

Abstract
Perturbations in myeloid cell differentiation are common in neoplasia, culminating in immature populations known as myeloid-derived suppressor cells (MDSCs). MDSCs favor tumor progression due to their ability to suppress host immunity or promote invasion and metastasis. They are thought to originate from the bone marrow as a result of exposure to stromal- or circulating tumor-derived factors (TDFs). Although great interest has been placed on understanding how MDSCs function, less is known regarding how MDSCs develop at a transcriptional level. Our work explores the premise that MDSCs arise because cancer cells, through the production of certain TDFs, inhibit the expression of interferon regulatory factor-8 (IRF8) that is ordinarily essential for controlling fundamental properties of myeloid cell differentiation. Our interest in IRF8 has been based on the following rationale. First, it is well-recognized that IRF8 is a ‘master regulator’ of normal myelopoiesis, critical not only for producing monocytes, dendritic cells (DCs), and neutrophils, but also for controlling the balance of all three major myeloid cell types. This became quite evident in IRF8−/− mice, whereby the loss of IRF8 leads to a disproportionate accumulation of neutrophils at the expense of monocytes and DCs. Second, we showed that such myeloid populations from IRF8−/− mice exhibit similar characteristics to MDSCs from tumor-bearing mice. Third, in a reciprocal fashion, we showed that enforced expression of IRF8 in the myeloid system significantly mitigates tumor-induced MDSC accumulation and improves immunotherapy efficacy. Altogether, these observations support the hypothesis that IRF8 is an integral negative regulator of MDSC biology.
Keywords: Regulatory myeloid cells, Cancer, Myelopoiesis, MDSCs, IRF8
Introduction
Patients with a variety of cancer types exhibit high circulating levels of myeloid populations, including those of the neutrophil series, including polymorphonuclear (PMN) neutrophils, myeloid-derived suppressor cells (MDSCs), and even progenitor-like populations, such as granulocyte–monocyte progenitors (GMPs) [1–10]. In fact, a recent seminal review concluded that among a wide range of potential cell-based biomarkers examined, the one that most associated with poor clinical response in patients with diverse solid malignancies was a high blood neutrophil to lymphocyte ratio [11]. Such myeloid populations are thought to aid tumor progression for multiple reasons, including their ability to suppress innate and adaptive immunity, and promote angiogenesis, invasion, and metastasis [1–5]. It is thought that these myeloid populations expand or acquire pro-tumor capabilities as a result of exposure to stromal- or circulating tumor-derived factors (TDFs). While there is strong consensus in the field that these myeloid populations are tumor-supportive, less is known about where and how they develop in the first place. Therefore, understanding the molecular bases by which neoplasia compromises myelopoiesis offers potentially new insights into the design or use of therapies that require an intact myeloid system. Thus, the goal of our work has been to build on an understanding of how neoplasia impairs myelopoiesis with a focus on the generation of MDSCs.
MDSCs comprise two major subsets: monocytic or polymorphonuclear (M-MDSC or PMN-MDSC, respectively), and over the last several years have attracted immense interest in the fields of cancer immunology and immunotherapy because they are highly protumorigenic [1–5, 12]. Intriguingly, the PMN-MDSC subset predominates in many tumor settings [1, 2, 4, 7, 10, 13–15]; yet, the mechanisms behind this MDSC subset bias have remained unclear. In general, in tumor-bearing hosts, dendritic cell (DC) differentiation is also disrupted, an observation underscored by Gabrilovich and colleagues [2, 16] and later by other laboratories [17–19]. Thus, myeloid differentiation in cancer is redirected from DCs to MDSCs, likely due to alterations in transcriptional networks. Our laboratory has focused on this facet of MDSC biology and recently identified previously unrecognized roles for interferon regulatory factor-8 (IRF8) in tumor-mediated MDSC production, especially of the PMN-MDSC subset. We also found that STAT3-activating cytokines known to facilitate PMN-MDSC production (such as granulocyte-colony stimulating factor; G-CSF) can inhibit IRF8 expression, thus establishing a novel STAT3-IRF8 axis in both IRF8 and MDSC biology [4, 10, 13, 18, 20–25]. These findings also provided a molecular underpinning for the importance of STATs, particularly STAT3, in MDSC biology [2–5, 12].
IRF8 is a well-recognized ‘master regulator’ of normal myelopoiesis, critical not only for producing lineage-committed monocytes, DCs and neutrophils, but also for controlling the balance of these major myeloid subsets [26–35]. This became quite evident in IRF8−/− mice [26, 35], as well as more recently in humans harboring a mutation in IRF8 [36, 37], whereby IRF8 loss led to a substantial and disproportionate accumulation of neutrophils at the expense of monocytes and DCs. Moreover, similar myeloid phenotypes are observed in mouse tumor models and cancer patients [2, 16–19], although the basis for this myeloid imbalance has remained poorly understood. Because of the unique role that IRF8 plays in myelopoiesis and the fact that PMN-MDSCs are a dominant MDSC subset, we have been testing the hypothesis that tumor-induced downregulation of IRF8 underlies a novel molecular basis for the robust PMN-MDSC response. Our more recent work is focusing on the precise origin of the PMN-MDSC response, with emphasis on the relevant myeloid progenitor stage(s) within the bone marrow. Altogether, we posit that myeloid cell skewing toward MDSCs, particularly the PMN-MDSC subset, is a result of dysregulation of IRF8 and/or its downstream targets. We believe that these advances will offer new insights into how to abrogate the MDSC response in cancer.
IRF8 is a negative regulator of MDSC production
The following is a synopsis of major findings from our work. We have been operating on the premise that disruption of the myeloid response in cancer is a critical reason that limits the potency of active or passive immunotherapies, which require competent antigen-presenting cells (APCs) for induction or maintenance of antitumor immunity. A key element of our work is defining transcriptional networks that ultimately impact the nature of the myeloid response in such cancer settings, with emphasis on IRF8 in MDSC-tumor biology. Altogether, we identified a previously unrecognized role for IRF8 in tumor-mediated MDSC production. The next section will provide a brief overview of IRF8 in myelopoiesis, followed by its novel role in MDSC biology.
IRF8 biology
IRF8 is a member of the interferon regulatory factor family of transcription factors, which is largely restricted to cells of the myeloid and lymphoid lineages, mainly monocytes/macrophages, certain DC subsets, and B cells [26–35]. Structurally, IRF8 is a 48-kD, 424 amino acid protein comprising two major elements, a well-conserved N-terminal DNA binding motif and a more variable C-terminal IRF association domain that enables interactions with other transcription factors, notably IRF-1, IRF-2, IRF-4, or PU.1. In the nucleus, such IRF8 heterocomplexes either activate or repress gene transcription by binding to specific DNA elements within the responsive promoter regions.
Within the myeloid lineage, steady-state IRF8 expression functions as a positive regulator for monocytes and DCs (particularly the CD8α, plasmacytoid and CD103+ subsets), whereas it conversely acts as a negative regulator of granulocyte differentiation, namely, neutrophils. IRF8 expression is thought to emerge at the granulocyte–monocyte progenitor (GMP) stage, which constitutes the branching point between monocytes/DCs and granulocyte differentiation. IRF8 expression then steadily rises during lineage commitment to monocytes/DCs and declines during lineage commitment to neutrophils. The mechanisms which govern IRF8 up or downregulation during normal myelopoiesis, however, remain unclear. Nonetheless, these observations have been elegantly unveiled and reinforced in IRF8−/− mice, which display significant alterations in myelopoiesis with marked increases in the proportion of neutrophils at the expense of monocytes (mainly Ly6C+) and several DC subsets, namely, plasmacytoid, CD8α+ and CD103+, as noted earlier. The precise mechanisms by which IRF8 act downstream to regulate myeloid differentiation and lineage commitment is still a very active area of investigation. Several seminal observations have revealed that IRF8 impacts the expression or function of integral genes associated with monocyte or neutrophil differentiation, such as KLF4 or C/EBPα, respectively [38, 39].
IRF8 expression in causally linked to MDSC development
We first demonstrated that IRF8 expression was significantly depressed in both MDSC subsets of tumor-bearing mice. However, IRF8 loss seemed to have a greater impact on the PMN-MDSC subset. This was based on the finding that IRF8 loss strongly favored the expansion of PMN-MDSCs [10], which is consistent with the role of IRF8 in myeloid differentiation [26–35]. Using a global IRF8−/− mouse model as a loss-of-function approach, we found that IRF8 deficiency led to a robust accumulation of MDSC-like cells that were heavily neutrophilic and mimicked what was observed in mammary tumor-bearing mice. This was observed at phenotypic and functional levels, as well as by differential gene expression analysis, wherein the gene expression profile of IRF8−/− mice resembled tumor-induced MDSCs significantly more so than the wild-type (WT) counterparts. Using a novel genetic gain-of-function approach, we showed that enforced IRF8 overexpression (using a CD11b-driven transgenic mouse model) reduced tumor-induced MDSC accumulation in the periphery and tumor tissue and significantly improved responses to second therapies, such as CTLA-4 blockade (described further below). Interestingly, although IRF8 overexpression reduced peripheral MDSC load (particularly the PMN-MDSC subset), the remaining MDSCs were still suppressive; thus, IRF8 seemed to exert a more profound impact on MDSCs at a quantitative level.
Next, we sought to extend our preclinical findings in mammary tumor models to breast cancer patients [10]. Similar to earlier reports reflecting various solid tumor types [4, 10, 13, 18, 20–25], we observed a significant increase in the percentage of MDSCs compared to healthy donor controls. Moreover, we found that high circulating MDSC percentages in patients with stage III/IV breast cancer at diagnosis predicted a poorer outcome. Surprisingly, no significant difference in IRF8 expression was observed between healthy donors and patients. However, when the patient IRF8 data were plotted in relation to their corresponding cell percentages, we observed a significant negative correlation between these two parameters. In contrast, no inverse correlation was observed with the controls. Taken collectively, these data support the hypothesis that MDSC frequency or expansion is IRF8-dependent and that higher MDSC frequencies coincide with poorer patient outcomes.
IRF8 downregulation in tumor-bearing hosts using a novel IRF8 reporter mouse model
Here, we made use a newly developed IRF8-EGFP reporter mouse [40] to verify IRF8 downregulation in tumor-bearing hosts. The mouse model was originally developed by Wang et al. [40] using a knock-in strategy, wherein the stop codon of exon 9 of the mouse IRF8 locus was replaced with an EGFP sequence that resulted in the production of an IRF8-EGFP fusion protein under the control of endogenous IRF8 regulatory elements. EGFP could be tracked in progeny under homozygous or heterozygous breeding conditions. The knock-in did not alter IRF8 biology, as myeloid differentiation or function remained intact [40]. IRF8 was expressed in the expected myeloid subsets of the bone marrow and peripheral hematopoietic compartments [40]. We observed no adverse hematologic effects in the peripheral blood, based on analyses of WBCs, RBCs, and platelets relative to the WT controls. Importantly, our data confirmed that IRF8 expression, as measured by changes in EGFP levels, was diminished in tumor-driven MDSCs or G-CSF-induced MDSCs (Fig. 1); albeit, this effect was more pronounced in the PMN-MDSC subset.
Fig. 1.

Use of an IRF8-EGFP reporter mouse model to track changes in IRF8 levels during pathologic insults. a EGFP levels (right y-axis) of blood CD11b+Gr-1+ cells (left y-axis) in non-tumor-bearing (NTB; n = 7) mice or 4T1 tumor-bearing mice (>250 mm3; n = 4). *P < 0.05 as determined by an unpaired t test. b EGFP levels of splenic CD11b+Ly6CloLy6G+ cells from PBS (grey-shaded area) or G-CSF (black line)-treated mice (10 µg/day for 5 days) of one of five representative mice per group
IRF8 expression is regulated by a novel STAT3-dependent pathway
A number of studies, including our work [4, 10, 13, 18, 20–25] and those elsewhere [2–5, 12], had established that tumor-derived G-CSF can drive PMN-MDSC production. Furthermore, it is well known that a major proximal target of G-CSF-mediated signaling is STAT3 activation [41, 42] and that STAT3 activation is important for MDSC accumulation [2–5, 12]. Our findings described above demonstrated that tumor-induced IRF8 downregulation promotes PMN-MDSC accumulation. Thus, a gap remained in our understanding of how IRF8 might be regulated. Based on this knowledge, we hypothesized that tumor-induced IRF8 downregulation occurred through an STAT3-dependent interaction.
To test this hypothesis, we made use of recombinant G-CSF protein as a tool to activate STAT3, followed by its effects on IRF8 expression. We found that G-CSF treatment of normal bone-marrow-derived CD11b+Gr-1+ cells in vitro rapidly reduced IRF8 expression, which was prevented using a selective STAT3 inhibitor, thereby unmasking an inverse link between STAT3 activity and IRF8 expression. Chromatin immunoprecipitation assays revealed direct interactions between activated STAT3 with IRF8 at the promoter level in response G-CSF treatment [10]. Altogether, these data provided evidence for a previously undescribed STAT3-IRF8 axis in IRF8 and myeloid biology, and that IRF8 represented a downstream target of STAT3.
IRF8 enhancement improves antitumor responses to second therapies
While MDSC load was reduced in IRF8-transgenic mice compared to WT mice, primary tumor growth was similar between both genotypes. We reasoned that despite the decrease in MDSC burden, the decrease was still above a biologic threshold to translate to effects on tumor growth. To test that notion, we added a therapeutic intervention to either further reduce PMN-MDSC burden (i.e., anti-Ly6G mAb) or potentiate T cell immunity (i.e., anti-CTLA-4 mAb). Indeed, we found that tumor growth in IRF8-transgenic mice was now significantly reduced compared to WT mice, demonstrating that IRF8 enhancement was bioactive and improved responses to a second therapy. Intriguingly, we showed that IRF8 overexpression (in the absence of a second therapy) significantly reduced spontaneous metastasis in both 4T1 orthotopic and PyMT-MMTV autochthonous tumor models [10]. Future studies are warranted to investigate in detail whether IRF8-mediated effects on spontaneous metastasis are dependent upon the adaptive immune response. Nonetheless, these data further showed that IRF8 was bioactive and, in some cancer settings, was sufficient to act alone to impact immune surveillance. The precise mechanisms underlying these anti-metastatic effects, however, require further study.
Where and how does IRF8 act downstream to impact PMN-MDSC production?
Although we identified a potential mechanism that regulates IRF8 expression, it still remains unclear what genes IRF8 regulates downstream to impact PMN-MDSC production. One approach to address this gap is to borrow knowledge from the field of ‘emergency’ granulopoiesis [42, 43]. Indeed, tumor-induced PMN-MDSC accumulation is now thought to be akin to the process of emergency granulopoiesis. Therefore, knowledge in this field may provide important insights into the identity of IRF8-regulated gene targets in PMN-MDSC biology. We [4, 10, 13, 18, 20–25] and other groups [2–5, 12] have now shown that G-CSF is produced in copious amounts by certain solid cancer types and, as an integral myelopoietic growth factor, can drive PMN-MDSC accumulation. Intriguingly, the characteristics of the resulting tumor-induced PMN-MDSC response are phenotypically, functionally, and molecularly similar to that induced by recombinant G-CSF administration [13], a pharmacologic mimic of emergency granulopoiesis [42, 43]. Thus, the transcriptional mechanisms which govern emergency granulopoiesis are seemingly homologous to those seen in PMN-MDSC-tumor biology.
Since emergency granulopoiesis is a process initiated at a progenitor level, we hypothesize that bone-marrow GMPs represent a significant source of PMN-MDSCs and that their development in cancer is regulated by IRF8. Recent data in our laboratory support this hypothesis and demonstrate that GMPs significantly expand in cancer models, just as effectively as the administration of G-CSF protein (Fig. 2) (unpublished observations). We observed similar findings in IRF8−/− mice (Fig. 2) (unpublished observations), suggesting that IRF8 acts at a myeloid progenitor level, such as the GMP stage to ultimately direct the PMN-MDSC response. Thus, in cancers, where PMN-MDSCs dominate, we posit that aphysiologic levels of STAT3-activating TDFs (i.e., G-CSF and others) engage various myeloid progenitors, including the GMPs (Fig. 3). Activated STAT3 protein then translocates to the nucleus where it signals to either inhibit IRF8 expression or prevent its induction, thereby shunting or fueling differentiation toward PMN-MDSCs. The major function of IRF8 in myeloid differentiation was long thought to occur at the GMP stage [26, 27, 29, 35], a bifurcation point that oversees the production and, very importantly, the balance of monocytes/DCs vs. granulocytes, as described earlier. Detailed flow cytometric analyses reveal that IRF8 expression levels begin to emerge at the GMP stage [40]. Thus, the production of PMN-MDSCs in tumor-bearing hosts may culminate from their selective expansion from the GMP stage. This will ensure that myeloid differentiation will be directed toward a robust granulocytic response. This does not preclude the possibility that PMN-MDSCs may also emerge at earlier or later progenitor stages.
Fig. 2.

Expansion of GMPs influenced by various pathologic insults. Percentage of GMPs (defined as Lineage (Lin)−Sca-1−ckit+CD16/32+CD150−) within the Lin−Sca-1−ckit+ fraction of the bone marrow of the following groups of mice: non-tumor-bearing (NTB) wild-type (n = 19), IRF8−/− (n = 3), 4T1 (n = 14) or AT-3 (n = 8) tumor-bearing (>250 mm3), or NTB wild-type mice following treatment with recombinant G-CSF protein (10 µg/day for 5 days; n = 5). Data compiled from multiple experiments. *P < 0.01 as determined by an unpaired t test
Fig. 3.
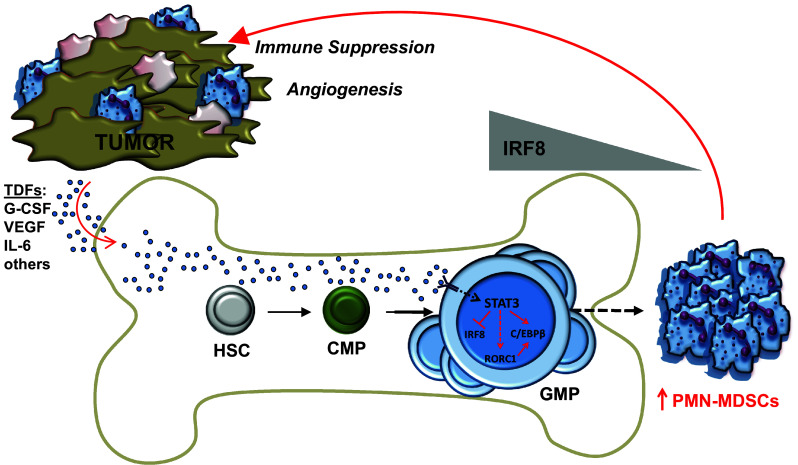
Model for the regulation of IRF8 in GMPs as an important molecular event underlying PMN-MDSC development. STAT3-activating TDFs (several of which are shown and are indicated by blue dots) circulate in the blood, then enter and permeate the bone marrow microenvironment to induce PMN-MDSCs by inhibiting IRF8 expression at the GMP stage. Exposure of GMPs to such TDFs not only expands the GMP pool, but skews myeloid differentiation toward a robust granulocytic response by ensuring that IRF8 levels remain low or downregulated (shown by the relative IRF8 gradient). This does not rule out the possibility that PMN-MDSCs may also emerge at earlier (e.g., HSC hematopoietic progenitor cells, CMP common myeloid progenitor cells) or later myeloid progenitor stages. In addition to IRF8, a number of other transcription factors (examples are shown) have been implicated in PMN-MDSC development/expansion. Although their precise relationship with IRF8 remains unresolved, it is known that C/EBPβ is a target of STAT3, while RORC1 may be a putative target of STAT3 signaling based on its role in emergency granulopoiesis (denoted by the dashed red line). Our model further posits that such molecular interactions converge at the GMP stage, although such interactions may occur at other progenitor stages as well. Once PMN-MDSCs accumulate in the periphery, they traffic to the tumor microenvironment, where they aid neoplastic growth through immune system-dependent (i.e., immune suppression) or independent mechanisms (i.e., angiogenesis). Pink cells broadly indicate other stromal cells of the tumor microenvironment
There is compelling data that G-CSF engagement promotes STAT3 phosphorylation, which in turn drives CCAAT/enhancer binding protein (C/EBP)-β transcription, a master regulator of emergency granulopoiesis [42–44] (in contrast, C/EBPα expression is required for steady-state granulopoiesis). Transcription of C/EBPβ then in cooperation with STAT3 drives MYC expression, which enables accelerated cell cycle progression, leading to increased neutrophil output. Both STAT3 and C/EBPβ expressions have been shown to be major players of PMN-MDSC formation, consistent with the notion that PMN-MDSC accumulation is a consequence of emergency granulopoiesis. Indeed, work by Bronte and colleagues has defined a link between STAT3 and C/EBPβ expression [45], while our laboratory demonstrated a link between STAT3 and IRF8 expression [10] (Fig. 3). Moreover, studies by Sica and colleagues [46] recently reported a novel role for the retinoic-acid-related orphan receptor protein, termed RORC1, in tumor-induced MDSC development (Fig. 3). RORC1 deficiency in the hematopoietic system abrogated MDSC development, including PMN-MDSC frequencies. Additional studies revealed that RORC1 positively regulated C/EBPβ expression and negatively regulated SOCS3 (suppressor of cytokine signaling-3) and Bcl3 expression, all players important for ensuring emergency or G-CSF-induced granulopoiesis. Finally, studies by Du and colleagues [47] previously identified an unrecognized role for the peroxisome proliferator-activated receptor-γ (PPARγ) in the expansion of MDSCs, including PMN-MDSCs (termed CD11b+Ly6G+ in that study) and various myeloid progenitors within the bone marrow. While several studies have now individually identified critical transcription factors and other molecules underlying PMN-MDSC development/expansion, what remains unresolved is the precise molecular relationship between IRF8 with C/EBPβ, RORC1, and/or PPARγ (and perhaps others) and whether these interactions selectively or preferentially occur in GMPs or other earlier/later stage myeloid progenitors. Indeed, future work is warranted to unravel these and other molecular interactions during coordination and oversight not only of the PMN-MDSC response, but also of the M-MDSC response.
Conclusions and future directions
In summary, our findings showed that IRF8 is an integral negative regulator of MDSCs, particularly of the PMN-MDSC subset, providing new insights into the fields of both IRF8 and MDSC biology (see model in Fig. 3). Using an IRF8−/− mouse model, we demonstrated that peripheral myeloid populations exhibited strikingly similar phenotypic, functional, and molecular features of PMN-MDSCs from tumor-bearing mice. Conversely, using a novel genetic gain-of-function approach produced in our laboratory, we showed that enforced IRF8 expression significantly mitigated PMN-MDSC accumulation in tumor-bearing mice and improved immunotherapeutic efficacy. Finally, the inverse relationship between IRF8 levels and MDSC frequency was observed in breast cancer patients, but not healthy controls.
Our work and those of other laboratories clearly provide the rationale to target MDSCs for therapeutic purposes [48]. While numerous approaches have been presented in the field, we would like to emphasize two of them as they relate to the concepts underscored in this review. The first approach is the induction phase, which is based on the intent to neutralize TDFs (e.g., G-CSF) that drive MDSC generation. The idea of blocking pathologic levels of physiologically relevant growth factors is conceptually analogous to those medical interventions that target circulating levels of TNF, for example, in autoimmune pathologies, such as rheumatoid arthritis or Crohn’s disease. In the tumor setting, this approach reflects the notion that if MDSCs are separated from their microenvironments, those typically rich in MDSC-sustaining growth factors, they rapidly differentiate or die in vitro. Thus, in vivo-based approaches, such as cytokine blockade, which separate MDSCs from their cognate survival/growth factors, are likely to impair MDSC viability and/or their persistence. The second approach is the bioactive phase, which is based on the intent to treat MDSCs as a myeloproliferative disorder, such as acute promyelocytic leukemia, and enforce their differentiation from immature to mature phenotypes using compounds, such as all-trans-retinoic acid (ATRA) [48–51]. It is interesting to speculate that such compounds may facilitate myeloid differentiation through regulating IRF8, either directly or indirectly. Nonetheless, targeting mechanisms of MDSC generation or MDSC differentiation represent sound strategies that will likely enhance immune surveillance or the efficacy of immunotherapies that require an intact myeloid system.
Acknowledgements
We gratefully thank all the current and past members of the Abrams Lab for their outstanding support and contributions to this work. This work was supported by R01CA140622 and R01CA172105 from the National Cancer Institute/NIH (to Scott Abrams) and NIH training grant T32CA085183 (to Colleen Netherby).
Abbreviations
- ATRA
All-trans-retinoic acid
- C/EBP
CCAAT/enhancer binding protein
- GMP
Granulocyte–monocyte progenitor
- IRF8
Interferon regulatory factor-8
- NTB
Non-tumor-bearing
- PMN
Polymorphonuclear
- PPARγ
Peroxisome proliferator-activated receptor-γ
- RORC1
Retinoic-acid-related orphan receptor C1
- TDF
Tumor-derived factor
- WT
Wild-type
Compliance with ethical standards
Conflict of interest
All authors declare no conflicts of interest related to this work.
Footnotes
This paper is a Focussed Research Review based on a presentation given at the conference Regulatory Myeloid Suppressor Cells: From Basic Discovery to Therapeutic Application which was hosted by the Wistar Institute in Philadelphia, PA, USA, 16th–19th June, 2016. It is part of a Cancer Immunology, Immunotherapy series of Focussed Research Reviews.
References
- 1.Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. 2015;98:913–922. doi: 10.1189/jlb.4RI0515-204R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Messmer MN, Netherby CS, Banik D, Abrams SI. Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol Immunother. 2015;64:1–13. doi: 10.1007/s00262-014-1639-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356–3364. doi: 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sagiv JY, Michaeli J, Assi S, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10:562–573. doi: 10.1016/j.celrep.2014.12.039. [DOI] [PubMed] [Google Scholar]
- 8.Wu WC, Sun HW, Chen HT, et al. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc Natl Acad Sci USA. 2014;111:4221–4226. doi: 10.1073/pnas.1320753111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez PC, Ernstoff MS, Hernandez C, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553–1560. doi: 10.1158/0008-5472.CAN-08-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waight JD, Netherby C, Hensen ML, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123:4464–4478. doi: 10.1172/JCI68189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Templeton AJ, McNamara MG, Seruga B, et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J Natl Cancer Inst. 2014;106:dju124. doi: 10.1093/jnci/dju124. [DOI] [PubMed] [Google Scholar]
- 12.Sonda N, Chioda M, Zilio S, Simonato F, Bronte V. Transcription factors in myeloid-derived suppressor cell recruitment and function. Curr Opin Immunol. 2011;23:279–285. doi: 10.1016/j.coi.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One. 2011;6:e27690. doi: 10.1371/journal.pone.0027690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solito S, Falisi E, Diaz-Montero CM, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011;118:2254–2265. doi: 10.1182/blood-2010-12-325753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–1766. [PubMed] [Google Scholar]
- 17.Capietto AH, Kim S, Sanford DE, et al. Down-regulation of PLCgamma2-beta-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer. J Exp Med. 2013;210:2257–2271. doi: 10.1084/jem.20130281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farren MR, Carlson LM, Netherby CS, et al. Tumor-induced STAT3 signaling in myeloid cells impairs dendritic cell generation by decreasing PKCbetaII abundance. Sci Signal. 2014;7:ra16. doi: 10.1126/scisignal.2004656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papaspyridonos M, Matei I, Huang Y, et al. Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat Commun. 2015;6:6840. doi: 10.1038/ncomms7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abrams SI, Waight JD. Identification of a G-CSF-Granulocytic MDSC axis that promotes tumor progression. Oncoimmunology. 2012;1:550–551. doi: 10.4161/onci.19334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paschall AV, Zhang R, Qi CF, et al. IFN regulatory factor 8 represses GM-CSF expression in T cells to affect myeloid cell lineage differentiation. J Immunol. 2015;194:2369–2379. doi: 10.4049/jimmunol.1402412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart TJ, Abrams SI. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J Immunol. 2007;179:2851–2859. doi: 10.4049/jimmunol.179.5.2851. [DOI] [PubMed] [Google Scholar]
- 23.Stewart TJ, Greeneltch KM, Reid JE, et al. Interferon regulatory factor-8 modulates the development of tumour-induced CD11b+ Gr-1+ myeloid cells. J Cell Mol Med. 2009;13:3939–3950. doi: 10.1111/j.1582-4934.2009.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart TJ, Liewehr DJ, Steinberg SM, Greeneltch KM, Abrams SI. Modulating the expression of IFN regulatory factor 8 alters the protumorigenic behavior of CD11b+ Gr-1+ myeloid cells. J Immunol. 2009;183:117–128. doi: 10.4049/jimmunol.0804132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waight JD, Banik D, Griffiths EA, Nemeth MJ, Abrams SI. Regulation of the interferon regulatory factor-8 (IRF-8) tumor suppressor gene by the signal transducer and activator of transcription 5 (STAT5) transcription factor in chronic myeloid leukemia. J Biol Chem. 2014;289:15642–15652. doi: 10.1074/jbc.M113.544320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holtschke T, Lohler J, Kanno Y, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/S0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 27.Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. 2000;13:155–165. doi: 10.1016/S1074-7613(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 28.Schiavoni G, Mattei F, Sestili P, et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med. 2002;196:1415–1425. doi: 10.1084/jem.20021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsujimura H, Nagamura-Inoue T, Tamura T, Ozato K. IFN consensus sequence binding protein/IFN regulatory factor-8 guides bone marrow progenitor cells toward the macrophage lineage. J Immunol. 2002;169:1261–1269. doi: 10.4049/jimmunol.169.3.1261. [DOI] [PubMed] [Google Scholar]
- 30.Aliberti J, Schulz O, Pennington DJ, et al. Essential role for ICSBP in the in vivo development of murine CD8alpha+ dendritic cells. Blood. 2003;101:305–310. doi: 10.1182/blood-2002-04-1088. [DOI] [PubMed] [Google Scholar]
- 31.Tsujimura H, Tamura T, Gongora C, et al. ICSBP/IRF-8 retrovirus transduction rescues dendritic cell development in vitro. Blood. 2003;101:961–969. doi: 10.1182/blood-2002-05-1327. [DOI] [PubMed] [Google Scholar]
- 32.Tsujimura H, Tamura T, Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J Immunol. 2003;170:1131–1135. doi: 10.4049/jimmunol.170.3.1131. [DOI] [PubMed] [Google Scholar]
- 33.Watowich SS, Liu YJ. Mechanisms regulating dendritic cell specification and development. Immunol Rev. 2010;238:76–92. doi: 10.1111/j.1600-065X.2010.00949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becker AM, Michael DG, Satpathy AT, Sciammas R, Singh H, Bhattacharya D. IRF-8 extinguishes neutrophil production and promotes dendritic cell lineage commitment in both myeloid and lymphoid mouse progenitors. Blood. 2012;119:2003–2012. doi: 10.1182/blood-2011-06-364976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamura T, Kurotaki D, Koizumi S. Regulation of myelopoiesis by the transcription factor IRF8. Int J Hematol. 2015;101:342–351. doi: 10.1007/s12185-015-1761-9. [DOI] [PubMed] [Google Scholar]
- 36.Hambleton S, Salem S, Bustamante J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365:127–138. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salem S, Langlais D, Lefebvre F, et al. Functional characterization of the human dendritic cell immunodeficiency associated with the IRF8(K108E) mutation. Blood. 2014;124:1894–1904. doi: 10.1182/blood-2014-04-570879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurotaki D, Osato N, Nishiyama A, et al. Essential role of the IRF8-KLF4 transcription factor cascade in murine monocyte differentiation. Blood. 2013;121:1839–1849. doi: 10.1182/blood-2012-06-437863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurotaki D, Yamamoto M, Nishiyama A, et al. IRF8 inhibits C/EBPalpha activity to restrain mononuclear phagocyte progenitors from differentiating into neutrophils. Nat Commun. 2014;5:4978. doi: 10.1038/ncomms5978. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, Yan M, Sun J, et al. A reporter mouse reveals lineage-specific and heterogeneous expression of IRF8 during lymphoid and myeloid cell differentiation. J Immunol. 2014;193:1766–1777. doi: 10.4049/jimmunol.1301939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116:2462–2471. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 43.Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine. 2008;42:277–288. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirai H, Zhang P, Dayaram T, et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 45.Marigo I, Bosio E, Solito S, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Strauss L, Sangaletti S, Consonni FM, et al. RORC1 regulates tumor-promoting “emergency” granulo-monocytopoiesis. Cancer Cell. 2015;28:253–269. doi: 10.1016/j.ccell.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 47.Wu L, Yan C, Czader M, et al. Inhibition of PPARgamma in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood. 2012;119:115–126. doi: 10.1182/blood-2011-06-363093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Haas N, de Koning C, Spilgies L, de Vries IJ, Hato SV. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology. 2016;5:e1196312. doi: 10.1080/2162402X.2016.1196312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kusmartsev S, Su Z, Heiser A, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2008;14:8270–8278. doi: 10.1158/1078-0432.CCR-08-0165. [DOI] [PubMed] [Google Scholar]
- 50.Lee JM, Seo JH, Kim YJ, Kim YS, Ko HJ, Kang CY. The restoration of myeloid-derived suppressor cells as functional antigen-presenting cells by NKT cell help and all-trans-retinoic acid treatment. Int J Cancer. 2012;131:741–751. doi: 10.1002/ijc.26411. [DOI] [PubMed] [Google Scholar]
- 51.Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother. 2013;62:909–918. doi: 10.1007/s00262-013-1396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]