcohol consumption disrupts myocardial protein balance and function in aged, but not adult, female F344 rats. Am J Physiol Regul Integr Comp Physiol. 2014;306:R23–33. doi: 10.1152/ajpregu.00414.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CH, Wu D, Frost RA, Jefferson LS, Kimball SR, Vary TC. Inhibition of muscle protein synthesis by alcohol is associated with modulation of eIF2B and eIF4E. Am J Physiol. 1999;277:E268–276. doi: 10.1152/ajpendo.1999.277.2.E268. [DOI] [PubMed] [Google Scholar]
- Li SY, Gilbert SA, Li Q, Ren J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J Mol Cell Cardiol. 2009;47:247–255. doi: 10.1016/j.yjmcc.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linford NJ, Beyer RP, Gollahon K, Krajcik RA, Malloy VL, Demas V, Burmer GC, Rabinovitch PS. Transcriptional response to aging and caloric restriction in heart and adipose tissue. Aging Cell. 2007;6:673–688. doi: 10.1111/j.1474-9726.2007.00319.x. [DOI] [PubMed] [Google Scholar]
- Ma H, Yu L, Byra EA, Hu N, Kitagawa K, Nakayama KI, Kawamoto T, Ren J. Aldehyde dehydrogenase 2 knockout accentuates ethanol-induced cardiac depression: role of protein phosphatases. J Mol Cell Cardiol. 2010;49:322–329. doi: 10.1016/j.yjmcc.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Preedy VR, Reilly ME, Patel VB, Richardson PJ, Peters TJ. Protein metabolism in alcoholism: effects on specific tissues and the whole body. Nutrition. 1999;15:604–608. doi: 10.1016/s0899-9007(99)00096-9. [DOI] [PubMed] [Google Scholar]
- Preedy VR, Richardson PJ. Ethanol induced cardiovascular disease. Br Med Bull. 1994;50:152–163. doi: 10.1093/oxfordjournals.bmb.a072873. [DOI] [PubMed] [Google Scholar]
- Preedy VR, Richardson PJ. Alcoholic cardiomyopathy: clinical and experimental pathological changes. Herz. 1996;21:241–247. [PubMed] [Google Scholar]
- Ren J, Davidoff AJ, Brown RA. Acetaldehyde depresses shortening and intracellular Ca2+ transients in adult rat ventricular myocytes. Cell Mol Biol (Noisy-le-grand) 1997;43:825–834. [PubMed] [Google Scholar]
- Ren J, Wang GJ, Petrovski P, Ren BH, Brown RA. Influence of hypertension on acetaldehyde-induced vasorelaxation in rat thoracic aorta. Pharmacol Res. 2002;45:195–199. doi: 10.1006/phrs.2002.0947. [DOI] [PubMed] [Google Scholar]
- Ren J, Wold LE. Mechanisms of alcoholic heart disease. Ther Adv Cardiovasc Dis. 2008;2:497–506. doi: 10.1177/1753944708095137. [DOI] [PubMed] [Google Scholar]
- Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549–564. doi: 10.1161/CIRCRESAHA.114.302022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq T, Salisbury JR, Richardson PJ, Preedy VR. Synthesis of ventricular mitochondrial proteins in vivo: effect of acute ethanol toxicity. Alcohol Clin Exp Res. 1993;17:894–899. doi: 10.1111/j.1530-0277.1993.tb00860.x. [DOI] [PubMed] [Google Scholar]
- Tiernan JM, Ward LC, Cooksley WG. Inhibition by ethanol of cardiac protein synthesis in the rat. Int J Biochem. 1985;17:793–798. doi: 10.1016/0020-711x(85)90266-6. [DOI] [PubMed] [Google Scholar]
- Umoh NA, Walker RK, Al-Rubaiee M, Jeffress MA, Haddad GE. Acute alcohol modulates cardiac function as PI3K/Akt regulates oxidative stress. Alcohol Clin Exp Res. 2014;38:1847–1864. doi: 10.1111/acer.12459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vary TC, Deiter G, Goodman SA. Acute alcohol intoxication enhances myocardial eIF4G phosphorylation despite reducing mTOR signaling. Am J Physiol Heart Circ Physiol. 2005;288:H121–128. doi: 10.1152/ajpheart.00440.2004. [DOI] [PubMed] [Google Scholar]
- Vary TC, Deiter G, Lantry R. Chronic alcohol feeding impairs mTOR(Ser 2448) phosphorylation in rat hearts. Alcohol Clin Exp Res. 2008;32:43–51. doi: 10.1111/j.1530-0277.2007.00544.x. [DOI] [PubMed] [Google Scholar]
- Vary TC, Jefferson LS, Kimball SR. Insulin fails to stimulate muscle protein synthesis in sepsis despite unimpaired signaling to 4E-BP1 and S6K1. Am J Physiol Endocrinol Metab. 2001a;281:E1045–1053. doi: 10.1152/ajpendo.2001.281.5.E1045. [DOI] [PubMed] [Google Scholar]
- Vary TC, Lang CH. Differential phosphorylation of translation initiation regulators 4EBP1, S6k1, and Erk 1/2 following inhibition of alcohol metabolism in mouse heart. Cardiovasc Toxicol. 2008;8:23–32. doi: 10.1007/s12012-008-9012-4. [DOI] [PubMed] [Google Scholar]
- Vary TC, Lynch CJ, Lang CH. Effects of chronic alcohol consumption on regulation of myocardial protein synthesis. Am J Physiol Heart Circ Physiol. 2001b;281:H1242–1251. doi: 10.1152/ajpheart.2001.281.3.H1242. [DOI] [PubMed] [Google Scholar]
- Vary TC, Nairn AC, Lang CH. Restoration of protein synthesis in heart and skeletal muscle after withdrawal of alcohol. Alcohol Clin Exp Res. 2004;28:517–525. doi: 10.1097/01.alc.0000121653.80502.54. [DOI] [PubMed] [Google Scholar]
- Vega-Rubin-de-Celis S, Abdallah Z, Kinch L, Grishin NV, Brugarolas J, Zhang X. Structural analysis and functional implications of the negative mTORC1 regulator REDD1. Biochemistry (Mosc) 2010;49:2491–2501. doi: 10.1021/bi902135e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersma M, Henning RH, Brundel BJ. Derailed Proteostasis as a Determinant of Cardiac Aging. Can J Cardiol. 2016;32:1166.e1111–1120. doi: 10.1016/j.cjca.2016.03.005. [DOI] [PubMed] [Google Scholar]
- Yuan F, Lei Y, Wang Q, Esberg LB, Huang Z, Scott GI, Li X, Ren J. Moderate ethanol administration accentuates cardiomyocyte contractile dysfunction and mitochondrial injury in high fat diet-induced obesity. Toxicol Lett. 2015;233:267–277. doi: 10.1016/j.toxlet.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Zhang B, Turdi S, Li Q, Lopez FL, Eason AR, Anversa P, Ren J. Cardiac overexpression of insulin-like growth factor 1 attenuates chronic alcohol intake-induced myocardial contractile dysfunction but not hypertrophy: Roles of Akt, mTOR, GSK3beta, and PTEN. Free Radic Biol Med. 2010;49:1238–1253. doi: 10.1016/j.freeradbiomed.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RH, Gao JY, Guo HT, Scott GI, Eason AR, Wang XM, Ren J. Inhibition of CYP2E1 attenuates chronic alcohol intake-induced myocardial contractile dysfunction and apoptosis. Biochim Biophys Acta. 2013;1832:128–141. doi: 10.1016/j.bbadis.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li SY, Brown RA, Ren J. Ethanol and acetaldehyde in alcoholic cardiomyopathy: from bad to ugly en route to oxidative stress. Alcohol. 2004;32:175–186. doi: 10.1016/j.alcohol.2004.01.005. [DOI] [PubMed] [Google Scholar]