

Abstract
Protein losing enteropathy (PLE) has been associated with more than 60 different conditions, including nearly all gastrointestinal diseases (Crohn’s disease, celiac, Whipple’s, intestinal infections, and so on) and a large number of non-gut conditions (cardiac and liver disease, lupus, sarcoidosis, and so on). This review presents the first attempt to quantitatively understand the magnitude of the PLE in relation to the associated pathology for three different disease categories: 1) increased lymphatic pressure (e.g., lymphangiectasis); 2) diseases with mucosal erosions (e.g., Crohn’s disease); and 3) diseases without mucosal erosions (e.g., celiac disease). The PLE with lymphangiectasis results from rupture of the mucosal lymphatics, with retrograde drainage of systemic lymph into the intestinal lumen with the resultant loss of CD4 T cells, which is diagnostic. Mucosal erosion PLE results from macroscopic breakdown of the mucosal barrier, with the epithelial capillaries becoming the rate-limiting factor in albumin loss. The equation derived to describe the relationship between the reduction in serum albumin (CP) and PLE indicates that gastrointestinal albumin clearance must increase by at least 17 times normal to reduce the CP by half. The strengths and limitations of the two quantitative measures of PLE (51Cr-albumin or α1-antitrypsin [αAT] clearance) are reviewed. αAT provides a simple quantitative diagnostic test that is probably underused clinically. The strong, unexplained correlation between minor decreases in CP and subsequent mortality in seemingly healthy individuals raises the question of whether subclinical PLE could account for the decreased CP and, if so, could the mechanism responsible for PLE play a role in the increased mortality? A large-scale study correlating αAT clearance with serum albumin concentrations will be required in order to determine the role of PLE in the regulation of the serum albumin concentration of seemingly healthy subjects.
Keywords: enteropathy, albumin, Crohn’s, celiac, lymphangiectasis, antitrypsin
Introduction
Protein losing enteropathy (PLE) refers to the abnormally rapid loss of serum proteins into the gut lumen. Since the first suggestion in 1947 that there was excessive loss of albumin in the stomach of patients with Menetrier’s disease, PLE has been observed in more than 60 different conditions, including nearly all gastrointestinal (GI) diseases (Crohn’s disease, celiac, Whipple’s, intestinal infections, and so on) and a large number of non-gut conditions (cardiac and liver diseases, lupus, sarcoidosis, and so on).1 While there is a vast literature concerning measurements of PLE in various diseases, there is a paucity of recent reviews other than those appearing in textbooks, such as the authoritative chapter by Greenwald.1 In this review, we have strived to present a unified analysis of the entire field of PLE with a focus on understanding how basic pathophysiologic mechanisms relate to the quantities of serum protein lost into the gut.
All blood proteins and, in some cases, blood cells are lost in PLE; however, the usual focus is on albumin. Because of the relatively long lifetime of albumin, even relatively slow leaks can appreciably increase the normal slow clearance and, hence, reduce the serum level of albumin. As discussed in our recent review of albumin metabolism, there is an unappreciated but astonishingly strong correlation between mortality in seemingly healthy individuals and relatively minor decreases in serum albumin concentration (CP) that remains well above the lower limit of normal (3.5 g/dL).2 For example, in a study of 1.7 million life insurance applicants followed for 12 years, apparently healthy 50–70-year-old males with a CP of 3.90–4.0 g/dL (mean of all subjects=4.4 g/dL) had a relative mortality ratio of 1.82.3 Our analysis strongly suggested that a low CP is a surrogate marker for underlying disease,2 and it seems possible that an understanding of the mechanism(s) causing reduced serum albumin might provide insights into the process(es) responsible for the increased mortality. As discussed below, the steady-state CP represents a balance between the rate of hepatic albumin synthesis versus catabolic, urinary, and intestinal (i.e., PLE) albumin clearances. It follows that one or more of these four factors must be altered in the subclinical conditions that lead to increased mortality of mildly hypoalbuminemic subjects. In this review, we examine the possibility that unrecognized PLE could be the mechanism responsible for this hypoalbuminemia.
A novel aspect of this review is the focus on understanding PLE quantitatively in terms of the underlying pathophysiology at the vascular and mucosal level. Figure 1 shows a schematic diagram of the intestinal mucosa that will be used as the basis for our discussion. There are two possible sources of the systemic protein that is lost into the gut: the mucosal capillaries and the lymphatics. As discussed below, the quantitative and clinical features of PLE differ markedly depending on which of these two sources is involved.
Figure 1.

Schematic diagram of normal intestinal mucosa.
Notes: The epithelial cells present a diffusion barrier between the interstitial space and the lumen. The albumin that slowly leaks from the plasma (concentration=CP) to the interstitial space (CTis) is removed by the lymphatics. The luminal albumin concentration (CLumen) is zero.
The “Albumin pharmacokinetics” section briefly discusses albumin pharmacokinetics and derives the quantitative relationship between CP and the GI protein clearance. The “Qualitative and quantitative measurements of PLE” section reviews the different approaches that have been developed to quantitate PLE, each of which has its strengths and limitations. A small protein loss occurs in normal subjects and the “Physiology of normal GI albumin clearance” section discusses the underlying physiology. Since more than 60 different pathologic conditions have been associated with PLE, the discussion is simplified by grouping these conditions into those thought to share common physiologic mechanisms. We will base our discussion on the organizational scheme of Greenwald that distinguishes three separate groupings: 1) increased lymphatic pressure (refer the “Increased lymphatic pressure” section); 2) mucosal erosions/ulcerations (refer the “Diseases with mucosal erosions” section); and 3) mucosa grossly intact (refer the “Diseases without mucosal erosions” section). These three classifications refer to intestinal disorders; gastric PLE is discussed as a separate problem (refer the “Gastric PLE” section). The “Genetic and animal models of PLE” section briefly discusses some animal and genetic models of PLE. Finally, in the “Summary and discussion: clinical implications of PLE” section, we briefly discuss the accepted as well as potential significance of PLE in clinical medicine.
Albumin pharmacokinetics
Analysis of albumin pharmacokinetics provides a rough estimate of the increase in the GI protein loss (PLE) required to account for varying degrees of hypoalbuminemia. In the steady state, the serum albumin concentration (CP) is determined by a balance between the rate of hepatic albumin synthesis versus the clearance via catabolism (ClCat), and urinary (ClUr) and GI (ClGI) losses:
| (1) |
where ClP is the total serum albumin clearance.2 In the normal human, the total body albumin is about 280 g with an absolute synthesis rate (i.e., turnover) of about 150 mg/kg/day or 10.5 g/day for a 70 kg human.2 This turnover corresponds to roughly 8.5% of plasma albumin or 4% of the total body albumin per day, with a total body albumin turnover time of about 25 days or a half-time of 17.3 days.2 Assuming a normal CP of 4.4 g/dL, this turnover corresponds to a serum albumin clearance (ClP) of about 240 mL/day for a 70 kg human. By far, the largest component of this clearance is catabolic (~90%), with urinary (ClUr) clearance of about 4% and GI clearance (ClGI) of 6% (15 mL/day, see below). Solving Equation 1 for ClGI:
| (2) |
Subtracting Equation 2 for a normal subject from that for a PLE subject and assuming that ClUr is unchanged in PLE:
| (3) |
Rearranging Equation 3:
| (4) |
A lower bound estimate for is obtained if one assumes that both Synthesis and ClCat remain normal in the subject with PLE in Equation 4:
| (5) |
Equation 5 predicts that a subject with PLE who has a serum albumin of half of the normal (CPN/CPPLE=2.0) and otherwise normal albumin kinetics will have an increase in GI albumin clearance equal to the normal total albumin clearance and, adding the baseline GI clearance , a total GI clearance of 255 mL/day/70 kg corresponding to a 5.61 g/day loss into the gut. However, this represents a low estimate of GI loss because as the serum albumin concentration (CP) falls, there is a variable homeostatic increase in the rate of albumin synthesis as well as a decrease in ClCat,2 both of which increase the estimate of (Equation 4) required to maintain the serum albumin at a given level. Measurements of the rates of synthesis in patients with severe PLE (serum albumin about half normal) and normal hepatic function showed ratios of SynthesisPLE/SynthesisN varying from 1.24 to 2.61.4–6 In a study of the response of a normal subject to a decrease in CP produced by plasmapheresis, lowering the CP by 20% increased the synthesis by 25%.2,7 In addition, there is a not commonly recognized reduction in ClCat as CP falls due to the competitive binding of albumin to the Fc IgG receptor (FcRn).8–10 FcRn was originally shown to bind IgG at acidic pH, diverting it from lysosomal degradation and prolonging the lifespan of IgG. Recently, it has been shown that this receptor also binds albumin, protecting it from lysosomal degradation. Since the amount of FcRn is insufficient to bind all albumin, only a fraction is protected by this mechanism. As CP falls, the fraction of albumin rescued increases and the ClCat decreases, with the rate of ClCat falling to near zero in subjects with very low serum albumin levels (congenital analbuminemia).2,8 As a rough approximation, we have assumed that the change in Synthesis and ClCat as CP falls is described by:
| (6) |
Substituting Equation 6 into Equation 4 provides a prediction of the increase in the steady-state GI albumin clearance (ClGI) in a patient with hypoalbuminemia resulting solely from PLE. This relation is plotted in Figure 2 (assuming , , and are 240, 215, and 15 mL/day, respectively). Using this more likely relationship between ClGI and CP, the otherwise “normal” subject with a CP of half of the normal would have to have a GI of 523 mL/day.
Figure 2.
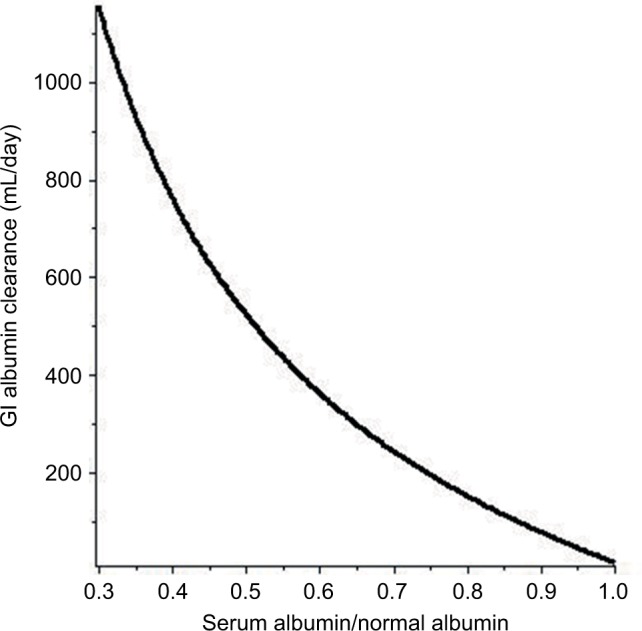
The predicted relationship between the increase in GI albumin clearance and the resulting steady-state serum albumin (serum albumin/normal albumin) in a PLE subject with normal renal and hepatic functions.
Abbreviations: GI, gastrointestinal; PLE, protein losing enteropathy.
Qualitative and quantitative measurements of PLE
Because most serum proteins lost into the intestine are rapidly hydrolyzed and the amino acids reabsorbed, analysis of intestinal or fecal contents cannot be used to diagnose PLE. In 1960, Steinfeld et al11 described the first attempt to quantitate PLE by measuring the percentage of an intravenous (IV) dose of 131I-polyvinylpyrrolidone (PVP) excreted in a 4-day stool collection. In a series of patients with regional enteritis or ulcerative colitis with low serum albumin, the percentage of the injected PVP excreted ranged from 2.3% to 7.4% (mean=4.1%), about 10 times the control value (range 0.16%–0.63%).11 Unfortunately, 131I-PVP measurements are only qualitative because PVP has a very heterogeneous size distribution (mean molecular weight of about 40 kDa), and the gut clearance may not be representative of albumin (molecular weight of 68 kDa).11,12
In 1961, Waldmann13 introduced what is considered to be the “gold standard”1 for quantitating PLE: measurement of the 51Cr fecal excretion following IV administration of 51Cr-albumin. The seemingly straightforward rationale underlying this measurement is that Cr+3 is not secreted or absorbed from the gut – thus, 51Cr excreted in feces over a given time period must quantitatively measure the rate that labeled albumin was lost into the GI tract. The product of this radioactivity excretion rate and the specific activity of serum albumin should thus provide a precise measure of the rate at which serum albumin leaked into the gut. Unfortunately, multiple factors tend to complicate this seemingly simple technique. Albumin has a high natural affinity for Cr, and labeling is carried out simply by incubating 51Cr+3 with albumin. However, this labeling, which results in the binding of about 7 Cr/albumin, is heterogeneous, with some loosely bound Cr that dissociates after IV injection and binds to other blood (e.g., endogenous albumin, globulins, and transferrin) and tissue proteins. Van Tongeren and Majoor12 and Kerr et al14 have carefully analyzed the pharmacokinetics of plasma 51Cr following an IV dose of 51Cr-albumin. There is an initial very rapid fall of serum radioactivity of about 90% over the first 4 days that markedly exceeds the rate of extravascular distribution measured simultaneously with 125I-labeled albumin. A similar difference in the disappearance of the two radioactive labels was observed when albumin was double-labeled with both 51Cr and 125I,14,15 indicating that the rapid clearance was not due to damage of albumin by Cr labeling, but rather that Cr was eluting from albumin. Since most of the rapidly disappearing 51Cr does not appear immediately in either urine or feces, it is presumably bound to extravascular tissue proteins. After about 4 days, the 51Cr decay becomes exponential, with a half time of roughly 6.6 days, about half that of 125I-albumin (14 days). Using starch gel electrophoresis, Kerr et al14 found that 15 minutes after the IV 51Cr-labeled albumin, only about 60% of the label was in the albumin fraction, with most of the remainder in the globulin fractions. By 6 days, 80% of the 51Cr activity was in the albumin fraction. The 51Cr serum disappearance rates following IV infusion of 51CrCl3 is virtually identical to that following IV infusion of prelabeled 51Cr-albumin,12 indicating that the 51Cr distributes rapidly to blood and tissue proteins, even when bound to albumin prior to the IV infusion.
These results clearly show that serum 51Cr is not a strict label for albumin. Since the clearance measurement utilizes the serum 51Cr concentration (not 51Cr-albumin), fecal radioactivity reflects a weighted average of the leak of various serum (and, possibly, tissue) Cr binding proteins, some of which may leak more rapidly than albumin. After about 4 days when the rapid serum 51Cr decay is completed, most of the serum 51Cr is albumin bound and the GI 51Cr clearance is, presumably, primarily a measure of GI albumin clearance (ClGI). However, the possibility remains that the gut clearance of a very small 51Cr-labeled protein could be manyfold faster than albumin; thus, even a low concentration of this serum protein could appreciably distort what is assumed to be albumin clearance. As noted, the exponential total clearance rate of 51Cr+3 4 days after infusion remains roughly twice that of iodine-labeled albumin. The extent to which rapid gut clearance of non-albumin-bound 51Cr+3 accounts for a fraction of this elevated total clearance remains to be determined.
Waldmann13 initially estimated PLE from a measurement of the percentage of the dose of the IV 51Cr-labeled albumin excreted during the first 4-day fecal sample following IV injection of the labeled compound. Because of the rapid initial serum 51Cr loss described above (90% over 4 days), this should be regarded as only a semi-quantitative measurement of GI albumin loss, and it does not provide an accurate measurement of GI albumin clearance (ClGI). Normal subjects excreted from 0.1% to 0.7% (mean 0.23%) of the infused dose, while patients with severe PLE (serum albumin less than half normal) excreted from 4% to 21% (100 times normal). Waldmann et al15 used this procedure in a later study of 180 subjects, 50 controls (normal CP and no evidence of GI disease) and 130 subjects were selected because they had significant hypoalbuminemia that could not be explained on the basis of either liver disease or proteinuria, with most having some sort of GI disease. Despite the semi-quantitative nature of the fecal loss measurements and the likelihood that Cp could be influenced by factors other than PLE, a remarkably good correlation was observed between 51Cr-albumin GI loss and CP. Nearly all patients with a CP <2.5 g/dL lost more than 6% of the dose/4 days in the stool (26 times normal).
In order to accurately determine the GI serum albumin clearance, it is necessary to relate the rate of fecal 51Cr-albumin excretion to the 51Cr-albumin concentration (or 51Cr-specific activity) in the serum at the time the 51C-albumin in the fecal sample was passing from the serum into the gut. Waldmann et al15 estimated this clearance by measuring the daily serum 51Cr concentration and the daily fecal 51Cr excretion over a 12-day period and determining the 51Cr clearance using the relation:
| (7) |
averaged over days 5–12. Normal subjects cleared about 15 mL of plasma/day (6.2% of the total normal albumin clearance of 240 mL/day), while PLE subjects cleared from 50 to 1,800 mL/day. Subjects with a serum albumin about half of the normal had clearances of about 600 mL/day, in good agreement with that predicted above in the “Albumin pharmacokinetics” section (Figure 2).
It should be stressed that accurate assessment of the clearance rate requires knowledge of the serum concentration of the cleared compound at the time the compound is being removed from the circulation (not the time when the compound is exiting from the body). This is a negligible problem with urinary clearance of compounds such as creatinine or sodium, since their serum concentrations remain roughly constant over the relatively short urine collection period. However, as noted above, serum 51Cr-albumin concentration declines by about 90% over the first 4 days following infusion and subsequently at a rate of roughly 6%/day. Clearance measurements made during the first few days after 51Cr-albumin infusion (which has been the case in many publications) are fraught with error since it is impossible to accurately predict the serum 51Cr-albumin concentration at the time the labeled protein leaked into the bowel – which might be several days prior to the passage of the fecal specimen in healthy subjects versus hours in subjects with diarrhea. Waldmann et al15 provided the most reliable clearance estimates utilizing individual fecal specimens obtained on days 5–12 after infusion of 51Cr-albumin and the serum 51Cr concentration obtained on the day prior to passage of the fecal sample (Equation 7), which assumes a 1-day transit time between gut leakage of protein and passage of the fecal sample. In normal controls where the plasma 51Cr concentration is falling by only about 6% per day, inaccuracies in the transit time assumption induce relatively small errors. However, in subjects with severe PLE in whom the plasma 51Cr concentration may decline by 50%/day, incorrect assumptions concerning the transit time lead to sizable errors in the estimated gut clearance of albumin. In many reports, the timing of the serum 51Cr-albumin measurement is not clearly stated.
The requisite radioactive exposure and the long experimental period of daily stool and serum samples (>4 days after infusion of the labeled albumin) markedly limit the clinical utility of 51Cr-albumin clearance measurements. The procedure currently used to quantitate PLE is the α1-antitrypsin (αAT) clearance measurement, first described by Bernier et al.16 Based on the ideal assumptions that αAT is neither absorbed from nor physiologically secreted into the intestine and is resistant to intestinal proteolysis, measurement of its fecal loss should be indicative of the rate of loss of similar-sized serum proteins. Since the molecular weight of αAT is 50 kDa, only slightly less than that of albumin (67 kDa) and, as shown below, there is only weak protein size discrimination in PLE, the fecal αAT loss should be similar to that of albumin. The ideal assumptions may not be perfectly satisfied because there may be some minor and varying rates of biliary secretion of αAT, some intraluminal proteolysis (see below), and the presence of several protein complexes of αAT in feces that differ from the serum form.17 However, these factors are generally regarded as minor, and αAT clearance has become a widely accepted measure of PLE. The αAT clearance is usually determined from the amount of αAT in a 1-day fecal collection divided by the plasma αAT. A major advantage of this methodology is that αAT is produced endogenously and the serum concentration remains relatively constant, hence the timing of the serum measurement is not critical as is the case with 51Cr-albumin. Alternative simpler approaches, which are only semi-quantitative and do not measure clearance, neglect the serum measurement and simply determine the fecal αAT concentration in a stool sample (mg/dry weight) or the amount of αAT in stool collected over a given period, for example, a 1-day collection. Plasma αAT has large normal variations with systematic variations observed in some diseases, such as inflammatory bowel disease (IBD) where the serum αAT is about twice normal.18 Thus, the clearance measurement is superior to the simpler approaches, as evidenced by a smaller normal range and better discrimination between normal and PLE patients.19,20
There have been three studies directly comparing 51Cr-albumin and αAT GI clearance in individual PLE patients, with disparate results. As discussed above, because of the initial rapid decline of plasma 51Cr-albumin, accurate timing of the serum 51C concentration used to calculate clearance is essential. Florent et al21 seemingly made the most accurate comparison in that they collected stools for 6 days beginning 2 days after the IV infusion of 51CrCl3 and used the daily fecal 51Cr and the serum 51Cr-albumin from the previous day to determine the clearance. They found a very strong linear correlation (r=0.96) between the αAT and 51Cr clearance, with the αAT clearance about half the 51Cr clearance. Quigley et al,17 using one serum 51Cr sample on day 3 and the average fecal 51Cr over multiple days, reported a weaker correlation (r=0.45) with the αAT clearance being ~60% of the 51Cr clearance. They determined a PLE diagnostic sensitivity and specificity of the αAT clearance (relative to 51Cr-albumin) of only 58% and 80%, respectively. Karbach et al19 found a stronger correlation (r=0.87), but with the αAT clearance about twice the 51Cr clearance, the opposite finding to that of Florent et al and Quigley et al17 who found the αAT clearance to be about half the 51Cr-albumin clearance. Karbach et al19 determined the 51Cr clearance from one bulk 3-day stool collection and a zero time plasma concentration estimated from the injected 51Cr-albumin dose divided by an estimation of serum volume, an obviously flawed methodology, given the previously described rapid initial decay of 51Cr concentration in the plasma. We believe the conclusion of Florent et al21 that αAT and 51Cr-albumin GI clearance are highly correlated with the αAT clearance about half that of the 51Cr-albumin clearance to be the best currently available result.
Since 51Cr-albumin clearance is no longer used and αAT clearance has become the current standard for quantitating PLE, we will heavily utilize αAT clearance measurements in this review. However, there is one situation in which αAT clearance is clearly an unreliable measure of PLE – when there is gastric protein loss such as that occurs in Menetrier’s disease – since αAT activity is almost completely destroyed during 1 hour of incubation in gastric juice at pH <3 maintained at 37°C.21 Florent et al22 have shown that if one suspects gastric PLE, the αAT clearance can provide a valid measure of protein loss if cimetidine IV infusion is employed to elevate the gastric pH. It seems likely that high-dose oral protein pump inhibitor administration could be used in a similar fashion to minimize acid denaturation of αAT in the stomach.
The normal average value for the αAT clearance is about 8 mL/day19 (about half the normal 51Cr-albumin clearance) with an upper limit of normal of 20 mL/day. Strygler et al20 have shown that this normal range is approximately doubled in normal subjects if rapid transit is induced with lactulose, sorbitol, sodium sulfate, or phenolphthalein, and they suggested that rapid transit decreases the time for luminal αAT degradation. While the concept that half the αAT entering the gut undergoes intestinal proteolysis is appealing in that it would explain the observation of Florent et al21 that the αAT clearance was about half the 51Cr-albumin clearance, it raises other questions. One problem is that this would predict that very clinically active IBD (with high PLE) with diarrhea and decreased transit times should have αAT clearances that approach 51Cr-albumin clearance. However, Florent et al21 observed that the GI αAT was half the 51Cr-albumin clearance for the entire range of pathologic clearances (15–400 mL/day). Second, although αAT is rapidly hydrolyzed at pH <3 in the stomach, hydrolysis is minimal during incubation for 2 hours in duodenal juice or 24 hours in feces;21 thus, it seems unlikely that there could be 50% hydrolysis during normal intestinal transit times. Finally, another possible explanation for why the normal GI αAT is half the 51Cr-albumin clearance is that a significant fraction (50%–100%) of the normal GI protein clearance occurs in the stomach (see below) and would be expected to be hydrolyzed. However, if this were the case, the αAT lost into the small intestine during PLE should not suffer the same hydrolysis, which is not consistent with the αAT results.21 In summary, the quantitative relationship between the αAT and 51Cr-albumin clearance is enigmatic. In any case, in the following sections, we will assume that the αAT clearance is an accurate indicator of GI protein loss and that, quantitatively, the αAT is about half the true (i.e., 51Cr-albumin) GI albumin clearance.
Another approach used to diagnose PLE is scintigraphy following administration of short lifetime tracer (Tc-99m, T1/2=6 hours or In-lll, T1/2=2.8 days)-labeled macromolecules (albumin, transferring, or dextran).1 This approach does not require fecal collection and has the unique advantage that it localizes the primary site of the protein leakage. In the most thorough protocols, imaging is acquired dynamically every minute for the first hour after IV injection, then statically for 3–7 hours (early phase) and at 24 hours (late phase). The early time course provides accurate identification of the initial leakage site and following the late phase, intestinal transit of the tracer provides more accurate gut (e.g., versus hepatic or renal) localization of the tracer, increasing the sensitivity.23,24 In a recent meta-analysis of scintigraphy’s PLE diagnostic accuracy, Khalesi et al24 found a high sensitivity (87%) and a somewhat lower specificity (62%) due to confusion with non-GI radioactive distribution. Scintigraphy is becoming an increasingly popular technique to diagnose and localize PLE.
Physiology of normal GI albumin clearance
As discussed above, the normal GI albumin clearance (ClGI) is about 15 mL/day. Although the mechanism and site of this normal loss have not been reviewed previously, it is generally assumed to represent baseline intestinal permeability. Given that the small intestine has about 25 m2 as the surface area25 and that the epithelial cells are rapidly turned over,26 it would not be surprising if there was some obligatory albumin leak. However, as discussed here, this minimal baseline leak may not represent intestinal permeability but rather albumin in the gastric, pancreatic, and/or hepatic secretions.
Using continuous gastric aspiration, Florent et al22 estimated the hourly rate of gastric albumin clearance in normal control subjects using either 51Cr-albumin or αAT with concomitant IV cimetidine. These gastric clearances were compared with simultaneous measurements of total daily GI clearance from fecal 51Cr. Surprisingly, the gastric clearance was roughly equal to the total GI clearance, suggesting that normal GI albumin clearance is mostly or entirely gastric.
There is also evidence that biliary secretion is the source of the normal protein loss into the gut. The most direct evidence in this regard is provided by the studies of 14C-PVP excretion in dogs by Ravin et al27 They showed that measurement of bile 14C-PVP equaled the fecal excretion and that diversion of bile completely eliminated the appearance of PVP in the stool. In humans, reports show the albumin concentration in hepatic bile to range from 0.5%28 to 2%29 of the plasma albumin. Using a human hepatic bile flow of 620 mL/day,30 this corresponds to bile GI albumin clearance of 3.1–12.4 mL/day.
Low amounts of albumin are also present in pancreatic secretion. Multigner et al31 reported that about 1% of the total protein in normal pancreatic juice was albumin. Using a total pancreatic juice protein of 12.8 mg/mL32 and a total pancreatic secretion of 1 L/day, this corresponds to a pancreatic GI albumin clearance of about 3 mL/day. According to these results, hepatic bile plus the pancreatic secretion can account for most of the total normal albumin clearance of 15 mL/day. These results are obviously contradictory, with Florent et al22 suggesting that the normal clearance is all gastric and Ravin et al27 suggesting that it is all hepatic. The gastric, hepatic, and pancreatic measurements use markedly different methodology and assumptions than the 24-hour fecal estimates of total GI albumin clearance and may not be directly comparable. However, these results suggest that the normal albumin clearance could be completely accounted for by the albumin in the normal biliary, pancreatic, or gastric secretions and is not necessarily the result of a low baseline intestinal permeability for albumin.
Increased lymphatic pressure
In his comprehensive review, Greenwald places 18 different diseases in the pathophysiologic category of increased lymphatic pressure.1 The three best characterized examples are primary intestinal lymphangiectasia, constrictive pericarditis, and the Fontan procedure. Analysis of these three conditions provides useful diagnostic criteria for assigning a lymphangiectatic etiology to a PLE condition, and these criteria will be used to investigate whether other diseases placed in this category by Greenwald are correctly assigned.
Primary intestinal lymphangiectasis is a rare disease, first described by Waldmann et al,5 in which there is a central blockage of lymph drainage producing generalized lymphedema (lower leg edema, pleural effusion, and chylous ascites) in addition to PLE.33 This condition is characterized by specific intestinal histologic (interstitial edema, dilated submucosal lymphatics) and endoscopic (edematous thickening of mucosal folds, nodular protrusions, diffuse white plaques, and white-tipped villi) findings.33–36 In one report, endoscopic biopsy of a “… 3 cm mulitlobulated cystic mass” in the duodenum “… resulted in an exudation of white, chylous material”,36 and there is also one report of direct duodenal sampling of a high-protein, high-fat chylous effusion in a fasting subject.37 Lymphography obtained by injection of contrast in a lymphatic channel in the foot demonstrates the retrograde passage of the contrast medium into the intestinal lymphatics and out into the small intestinal lumen.38–41 In addition to the severe hypoalbuminemia (~2 g/dL), other characteristic abnormalities include low immunoglobulin levels (IgG, IgA, and IgM) and uniquely very low CD4 T cell counts.33 Substitution of medium-chain triglycerides which are transported directly into portal blood rather than the lymphatic system reduces intestinal lymphatic flow with an associated partial or complete clearance of the symptoms and signs of PLE.42,43
Wochner et al6 measured the rate of albumin synthesis (bicarbonate-14C labeling), albumin catabolism or turnover (131I-albumin time constant), and intestinal loss (51Cr-albumin in 4-day feces) in four subjects with primary lymphangiectasis. The serum albumin was about half normal and the albumin synthesis rate was about 40% greater than normal. Using the steady-state assumptions in Equations 4 and 6 (plotted in Figure 2), the GI albumin clearance should have been 500 mL/day, consistent with the experimental 51Cr-albumin fecal excretion corresponding to a of 420 mL/day, which is 30 times normal.
Constrictive pericarditis, in rare cases, can produce large, nonfluctuating increases in venous pressure (20–30 mm Hg) that are accompanied by PLE that has a clinical presentation (symptoms, mucosal histopathology, and laboratory findings) nearly identical to that of primary lymphangiectasis.44–47 All these findings are normalized when the venous pressure elevations are reversed by pericardiectomy, proving that the high venous pressure leads to the increased lymphatic pressure and lymph leakage into the intestinal lumen. Wilkinson et al47 described one subject with a venous pressure of 24 mmHg, a serum albumin of half normal, a normal albumin synthesis rate, and a 21.4% 4-day fecal 51Cr-albumin loss. After surgery, the serum albumin returned to normal and the 4-day fecal 51Cr-albumin loss fell to 0.59% (a 36-fold decrease).
The assumed pathophysiology which produces the PLE associated with constrictive pericarditis is shown in Figure 3. High systemic venous pressure produces increased intestinal lymphatic pressure which, in select cases, causes localized rupture of the intestinal lymphatics into the lumen that drains and decompresses the entire systemic lymphatic system. The albumin GI clearance of 420 mL/day reported by Wochner et al6 would correspond to the intestinal loss of about a liter of lymph/day, assuming a lymph albumin concentration 45% of plasma.
Figure 3.

Schematic diagram illustrating the pathophysiology for the PLE produced by increased lymphatic pressure resulting from increased venous pressure (PVein).
Note: The lymphatic ruptures into the intestinal lumen, allowing decompression and retrograde drainage of the systemic lymph.
Abbreviation: PLE, protein losing enteropathy.
Markedly increased central venous pressure influences lymphatic pressure by two mechanisms: first, the high venous pressure causes an imbalance in Starling forces, such that there is markedly increased capillary filtration and lymph production in the gut mucosa. This imbalance is further increased if the lymphatics rupture into the intestinal tract, markedly lowering plasma albumin and oncotic pressure. Petersen and Hastrup46 made quantitative measurements of thoracic duct lymph flow and albumin concentration in one constrictive pericarditis subject with severe PLE and in normal controls. The thoracic duct flow in the PLE subject was 4.6 times greater than normal (4.44 versus 0.96 L/day). If the lymph flow resistance is unchanged in the PLE subject, this increased flow should, by itself, increase the downstream lymph pressure by a factor of 4.6. Second, the high pressure in the innominate vein (into which the thoracic duct drains) increases the resistance to drainage of the thoracic duct lymph flow. The importance of the latter mechanism, which is not widely appreciated, is directly supported by the observations that overt PLE is rarely seen in patients with severe portal hypertension with portal pressures that are routinely comparable or greater to those in constrictive pericarditis, but relatively normal systemic venous pressures (see below).
Surprisingly, the major focus of recent PLE clinical investigation has been the study of the mechanism and treatment of PLE associated with the Fontan procedure, a surgical technique that involves diverting the venous blood directly to the pulmonary artery in children with a single ventricle. This procedure markedly increases the central venous pressure and in 3%–18% of subjects, PLE appears from 2 to 10 years after the surgery.48 This rare form of PLE (prevalence of <2,000 in USA49) has been intensively investigated because it is a leading cause of morbidity and mortality in patients undergoing the Fontan procedure.50,51 There is additional interest in this syndrome because it represents the best characterized “experimental model” of PLE. While all patients who have undergone a Fontan procedure have elevated venous pressure, the finding that only a minority develop PLE has led to speculation as to the etiology of the PLE. Initially, it was suggested that the decreased mesenteric blood flow associated with the procedure was the primary event leading to intestinal mucosal damage and protein leakage.52 However, the mucosal histology in PLE of these subjects is identical to that found in primary intestinal lymphangiectasia and constrictive pericarditis,52–57 and it is now generally accepted that the PLE is the result of the high systemic venous pressure that leads to lymphatic rupture in the gut. Direct evidence that PLE in Fontan subjects results from the increased lymphatic pressure is provided by the recent description of dramatic improvement of PLE following diversion of the innominate vein (which drains the thoracic duct) to the low-pressure atrium.55 The deleterious effect of the resultant shunting of blood past the lung with the concomitant arterial desaturation is considered to be of minor clinical importance relative to the benefit that is achieved with the alleviation of the PLE.
It is not clear why only some Fontan subjects develop PLE. There is a correlation of PLE with the central venous pressure at the time of development of PLE, but this correlation disappears during the chronic phase.58 However, this correlation is weak, with some patients developing PLE even though they have a “good” surgical result (i.e., right atrial pressure <15 mmHg).51 The most likely explanation is that the lymphatic system is operating near capacity in most subjects following the Fontan procedure and either some predisposing defect (e.g., congenital lymphatic malformation) or an event such as an infection56,59,60 results in the rupture of the lymphatics in a localized area of the intestine.51 Direct support for the localization of the leak is provided by the observations that scintigraphy often demonstrates localized areas of leaks and resection of the identified region alleviates the PLE.56
This observation that only a subset of Fontan patients has overt clinical signs of PLE (e.g., edema, effusions, and so on) raises the question as to whether the remainder of the subjects have subclinical PLE. The vast majority of this well-studied group of patients had normal serum albumin61–63 and stool αAT clearance or concentration that was normal61,64 or only slightly elevated (~2-fold).62,63 Thus, the relatively high venous pressure (~12 mmHg) routinely observed in Fontan subjects usually does not produce appreciable albumin leakage into the gut, that is, PLE is an all or none phenomenon in these subjects. It is necessary to postulate that some unidentified abnormality other than elevated venous pressure must render a subset of Fontan subjects susceptible to chronic lymphatic rupture. The PLE observed in Fontan subjects has been reported to be triggered by an enteric infection.59,60 It seems possible that such tissue injury initiates PLE, which then becomes self-sustaining, possibly in part secondary to the enhanced lymph production engendered by the hypoalbuminemia that results from protein loss into the gut.
The occurrence of lymphatic PLE produced by the increased systemic venous pressure with constrictive pericarditis or the Fontan procedure raises the question of whether PLE is also associated with the very common problem of congestive heart failure (CHF). Although there are case reports of PLE in patients with CHF,65 it is a very rare association. Berkowitz and Segal66 measured αAT clearance in 25 patients with severe heart failure and found only one abnormally high value (four times normal; data plotted in Figure 4).
Figure 4.

Fecal α-antitrypsin clearance in various disease states.
Notes: The “normal” clearance corresponds to the control data for the Crohn’s disease study (mean=8.3 mL/day, range of 1.5–19.2 mL/day). In patients with heart failure, the clearance is normal except for one patient. In contrast, most subjects with Crohn’s or celiac disease have a statistically significant increase in clearance.
Unlike the situation with constrictive pericarditis or the Fontan procedure, the systemic venous pressure draining the thoracic duct is close to normal in patients with portal hypertension secondary to liver disease or portal vein obstruction. The markedly increased portal vein pressure leads to increased liver sinusoidal and intestinal capillary fluid filtration, which is balanced by an increase in thoracic duct flow, which theoretically could also produce lymphatic PLE. Dumont and Mulholland67 measured thoracic duct pressures and flows in 20 patients with long-standing liver dysfunction (17 with ascites) and all had increased pressures and flows, from 2 to 10 times normal. Hypoalbuminemia is routinely associated with portal hypertension and an important clinical question is with what frequency does PLE cause/contribute to this hypoalbuminemia. A few case reports have documented that portal hypertension can cause PLE by a clear-cut lymphangiectatic mechanism, but this association appears to be rare. Stanley et al68 described a patient with portal hypertension with PLE documented by whole gut lavage (αAT 100 times normal) and edematous villi with distended lymphatics on biopsy, all of which returned to normal following a transjugular intrahepatic portosystemic shunt. Wong et al69 described a patient with severe liver disease and portal hypertension with a serum albumin one-fourth of the normal, with the classical lymphangiectactic intestinal biopsy and PLE documented by an αAT clearance of 10-fold normal which was reversed by liver transplantation. There are surprisingly few large-scale clinical studies of the association of PLE with liver cirrhosis and portal hypertension. Georgopoulos et al70 studied 12 patients with moderate to severe alcoholic cirrhosis and portal hypertension. All had low serum albumin (mean of two-thirds of normal) and 9 of the 12 had ascites. None of them had PLE as defined by an increased whole gut lavage αAT. In an older study, Davcev et al71 studied 18 patients with advanced liver cirrhosis (alcoholic and viral origin) and used 131I-PVP to measure PLE. Two of the 18 had significantly increased PVP secretion and both of these had classical intestinal mucosal lymphangiectasia on biopsy.
These results for portal hypertension and CHF are similar to those described above for the Fontan procedure, with PLE occurring only in a very small subset of patients. Thus, the PLE produced by increased venous pressure seems to be an all or none phenomenon, with no PLE occurring until the downstream lymphatic pressure gets high enough to rupture the lymphatics locally into the intestine, draining and decompressing the entire lymphatic system. The fact that most Fontan or CHF patients with markedly increased systemic venous pressure or cirrhotic patients with large increases in portal vein pressure do not have PLE indicates that increased mucosal capillary pressure (and the concomitant increase in mucosal interstitial pressure) does not by itself produce PLE.
Greenwald1 assigns several PLE disease entities to multiple pathophysiologic categories. For example, Whipple’s disease and systemic lupus erythematosus (SLE) are in both the “No mucosal erosion” and “Increased lymph pressure” (i.e., lymphangiectasis) classes and Crohn’s disease is in both the “Mucosal erosion” and “Increased lymph pressure” classes. This is an indication of the poorly understood pathology of PLE and the difficultly of assigning a definite mechanism. One would predict that there should be marked differences in the hematologic findings in the three categories of PLE since in lymphangiectasia there is bulk loss of lymph, including the lymphoid cells, whereas the other two classifications have negligible cellular loss. As predicted, the characteristic findings in the three classical increased lymph pressure conditions discussed above (primary lymphangiectasis, constrictive pericarditis, Fontan procedure) are lymphopenia and a dramatic decrease in CD4 T cells (about 10% of normal).53,72,73 This selective depletion of CD4 cells presumably reflects the tendency of CD4 cells to circulate in lymph, whereas other immunologic cells are more tissue bound.72 Thus, the low CD4 cell count is a direct quantitative measure of bulk lymph loss into the intestine and the CD4 cell count can be used as a simple hematologic marker of lymphangiectasis. Surprisingly, this simple diagnostic criterion is not widely recognized.
One would also predict that, since there is bulk loss of lymph in lymphangiectasis, the rate of loss of plasma proteins should be proportional to the lymph protein concentration. Bendixen et al74 simultaneously measured the steady-state 125I-albumin and 131I-IgG loss in seven lymphangiectasis patients. The rate of IgG loss was 75% that of albumin, just what one would predict from intestinal lymph IgG concentration, that is, 75% of the albumin concentration.75,76 The rate of protein loss does not discriminate between the three PLE classes because, as discussed below, in both the “No mucosal erosion” (e.g., celiac disease) and the “Mucosal erosion” (e.g., Crohn’s disease), there is minimal size discrimination and the PLE loss rate is also proportional to the lymph concentration.
Diseases with mucosal erosions
Greenwald lists more than 20 diseases in this category, including malignancies (e.g., lymphoma, Kaposi’s sarcoma), infectious diseases (e.g., Clostridium difficile, Shigella), sarcoidosis, and IBD (e.g., Crohn’s disease, ulcerative colitis).1 Crohn’s disease is, by far, the best characterized and the most intensely investigated, and we will focus on this condition as the classic example of this PLE class. Crohn’s disease is characterized by local areas of chronic inflammation which lead to surface erosions and ulcers.77 Even in the early, mild form of the disease, focal erosions are present and, on endoscopy, these erosions may be visibly oozing a serous material.78 These erosions represent a clear breakdown of the normal mucosal permeability barrier and are an obvious candidate for the site of the PLE. This situation is schematically illustrated in Figure 5, where there is a breakdown of the mucosal barrier allowing free passage of interstitial protein into the intestine. Since the Crohn’s disease involves both the small bowel and/or colon, the details of the albumin permeability and surface area will obviously differ markedly depending on the location of the erosions.
Figure 5.

Schematic diagram of the PLE occurring in diseases with mucosal erosions.
Note: Because of the large breaks in the epithelial permeability barrier, the capillaries become the rate-limiting step in the leak of serum albumin and the interstitial albumin (CTis) locally equilibrates with the lumen (CTiss=CLumen=0).
Abbreviation: PLE, protein losing enteropathy.
As indicated in Figure 5, the rate of albumin loss at these erosion sites probably becomes limited by the normal capillary albumin permeability. In normal subjects, the flux of albumin across the capillary (JCapAlb) is balanced by the returning intestinal lymph flow rate (JLymph) times the lymph concentration (CLymph) which is equal to the interstitial albumin concentration (CTis):
| (8) |
where and SCap and PCap are the capillary surface area and albumin permeability, respectively. As shown in Equation 8, the lymph/plasma concentration ratio (CLymph/CP) provides a measure of the capillary permeability (PCap). The total thoracic duct lymph flow is about 2 L/day, of which about half (≈1 L/day) is intestinal (mesenteric).79,80 Since CLymph=CTis≈2.2 g/dL, about half CP,75 the normal JCapAlb is about 22 g/day, corresponding to a clearance (=JCapAlb/CP) of 500 mL/day (about twice the total rate of normal albumin synthesis). As illustrated in Figure 5, in the regions where the epithelial barrier is lost, the CTis should fall to zero, so that the diffusional gradient (CP–CTis) is doubled and, from Equation 8, the maximum possible PLE clearance if the entire mucosal barrier was lost would be 1,000 mL/day, twice the normal JCapAlb. Thus, the PLE albumin clearance should be proportional to the fraction of the total intestinal mucosa that is involved in the Crohn’s disease, varying from 0 for completely normal intestine to a maximum 1,000 mL/day if the entire intestinal mucosal surface were involved. This rough estimate assumes that the capillary permeability remains normal in the ulcerated area.
This description of GI albumin clearance can be quantitatively compared with the measurements of αAT clearance in a large series of patients with active Crohn’s disease plotted in Figure 4 (data from Karbach et al19). It can be seen that the αAT clearance is from 10 mL/day up to 200 mL/day greater than the normal clearance. Using the above estimate of mucosal capillary permeability, this would correspond to a loss from 1% to 20% of the normal intestinal mucosal barrier. It is important to recognize the marked difference in the pattern of αAT clearance in Crohn’s disease versus patients with increased lymphatic pressure (e.g., cirrhosis, heart failure, Fontan). In the latter, the αAT clearance is normal in the great majority of patients and becomes markedly abnormal only when the lymphatics rupture with leak of gross lymph into the intestine. In contrast, in active Crohn’s disease, all patients have a greater than normal αAT, as one would predict if the clearance were proportional to the fraction of intestine involved.
If the capillary is the rate-limiting barrier in diseases with mucosal erosions, the rate of PLE of albumin (68 kDa) versus IgG (150 kDa) should be determined by the normal capillary permeability. In direct measurements of the rabbit small intestinal capillary permeability using I-labeled albumin and IgG, Bill reported that the albumin permeability was about twice that of IgG.81 Estimates of the intestinal capillary permeability from the steady-state intestinal lymph/plasma ratio (CLymph/CP, Equation 8) suggest less size selectivity. Vaerman and Heremans76 measured the dog mesenteric lymph/plasma ratio for endogenous proteins varying in molecular weight from 44 kDa (orosomucoid) to 890 kDa (IgM). Although they did find a correlation between molecular weight and lymph/plasma ratio, the selectivity was surprisingly small, with IgG and IgM ratios of about 75% and 30% that of albumin, respectively. Beh et al75 found that, in sheep, albumin and IgG had nearly identical intestinal lymph/plasma ratios, indicating no size discrimination. These lymph/plasma permeability estimates assume that there is negligible local intestinal protein synthesis, which seems to be the case with the exception of IgA.76
These results indicate that there is minimal size discrimination in the intestinal capillary permeability of albumin versus IgG and, if this is the limiting barrier in PLE, one would predict that albumin and IgG should be lost at similar rates in PLE. Kingham and Loehry82 determined the selectivity of small intestinal loss into perfused small intestinal segments with radiologically identified Crohn’s disease using IV-injected 125I-PVP with a size range of 8–80 kDa. In normal subjects, the small intestinal epithelial permeability of the largest PVP (80 kDa) was about a 1,000-fold less than that of the smallest (8 kDa). In contrast, in Crohn’s disease, there was only about a 10-fold difference and, in the size range of about 40–80 kDa, there was almost no size discrimination, in agreement with the above prediction based on the finding of a normal albumin versus IgG capillary permeability. Bendixen et al74 simultaneously measured the steady-state 125I-albumin and 131I-IgG synthesis rates and the PLE rate using 59Fe-dextrin in 10 cases of Crohn’s disease. They found that the IgG and albumin synthesis rates were increased in proportion to the 59Fe-dextrin fecal clearance. The increase in the synthesis rate was about 45% greater for IgG than albumin, which they interpreted as indicating that there was increased catabolism of IgG in Crohn’s disease, in addition to the IgG PLE loss.
In contrast to the situation with lymphangiectasia, the increase in permeability produced by mucosal erosions should be bidirectional. That this is the case is confirmed by measurements of the rate of intestinal absorption of solutes that are normally impermeable, such as polyethylene glycol (PEG),83 ethylene glycol tetraacetic acid (EGTA),84 or lactulose.85,86 Probably, the best available measurement of intestinal permeability is the ratio of the absorption of orally administered lactulose/mannitol or lactulose/l-rhamnose, determined from a 5-hour urine collection. Lactulose normally has a very low permeability, while mannitol and l-rhamnose are inert markers that have a finite permeability through the normal tight junction complex (Figure 1) and correct for changes in transit time (e.g., diarrhea) and surface area (e.g., in celiac disease). In normal subjects, the lactulose/mannitol ratio is 0.02, increasing to about 0.07 in Crohn’s disease.86 Figure 6 shows a plot of the lactulose/mannitol ratio for controls versus patients with moderate to severe Crohn’s disease (data from Murphy et al86). The distribution of the increase in lactulose permeability is similar to the increase in αAT clearance in Crohn’s disease, as plotted in Figure 4. This would be predicted for erosions that produce a large, nonselective increase in epithelial permeability (Figure 5) that cannot distinguish between lactulose versus αAT (or albumin). The nonselectivity of the leak is also demonstrated by measurements of the absorption of PEG 600 from isolated, perfused ileal segments in humans.83 PEG 600 has a distribution of molecular weights varying from 590 to 942 Da. In normal subjects, the absorption decreases from 0.45% for 590 Da to 0.05% for the 942 Da PEG. In ileal loops with Crohn’s disease, the absorption rate of the largest PEG (942 Da) increases 25-fold to 1.25% and is not dependent on the molecular size, confirming the nonselectivity of the erosion permeability defect.
Figure 6.

The lactulose/mannitol permeability ratio in Crohn’s and celiac diseases.
Greenwald places Crohn’s disease in both the “Mucosal erosion” and “Increased lymph pressure” categories because the local inflammatory response may be associated with lymphangiogenesis, focal lymphatic obstruction, submucosal edema, and dilation of lacteals.87–89 However, this local pathologic response differs markedly from the “Increased lymph pressure” events described above, where there is a central blockage of the systemic lymph flow, producing a rupture in which the total body lymph leaks into the intestine. It is unlikely that a local lymph obstruction could produce enough pressure to produce this rupture and, if it did, it would result in only small, local lymph losses. Consistent with this, CD4 T cell levels which, as discussed above, are markedly reduced in the “Increased lymph pressure diseases” are normal in Crohn’s disease,90 ruling out any large lymphatic leak.
Diseases without mucosal erosions
Greenwald places more than 20 diseases in this category, including celiac disease, Whipple’s disease, SLE, cobalamin deficiency, and so on.1 Although all these diseases have varying and clearly defined changes in the intestinal epithelium, the mucosa appears grossly intact and there is no obvious break in the diffusional barrier. As shown schematically in Figure 7, it is usually assumed that there is some defect in the tight junction complex.91
Figure 7.

Schematic diagram of the PLE occurring in diseases without mucosal erosions.
Notes: It is assumed that the increased permeability results from a defect in the epithelial tight junctions. The epithelium is the rate-limiting step in the albumin leak and the interstitial albumin (CTis) is greater than that in the lumen (CLumen=0).
Abbreviation: PLE, protein losing enteropathy.
We will focus on celiac disease because it is the best characterized and a classic example of this form of PLE. In contrast to the localized focal erosions in Crohn’s disease, the pathologic mucosal changes in celiac disease are diffuse and widespread, usually involving most of the jejunum. It seems likely that, in this case, the mucosal permeability is rate limiting (Figure 7), unlike the case for Crohn’s disease where the capillaries are limiting, and the net albumin leak rate (JAlb) is described by:
| (9) |
where PEpith and SEpith are the permeability and surface area, respectively, of the affected mucosa and CTis is the interstitial albumin concentration.
Figure 4 shows the distribution of αAT clearance in untreated and treated celiac patients (data from Bai et al18). The clearance is significantly increased in most patients, varying from 20 to 90 mL/day, similar to but less than in severe Crohn’s disease. In celiac patients treated with a gluten-free diet, the αAT clearance returns to normal (Figure 4). Again, one would expect the increased permeability to be bidirectional, and Figure 6 shows the ratio of the lactulose/l-rhamnose permeability for treated and untreated celiacs (data from Stenhammar et al92). (These ratios have been scaled in Figure 6, so that the normal lactulose/l-rhamnose ratio is equal to that of the normal lactulose/mannitol ratio in the Crohn’s disease study.) Again, the increase in the lactulose permeability in celiac patients has a quantitatively similar distribution as the increased αAT clearance (compare Figures 4 and 6). This strongly suggests that the breakdown of the mucosal permeability barrier has a large enough pore size that it does not discriminate between lactulose and αAT (or albumin) and the magnitude of the leak is presumably proportional to the fraction of tight junctions that are defective (PEpith) and the surface area of the intestine that is involved (SEpith), as shown in Equation 9. Direct evidence for the relative nonselectivity of the leak in celiac disease is provided by measurements of the rate of leak of IV-injected 125I-PVP using the same techniques described above for Crohn’s disease.82 The loss of the normal size selectivity in celiac disease was nearly identical to that in Crohn’s, with only about a 10-fold difference in clearance between the smallest (8,000) and largest (80,000) PVP and, in the size range of about 40,000–80,000, there was almost no size discrimination.
More than 20 other diseases are usually placed in this category of diseases without mucosal erosions.1 We will discuss two of these in order to illustrate the markedly different pathologies in this category. Patients with active Whipple’s disease often have severe PLE that is responsible for marked hypoalbuminemia with a CP of <2.5 g/dL and GI albumin clearance 20 times greater than normal.93 Although the villi in these patients may be markedly abnormal and distended by large lipid droplets, the epithelial layer appears intact and there is no obvious site of protein leakage.94 Similarly, although rare, SLE can be associated with severe PLE with αAT clearance of 800 mL/day (100 times normal) and a CP of 1.5 g/dL.95,96 As demonstrated by 99mTc scintigraphy, the leak in SLE occurs over most of the small intestine.96 Small bowel biopsies in SLE patients with PLE may show mild mucosal edema and inflammatory infiltrate, but otherwise they are relatively normal with no evidence of mucosal ulceration or lymphangiectasis.95 Greenwald placed Whipple’s and SLE in both the “nonerosion” and “increased lymph pressure” classes. This latter classification is because of the lymphopenia and dilated intestinal mucosal lymphatics seen in Whipple’s disease93 and the systemic edema along with the appearance of lymphangiectasis and intestinal mucosal edema on biopsy in some, but not all, SLE patients.96 However, since neither Whipple’s93,97,98 nor SLE96,99 patients have the reduced CD4 counts that are characteristic of lymphatic leaks (see above), it seems unlikely that their PLE mechanism is the result of ruptured lymphatics.
These three diseases (celiac, Whipple’s, and SLE) are typical of the diseases in this class in that, although there may be some pathologic changes in the mucosa, the epithelial permeability barrier appears intact at the light microscopic level. This has led to the standard assumption of some sort of tight junction defect that allows relatively free passage of molecules as large as albumin (Figure 7). Clayburgh et al100 have discussed the evidence that tight junctions are dynamic structures whose permeability can be altered by bacterial infection or inflammatory immune responses. The immature cells in the crypts of normal mucosa occasionally have disorganized tight junction strands that disappear as the cells mature. Because of the shortened lifetime and increased turnover of the cells in celiac disease, there is a significantly increased fraction of this disorganization and an increase in the number of strand discontinuities.101,102 However, it should be emphasized that these changes are relatively minor and that there is no direct evidence that the leak occurs through tight junctions. A later section describes some experimental models that suggest that tight junction pathology can lead to PLE analogous to that seen, for example, in celiac disease.
Gastric PLE
Menetrier’s disease (hypoproteinemic hypertrophic gastropathy) was the first disease entity in which PLE was documented and quantitated.103 It is characterized by massive hyperplasia of the surface and mucus cells, with grossly enlarged folds in the fundus and body of the stomach. Although, as the name implies, hypoproteinemia (albuminemia) is a defining feature of the classical syndrome, patients can present with similar gross pathology without the PLE, and it is not clear if these cases represent completely different diseases.104 Meuwissen et al105 emphasize the importance of documenting the decreased acid secretion that is characteristic of Menetrier’s, in order to distinguish it from Zollinger–Ellison syndrome. The etiology is unknown, although there have been suggestions of associated infections such as Helicobacter pylori, herpes simplex virus, or cytomegalovirus.106 Efficacy of treatments with H. pylori eradication, antibiotics, prednisone, and octreotide is inconsistent.106 Although increased epidermal growth factor (EGF) ligands have been implicated in its pathology, treatment with EGF-blocking monoclonal antibodies has been disappointing.107,108 The only definitive treatment is gastrectomy, which relieves all the symptoms, including hypoproteinemia. There is a nearly identical PLE disorder in children that, unlike the adult form, is acute, resolves spontaneously, and has a strong association with cytomegalovirus infection.109
The PLE in Menetrier’s is clearly localized to the stomach. The early studies of Citrin et al103 and Jarnum and Jensen110 measured the appearance of iodine-labeled albumin and/or globulin in the stomach using extended periods of continuous gastric aspiration and compared this to measurement of total body albumin or globulin clearance. These are underestimates because of the rapid proteolysis that occurs in gastric secretions (hence the incentive for the later development of the Cr51-albumin technique). This proteolysis is variable and is decreased in patients with achlorhydria.110 Despite this underestimate, most of the increased albumin turnover in the Menetrier’s subject could be accounted for by the gastric loss. For example, in one patient studied by Citrin et al,103 the total albumin turnover was 17.6 g/day (7.1 g/day greater than the normal value of 10.5 g/day) and the concentration of 131I albumin in the 2 L/day of aspirated gastric juice was 4 g/L, corresponding to a gastric loss of 8 g/day. Jarnum and Jensen110 found that the rate of gastric albumin loss into the aspirate was four times greater when secretion was stimulated with histamine versus hexamethonium inhibition. Also, Reinhart et al111 observed a similar 4-fold increase in 51Cr secretion above the basal level with IV pentagastrin, indicating that albumin loss is directly related to the secretion rate. Following IV administration of 51Cr-albumin in Menetrier’s subjects, Florent et al21 compared the total GI protein loss determined from daily fecal 51Cr to the gastric 51Cr loss in a 90-minute gastric aspirate. They found that the gastric loss (extrapolated to 24 hours) represented about half the total daily GI loss. However, since the aspiration is by necessity in fasting subjects with reduced gastric secretion rates, this discrepancy is not surprising.
The major new development in the field of Menetrier’s is its association with EGF receptor ligands such as transforming growth factor α (TGF-α).112 Enhanced production of TGF-α has been found in Menetrier’s subjects.112 More convincingly, transgenic mice overexpressing TGF-α develop severe gastric adenomatous hyperplasia that is strikingly similar to Menetrier’s.113 Unfortunately, treatments of the Menetrier’s with TGF-blocking monoclonal antibodies have had only limited success.107,108 The transgenic mice produce increased mucus-laden secretions, but do not secrete albumin. As discussed above, there is a human variant of Menetrier’s that is not associated with PLE; thus, the protein loss is not of necessity associated with the gross pathologic features that characterize Menetrier’s.
The mechanism of the gastric protein loss in Menetrier’s is uncertain. One can rule out a lymphangiectasia mechanism because Miura et al114 carried out lymphographic studies in four Menetrier’s patients and found either no pathology or only minor local changes. There are no obvious gastric erosions or ulcers; Greenwald1 puts it in the “Diseases without erosions” category and it is usually assumed that there is a tight junction leak, similar to the mechanism described above for cystic fibrosis. Consistent with this, Oderda et al115 reported that the gastric epithelial tight junctions’ width is increased from the normal 7.43 nm to about 10 nm. As discussed above for celiac disease, even if this small increase in tight junction width was enough to markedly increase the epithelial albumin permeability, the leak would still be limited by the relatively low normal albumin permeability of the gastric capillaries (Figure 7). It seems unlikely that the massive protein leaks seen in some cases of Menetrier’s (clearances of 300–600 mL/day) could be explained by this small increase in gap junction width. Kelly et al116 measured both gastric albumin clearance (continuous gastric aspiration after IV 51Cr-albumin) and gap junction width in seven Menetrier’s patients. The severity of the PLE as determined by serum albumin (CP) was strongly correlated with the gastric albumin loss. For example, the patient with a CP that was half normal had a huge gastric albumin clearance of 800 mL/day, while three patients with nearly normal CP had only slightly increased albumin clearance. It is of interest that all the seven patients had the same increase in tight junction width, that is, this width was not correlated with the degree of gastric PLE, again suggesting that this may not be the leak mechanism. Also, Jarnum and Jensen110 found that IgG and albumin had similar rates of gastric clearance, which would not be expected for sieving through 10 nm tight junctions because IgG is about twice the size of albumin. Menetrier’s is characterized by a massive increase in mucus secretion and there may be some sort of coupling between secretion and albumin loss.
The only other disease in which gastric PLE has been implicated comes from the studies of Becheur et al117 in 26 patients with alcoholic cirrhosis. They measured gastric clearance of αAT using the method of Florent et al22 described above (continuous gastric aspiration with concomitant IV cimetidine to reduce αAT proteolysis). Although there was no significant difference from control for the group as a whole, three subjects had mildly elevated gastric clearances of 68, 98, and 157 mL/day. All three patients had severe hypoalbuminemia, less than half normal, which, as discussed above, would require total GI clearances of at least 240 mL/day if they had normal liver function. Thus, even in the subjects with the highest loss rate, the gastric GI loss is not large enough to explain the decreased serum albumin and some other factor, for example, decreased hepatic albumin synthesis, must also be present. The mechanism of this increased gastric clearance is not clear. Presumably, the increased portal vein pressure in these cirrhotics leads to some pathologic break in the epithelial barrier.
Genetic and animal models of PLE
The most exciting area of investigation of PLE concerns the physiologic role of heparin and its possible therapeutic implications. Interest in heparin originated in 1997 with the report of Donnelly et al118 that IV heparin (10,000 units every 12 hours) dramatically reversed PLE in three patients post-Fontan procedure. Subsequent studies have not supported this observation. Although there have been scattered reports of the effectiveness of heparin in the treatment of PLE in Crohn’s disease and ulcerative colitis,119,120 there have been many negative studies.121 Nevertheless, interest in this area was rekindled in 2008 by the report of Bode et al122 of a series of in vitro and in vivo mouse studies that clearly demonstrate the importance of heparin and heparan sulfate proteoglycan in modifying intestinal permeability. They used 51Cr-albumin and αAT to measure in vivo protein loss, with both methods giving nearly identical results. Knockout of syndecan-1, the predominant heparan sulfate proteoglycan on the basolateral surface of intestinal epithelial cells, increased the rate of protein leak by a small (about 2-fold), but statistically significant, amount. Combining the knockout with inflammatory cytokines (e.g., tumor necrosis factor [TNF]-α and interferon-γ) and/or increased venous pressure produced a synergistically greater leak. (TNF-α in wild-type mice produced about a 3-fold increase in protein leak.) Daily injections of high-molecular weight heparin reversed most of this increased leak. As a possible human analog of this knockout, Murch et al123 described three infants with massive congenital PLE who had a marked absence of heparan sulfate glycosaminoglycans on the enterocyte basolateral membrane. These results along with more recent results124 showing the importance of syndecan-1 in regulating intestinal permeability demonstrate that defects in tight junctions can partially mimic the increased permeability seen in PLE diseases without mucosal erosions (e.g., celiac disease).
There are a number of other rare genetic diseases that manifest as severe diarrhea with PLE and hypoalbuminemia in infancy. Both microvillus inclusion disease125 and congenital tufting enteropathy126 have major defects in the enterocytes, unlike what is seen in noncongenital acquired PLE described above. In contrast, the epithelium has a relatively normal appearance in the PLE associated with the recently described DGAT1 mutation that disrupts an intestinal epithelial luminal membrane transferase protein involved in triglyceride absorption.127 The epithelium is completely normal in the PLE resulting from the “plasmalemma vesicle associated protein” mutation that produces a massive breakdown in the fenestrations in epithelial capillaries, preventing normal water absorption.128 None of these genetic diseases seem to be useful models of the pathology involved in the usual clinical forms of PLE.
More than 60 animal models of IBD have been described using a range of approaches, including chemical irritants, immune antigens, and genetic alterations.100,129 However, these models are not useful for the purposes of this review in that they focus on the factors involved in the induction of IBD with little attention directed to understanding the physiology of the PLE mechanisms associated with the inflammation.
Summary and discussion: clinical implications of PLE
As evidenced by the foregoing, PLE is a complex, relatively common entity that occurs in a variety of GI as well as non-GI conditions. Thus, it is surprising that many, if not most, practicing physicians are unaware of PLE, and many who are familiar with the existence of this problem are not aware that it can be relatively easily diagnosed and quantified via αAT clearance measurements. This lack of awareness presumably reflects the fact that PLE is not a disease, but rather a phenomenon that “silently” complicates a variety of diseases. Treatment is almost always directed at correction of the underlying condition. Only very rarely, such as in some intestinal lymphagiectasis or post-Fontan procedures, is treatment specifically directed towards reducing the PLE.
Given the lack of general awareness of PLE, it is worthwhile to review the accepted and potential clinical value of measurements of protein loss into the gut lumen. Since PLE has been reported to occur in 60 different conditions, the finding of PLE usually does not aid in the diagnosis of any given disease. A potential diagnostic usage of PLE measurements might be as a screening tool to differentiate irritable bowel syndrome (IBS) from all other maladies of the GI tract, with excessive loss of protein excluding IBS. However, while it is commonly assumed that intestinal protein permeability is normal in IBS, there have not been any systematic measurements of PLE in IBS. Increased permeability to small molecules (i.e., lactulose) has been described in IBS130 and, since PLE occurs in diseases with anatomically normal-appearing mucosa, it is not a foregone conclusion that IBS patients do not have PLE.
Suspicion of the existence of PLE is usually prompted by the finding of marked hypoalbuminemia not attributable to liver disease. As discussed, relatively enormous elevations of the normal gut protein loss are required if PLE is to be deemed the sole source of very low albumin levels. For example, to account for a 50% reduction of normal serum albumin if all other aspects of albumin kinetics remain normal, there needs to be an increase in the GI clearance equal to the normal total clearance, corresponding to a total GI protein loss of about 5.61 g/day (refer Equation 5). This would require a gut clearance of albumin of about 255 mL/day, roughly 17 times the normal mean intestinal albumin clearance of 15 mL/day. As discussed, if the normal albumin homeostatic mechanisms come into play, even greater gut clearances would be required (Figure 2). The quantitative relationship between serum albumin and PLE has been investigated in two papers. Waldmann et al15 found that PLE (assessed by fecal excretion of 51Cr-albumin) almost perfectly accounted for the hypoalbuminemia observed in patients with not very well-delineated disease states. The only other large screening study of PLE is the αAT clearance measurements of Strygler et al20 in 87 patients selected for evaluation of “chronic idiopathic diarrhea, malabsorption, or hypoalbuminemia of unknown origin”. If, as discussed previously, the true GI albumin clearance (ClGI) was twice the αAT clearance, PLE ClGI could also account for most of the observed hypoalbuminemia in the Strygler et al20 subjects, although the correlation was not as strong as Waldmann et al15 found using 51Cr. The finding that gut losses could roughly account for the low serum albumin of the subjects in these two studies raises the question as to how often unrecognized gut protein losses, subtle to massive, are responsible for reductions in serum albumin observed in patients with a variety of diseases that seemingly do not involve the bowel. For example, two likely candidates of common pathologies that might have associated PLE are chronic heart failure and cirrhosis. However, as discussed above, although PLE has been reported with these two diseases, it is a rare and unusual occurrence.
A number of studies have assessed PLE in inflammatory bowel disease (IBD), primarily Crohn’s disease. While very sizable PLE has been noted with both 51Cr-albumin and αAT measurements, there is a surprising lack of correlation between protein loss and disease activity with PLE often observed in inactive disease.19,131 In addition, PLE measurements had only limited success in predicting relapse of quiescent disease.132 Thus, it appears that αAT excretion offers no advantages over the much simpler measurements of disease activity presently in use, such as the C-reactive protein and the erythrocyte sedimentation rate.
One aspect of PLE in Crohn’s disease that merits study is the possibility that the magnitude of PLE influences the efficacy of therapy with anti-TNF globulins. The serum half-time of the anti-TNF agents is reduced in severe Crohn’s disease and this reduction in half-time correlates with low serum albumin levels.133 In addition, PLE in very active Crohn’s is sufficient to materially alter the normal 7–10-day half-time of infliximab. As discussed above, there is very little size selectivity in PLE, with albumin and IgG having similar leak rates. Thus, it seems possible or even likely that gut loss plays a major role in the more rapid turnover of anti-TNF globulins observed in severe Crohn’s disease. Brandse et al134 measured infliximab concentrations in the feces of patients with ulcerative colitis. While frequently detectable immediately after the infusion, infliximab concentrations were neither quantitatively sufficient to account for a reduced half-time of this compound nor was there a significant correlation between the fecal concentration and the observed half-time. These findings do not rule out an appreciable role of PLE in infliximab turnover since this compound could be rapidly and variably destroyed in the fecal stream. Also, the measurement of fecal concentration, rather than daily excretion, might have obscured meaningful correlations between gut loss and serum half-times. Study of the relationship between αAT clearance and infliximab turnover would provide more reliable, albeit indirect, information on the relationship between PLE and infliximab loss into the gut. The finding of a significant correlation between PLE and infliximab concentrations could be used as the basis for greater or more frequent dosing with anti-TNF agents in patients with high αAT clearances.
Finally, there is an unexplained strong correlation between mortality of apparently healthy subjects and their serum albumin concentrations, even when the serum albumin is well within the accepted normal range (>3.5 g/L).2 This observation raises the totally unexplored questions of whether PLE could account for this relative hypoalbuminemia and, if so, could the mechanism responsible for PLE play a role in the increased mortality. For example, the above described increase in PLE observed with TNF-α administration122 in experimental animals suggests that the release of inflammatory cytokines could provide a potential link between PLE and increased mortality of seemingly healthy subjects. The increases in PLE required to produce the modest hypoalbuminemia associated with the increased mortality are predicted by Equations 4–6 and plotted in Figure 2. For example, gut albumin clearance would have to increase from about the normal value of 15 mL/day to at least 40 mL/day (Equation 5) to account for a serum albumin decline from 4.5 to 4.1 g/dL. Several studies of PLE in groups of “normal” controls found that there were outliers who might have this level of PLE,17,20,131 particularly if αAT clearance is only half the true clearance as a result of the intestinal proteolysis discussed above. Although no such study is likely to be performed, the “dream” study would compare the serum albumin concentrations of healthy subjects with their fecal αAT clearance obtained following administration of high-dose protein pump inhibitor and polyethylene glycol to minimize gastric and intestinal destruction of antitrypsin, respectively. Such a study would provide the first, presumably, accurate assessment of the true intestinal αAT clearance in “healthy” individuals and its possible correlation with variations in serum albumin.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Greenwald DA. Protein-losing gastroenteropathy. In: Feldman M, Friedman LS, Brandt LJ, editors. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Vol. 1. Philadelphia, PA: Saunders; 2016. pp. 464–470. [Google Scholar]
- 2.Levitt DG, Levitt MD. Human serum albumin homeostasis: a new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int J Gen Med. 2016;9:229–255. doi: 10.2147/IJGM.S102819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fulks M, Stout RL, Dolan VF. Albumin and all-cause mortality risk in insurance applicants. J Insur Med. 2010;42(1):11–17. [PubMed] [Google Scholar]
- 4.Schomerus H, Mayer G. Synthesis rates of albumin and fibrinogen in patients with protein-losing enteropathy and in a patient recovering from protein malnutrition. Digestion. 1975;13(4):201–208. doi: 10.1159/000197709. [DOI] [PubMed] [Google Scholar]
- 5.Waldmann TA, Steinfeld JL, Dutcher TF, Davidson JD, Gordon RS., Jr The role of the gastrointestinal system in “idiopathic hypoproteinemia”. Gastroenterology. 1961;41:197–207. [PubMed] [Google Scholar]
- 6.Wochner RD, Weissman SM, Waldmann TA, Houston D, Berlin NI. Direct measurement of the rates of synthesis of plasma proteins in control subjects and patients with gastrointestinal protein loss. J Clin Invest. 1968;47(5):971–982. doi: 10.1172/JCI105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen SB, Rossing N. Metabolism of albumin and gammaG-globulin during albumin infusions and during plasmapheresis. Scand J Clin Lab Invest. 1967;20(2):183–184. [Google Scholar]
- 8.Bennhold H, Kallee E. Comparative studies on the half-life of I 131-labeled albumins and nonradioactive human serum albumin in a case of analbuminemia. J Clin Invest. 1959;38(5):863–872. doi: 10.1172/JCI103868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freeman T, Gordon AH. Albumin catabolism in hypoproteinaemic states studies with 131-I-albumin. Bibl Haematol. 1965;23:1108–1115. [PubMed] [Google Scholar]
- 10.Roopenian DC, Low BE, Christianson GJ, Proetzel G, Sproule TJ, Wiles MV. Albumin-deficient mouse models for studying metabolism of human albumin and pharmacokinetics of albumin-based drugs. MAbs. 2015;7(2):344–351. doi: 10.1080/19420862.2015.1008345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinfeld JL, Davidson JD, Gordon RS, Jr, Greene FE. The mechanism of hypoproteinemia in patients with regional enteritis and ulcerative colitis. Am J Med. 1960;29:405–415. doi: 10.1016/0002-9343(60)90036-x. [DOI] [PubMed] [Google Scholar]
- 12.van Tongeren JH, Majoor CL. Demonstration of protein-losing gastroenteropathy. The disappearance rate of 51Cr from plasma and the binding of 51Cr to different serum proteins. Clin Chim Acta. 1966;14(1):31–41. doi: 10.1016/0009-8981(66)90061-1. [DOI] [PubMed] [Google Scholar]
- 13.Waldmann TA. Gastrointestinal protein loss demonstrated by Cr-51-labeled albumin. Lancet. 1961;2(7194):121–123. doi: 10.1016/s0140-6736(61)92646-0. [DOI] [PubMed] [Google Scholar]
- 14.Kerr RM, Du Bois JJ, Holt PR. Use of 125-I- and 51-Cr-labeled albumin for the measurement of gastrointestinal and total albumin catabolism. J Clin Invest. 1967;46(12):2064–2082. doi: 10.1172/JCI105694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waldmann TA, Wochner RD, Strober W. The role of the gastrointestinal tract in plasma protein metabolism. Studies with 51Cr-albumin. Am J Med. 1969;46(2):275–285. doi: 10.1016/0002-9343(69)90011-4. [DOI] [PubMed] [Google Scholar]
- 16.Bernier JJ, Florent C, Desmazures C, Aymes C, L’Hirondel C. Diagnosis of protein-losing enteropathy by gastrointestinal clearance of alpha1-antitrypsin. Lancet. 1978;2(8093):763–764. doi: 10.1016/s0140-6736(78)92650-8. [DOI] [PubMed] [Google Scholar]
- 17.Quigley EM, Ross IN, Haeney MR, Holbrook IB, Marsh MN. Reassessment of fecal alpha-1-antitrypsin excretion for use as screening test for intestinal protein loss. J Clin Pathol. 1987;40(1):61–66. doi: 10.1136/jcp.40.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai JC, Sambuelli A, Niveloni S, et al. Alpha 1-antitrypsin clearance as an aid in the management of patients with celiac disease. Am J Gastroenterol. 1991;86(8):986–991. [PubMed] [Google Scholar]
- 19.Karbach U, Ewe K, Bodenstein H. Alpha 1-antitrypsin, a reliable endogenous marker for intestinal protein loss and its application in patients with Crohn’s disease. Gut. 1983;24(8):718–723. doi: 10.1136/gut.24.8.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strygler B, Nicar MJ, Santangelo WC, Porter JL, Fordtran JS. Alpha 1-antitrypsin excretion in stool in normal subjects and in patients with gastrointestinal disorders. Gastroenterology. 1990;99(5):1380–1387. doi: 10.1016/0016-5085(90)91165-3. [DOI] [PubMed] [Google Scholar]
- 21.Florent C, L’Hirondel C, Desmazures C, Aymes C, Bernier JJ. Intestinal clearance of alpha 1-antitrypsin. A sensitive method for the detection of protein-losing enteropathy. Gastroenterology. 1981;81(4):777–780. [PubMed] [Google Scholar]
- 22.Florent C, Vidon N, Flourie B, et al. Gastric clearance of alpha-1-antitrypsin under cimetidine perfusion. New test to detect protein-losing gastropathy? Dig Dis Sci. 1986;31(1):12–15. doi: 10.1007/BF01347903. [DOI] [PubMed] [Google Scholar]
- 23.Chau TN, Mok MY, Chan EY, et al. Evaluation of performance of measurement of fecal alpha(1)-antitrypsin clearance and technetium-99m human serum albumin scintigraphy in protein-losing enteropathy. Digestion. 2011;84(3):199–206. doi: 10.1159/000327914. [DOI] [PubMed] [Google Scholar]
- 24.Khalesi M, Nakhaei AA, Seyed AJ, et al. Diagnostic accuracy of nuclear medicine imaging in protein losing enteropathy: systematic review and meta-analysis of the literature. Acta Gastroenterol Belg. 2013;76(4):413–422. [PubMed] [Google Scholar]
- 25.Wilson JP. Surface area of the small intestine in man. Gut. 1967;8(6):618–621. doi: 10.1136/gut.8.6.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watson AJ, Chu S, Sieck L, et al. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. 2005;129(3):902–912. doi: 10.1053/j.gastro.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 27.Ravin HA, Seligman AM, Fine J. Polyvinyl pyrrolidone as a plasma expander; studies on its excretion, distribution and metabolism. N Engl J Med. 1952;247(24):921–929. doi: 10.1056/NEJM195212112472403. [DOI] [PubMed] [Google Scholar]
- 28.Keulemans YC, Mok KS, de Wit LT, Gouma DJ, Groen AK. Hepatic bile versus gallbladder bile: a comparison of protein and lipid concentration and composition in cholesterol gallstone patients. Hepatology. 1998;28(1):11–16. doi: 10.1002/hep.510280103. [DOI] [PubMed] [Google Scholar]
- 29.Mullock BM, Shaw LJ, Fitzharris B, et al. Sources of proteins in human bile. Gut. 1985;26(5):500–509. doi: 10.1136/gut.26.5.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyer JL, Bloomer JR. Canalicular bile secretion in man. Studies utilizing the biliary clearance of (14C)mannitol. J Clin Invest. 1974;54(4):773–781. doi: 10.1172/JCI107817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Multigner L, Figarella C, Sahel J, Sarles H. Lactoferrin and albumin in human pancreatic juice: a valuable test for diagnosis of pancreatic diseases. Dig Dis Sci. 1980;25(3):173–178. doi: 10.1007/BF01308135. [DOI] [PubMed] [Google Scholar]
- 32.Hayakawa T, Harada H, Noda A, Kondo T. Lactoferrin in pure pancreatic juice in chronic pancreatitis. Am J Gastroenterol. 1983;78(4):222–224. [PubMed] [Google Scholar]
- 33.Vignes S, Bellanger J. Primary intestinal lymphangiectasia (Waldmann’s disease) Orphanet J Rare Dis. 2008;3:5. doi: 10.1186/1750-1172-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takenaka H, Ohmiya N, Hirooka Y, et al. Endoscopic and imaging findings in protein-losing enteropathy. J Clin Gastroenterol. 2012;46(7):575–580. doi: 10.1097/MCG.0b013e31823832ac. [DOI] [PubMed] [Google Scholar]
- 35.Aoyagi K, Iida M, Yao T, Matsui T, Okada M, Fujishima M. Intestinal lymphangiectasia: value of double-contrast radiographic study. Clin Radiol. 1994;49(11):814–819. doi: 10.1016/s0009-9260(05)81974-9. [DOI] [PubMed] [Google Scholar]
- 36.Salomons HA, Kramer P, Nikulasson S, Schroy PC. Endoscopic features of long-standing primary intestinal lymphangiectasia. Gastrointest Endosc. 1995;41(5):516–518. doi: 10.1016/s0016-5107(05)80016-1. [DOI] [PubMed] [Google Scholar]
- 37.Stoelinga GB, van MJ, Slooff JP. Chylous effusions into the intestine in a patient with protein-losing gastroenteropathy. Pediatrics. 1963;31:1011–1018. [PubMed] [Google Scholar]
- 38.Mistilis SP, Skyring AP, Stephen DD. Intestinal Lymphangiectasia Mechanism of Enteric Loss of Plasma-Protein and Fat. Lancet. 1965;1(7376):77–79. doi: 10.1016/s0140-6736(65)91657-0. [DOI] [PubMed] [Google Scholar]
- 39.So Y, Chung JK, Seo JK, et al. Different patterns of lymphoscintigraphic findings in patients with intestinal lymphangiectasia. Nucl Med Commun. 2001;22(11):1249–1254. doi: 10.1097/00006231-200111000-00013. [DOI] [PubMed] [Google Scholar]
- 40.Soucy JP, Eybalin MC, Taillefer R, Levasseur A, Jobin G. Lymphoscintigraphic demonstration of intestinal lymphangiectasia. Clin Nucl Med. 1983;8(11):535–537. doi: 10.1097/00003072-198311000-00006. [DOI] [PubMed] [Google Scholar]
- 41.Gold RH, Youker JE. Idiopathic intestinal lymphangiectasis (primary protein-losing enteropathy). Lymphographic verification of enteric and peritoneal leakage of chyle. Radiology. 1973;109(2):351–352. [PubMed] [Google Scholar]
- 42.Desai AP, Guvenc BH, Carachi R. Evidence for medium chain triglycerides in the treatment of primary intestinal lymphangiectasia. Eur J Pediatr Surg. 2009;19(4):241–245. doi: 10.1055/s-0029-1216389. [DOI] [PubMed] [Google Scholar]
- 43.Lai Y, Yu T, Qiao XY, Zhao LN, Chen QK. Primary intestinal lymphangiectasia diagnosed by double-balloon enteroscopy and treated by medium-chain triglycerides: a case report. J Med Case Rep. 2013;7:19. doi: 10.1186/1752-1947-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meijers BK, Schalla S, Eerens F, et al. Protein-losing enteropathy in association with constrictive pericarditis. Int J Cardiovasc Imaging. 2006;22(3–4):389–392. doi: 10.1007/s10554-005-9067-2. [DOI] [PubMed] [Google Scholar]
- 45.Nelson DL, Blaese RM, Strober W, Bruce R, Waldmann TA. Constrictive pericarditis, intestinal lymphangiectasia, and reversible immunologic deficiency. J Pediatr. 1975;86(4):548–554. doi: 10.1016/s0022-3476(75)80145-4. [DOI] [PubMed] [Google Scholar]
- 46.Petersen VP, Hastrup J. Protein-losing enteropathy in constrictive pericarditis. Acta Med Scand. 1963;173:401–410. doi: 10.1111/j.0954-6820.1963.tb17423.x. [DOI] [PubMed] [Google Scholar]
- 47.Wilkinson P, Pinto B, Senior JR. Reversible protein-losing enteropathy with intestinal lymphangiectasia secondary to chronic constrictive pericarditis. N Engl J Med. 1965;273(22):1178–1181. doi: 10.1056/NEJM196511252732202. [DOI] [PubMed] [Google Scholar]
- 48.Johnson JN, Driscoll DJ, O’Leary PW. Protein-losing enteropathy and the Fontan operation. Nutr Clin Pract. 2012;27(3):375–384. doi: 10.1177/0884533612444532. [DOI] [PubMed] [Google Scholar]
- 49.Schumacher KR, Stringer KA, Donohue JE, et al. Social media methods for studying rare diseases. Pediatrics. 2014;133(5):e1345–e1353. doi: 10.1542/peds.2013-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.John AS, Johnson JA, Khan M, Driscoll DJ, Warnes CA, Cetta F. Clinical outcomes and improved survival in patients with protein-losing enteropathy after the Fontan operation. J Am Coll Cardiol. 2014;64(1):54–62. doi: 10.1016/j.jacc.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 51.Mertens L, Hagler DJ, Sauer U, Somerville J, Gewillig M. Protein-losing enteropathy after the Fontan operation: an international multicenter study. PLE study group. J Thorac Cardiovasc Surg. 1998;115(5):1063–1073. doi: 10.1016/s0022-5223(98)70406-4. [DOI] [PubMed] [Google Scholar]
- 52.Rychik J. Protein-losing enteropathy after Fontan operation. Congenit Heart Dis. 2007;2(5):288–300. doi: 10.1111/j.1747-0803.2007.00116.x. [DOI] [PubMed] [Google Scholar]
- 53.Magdo HS, Stillwell TL, Greenhawt MJ, et al. Immune abnormalities in Fontan protein-losing enteropathy: a case-control study. J Pediatr. 2015;167(2):331–337. doi: 10.1016/j.jpeds.2015.04.061. [DOI] [PubMed] [Google Scholar]
- 54.Meadows J, Jenkins K. Protein-losing enteropathy: integrating a new disease paradigm into recommendations for prevention and treatment. Cardiol Young. 2011;21(4):363–377. doi: 10.1017/S1047951111000102. [DOI] [PubMed] [Google Scholar]
- 55.Antonio M, Gordo A, Pereira C, Pinto F, Fragata I, Fragata J. Thoracic duct decompression for protein-losing enteropathy in failing Fontan circulation. Ann Thorac Surg. 2016;101(6):2370–2373. doi: 10.1016/j.athoracsur.2015.08.079. [DOI] [PubMed] [Google Scholar]
- 56.Connor FL, Angelides S, Gibson M, et al. Successful resection of localized intestinal lymphangiectasia post-Fontan: role of (99m)technetium-dextran scintigraphy. Pediatrics. 2003;112(3 Pt 1):e242–e247. doi: 10.1542/peds.112.3.e242. [DOI] [PubMed] [Google Scholar]
- 57.Lin WS, Hwang MS, Chung HT, et al. Protein-losing enteropathy after the Fontan operation: clinical analysis of nine cases. Chang Gung Med J. 2006;29(5):505–512. [PubMed] [Google Scholar]
- 58.Ohuchi H, Yasuda K, Miyazaki A, et al. Hemodynamic characteristics before and after the onset of protein losing enteropathy in patients after the Fontan operation. Eur J Cardiothorac Surg. 2013;43(3):e49–e57. doi: 10.1093/ejcts/ezs714. [DOI] [PubMed] [Google Scholar]
- 59.Lenz D, Hambsch J, Schneider P, et al. Protein-losing enteropathy in patients with Fontan circulation: is it triggered by infection? Crit Care. 2003;7(2):185–190. doi: 10.1186/cc2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ryerson L, Goldberg C, Rosenthal A, Armstrong A. Usefulness of heparin therapy in protein-losing enteropathy associated with single ventricle palliation. Am J Cardiol. 2008;101(2):248–251. doi: 10.1016/j.amjcard.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 61.Davis CA, Driscoll DJ, Perrault J, et al. Enteric protein loss after the Fontan operation. Mayo Clin Proc. 1994;69(2):112–114. doi: 10.1016/s0025-6196(12)61035-0. [DOI] [PubMed] [Google Scholar]
- 62.Fujii T, Shimizu T, Takahashi K, et al. Fecal alpha1-antitrypsin concentrations as a measure of enteric protein loss after modified Fontan operations. J Pediatr Gastroenterol Nutr. 2003;37(5):577–580. doi: 10.1097/00005176-200311000-00014. [DOI] [PubMed] [Google Scholar]
- 63.Thorne SA, Hooper J, Kemp M, Somerville J. Gastro-intestinal protein loss in late survivors of Fontan surgery and other congenital heart disease. Eur Heart J. 1998;19(3):514–520. doi: 10.1053/euhj.1997.0777. [DOI] [PubMed] [Google Scholar]
- 64.Ostrow AM, Freeze H, Rychik J. Protein-losing enteropathy after Fontan operation: investigations into possible pathophysiologic mechanisms. Ann Thorac Surg. 2006;82(2):695–700. doi: 10.1016/j.athoracsur.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 65.Chan FK, Sung JJ, Ma KM, Leung YL, Yeung VT. Protein-losing enteropathy in congestive heart failure: diagnosis by means of a simple method. Hepatogastroenterology. 1999;46(27):1816–1818. [PubMed] [Google Scholar]
- 66.Berkowitz I, Segal I. Protein-losing enteropathy in congestive cardiac failure: an entity of minor clinical significance. Am J Gastroenterol. 1990;85(2):154–156. [PubMed] [Google Scholar]
- 67.Dumont AE, Mulholland JH. Alterations in thoracic duct lymph flow in hepatic cirrhosis: significance in portal hypertension. Ann Surg. 1962;156(4):668–675. doi: 10.1097/00000658-196210000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stanley AJ, Gilmour HM, Ghosh S, Ferguson A, McGilchrist AJ. Transjugular intrahepatic portosystemic shunt as a treatment for protein-losing enteropathy caused by portal hypertension. Gastroenterology. 1996;111(6):1679–1682. doi: 10.1016/s0016-5085(96)70033-1. [DOI] [PubMed] [Google Scholar]
- 69.Wong WM, Hui CK, Yuen MF, et al. Reversal of protein-losing enteropathy by liver transplantation. J Clin Gastroenterol. 2003;36(1):86–87. doi: 10.1097/00004836-200301000-00030. [DOI] [PubMed] [Google Scholar]
- 70.Georgopoulos P, Mowat C, McMillan DC, Kingstone K, Ghosh S, Stanley AJ. Is portal hypertension associated with protein-losing enteropathy? J Gastroenterol Hepatol. 2005;20(1):103–107. doi: 10.1111/j.1440-1746.2004.03475.x. [DOI] [PubMed] [Google Scholar]
- 71.Davcev P, Vanovski B, Sestakov D, Tadzer I. Protein-losing enteropathy in patients with liver cirrhosis. Digestion. 1969;2(1):17–22. doi: 10.1159/000196916. [DOI] [PubMed] [Google Scholar]
- 72.Fuss IJ, Strober W, Cuccherini BA, et al. Intestinal lymphangiectasia, a disease characterized by selective loss of naive CD45RA + lymphocytes into the gastrointestinal tract. Eur J Immunol. 1998;28(12):4275–4285. doi: 10.1002/(SICI)1521-4141(199812)28:12<4275::AID-IMMU4275>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 73.Muller C, Wolf H, Gottlicher J, Zielinski CC, Eibl MM. Cellular immunodeficiency in protein-losing enteropathy. Predominant reduction of CD3+ and CD4+ lymphocytes. Dig Dis Sci. 1991;36(1):116–122. doi: 10.1007/BF01300099. [DOI] [PubMed] [Google Scholar]
- 74.Bendixen G, Jarnum S, Soltoft J, Westergaard H, Weeke B, Yssing M. IgG and albumin turnover in Crohn’s disease. Scand J Gastroenterol. 1968;3(5):481–489. doi: 10.3109/00365526809179907. [DOI] [PubMed] [Google Scholar]
- 75.Beh KJ, Watson DL, Lascelles AK. Concentrations of immunoglobulins and albumin in lymph collected from various regions of the body of the sheep. Aust J Exp Biol Med Sci. 1974;52(1):81–86. doi: 10.1038/icb.1974.6. [DOI] [PubMed] [Google Scholar]
- 76.Vaerman JP, Heremans JF. Origin and molecular size of immunoglobulin-A in the mesenteric lymph of the dog. Immunology. 1970;18(1):27–38. [PMC free article] [PubMed] [Google Scholar]
- 77.Sands BE, Siegel CA. Crohn’s Disease. In: Feldman M, Friedman AN, Brandt LJ, editors. Sleisenger and Fordtran’s Gastroinstestinal and Liver Disease. 10th ed. Vol. 2. Saunders; 2016. pp. 1990–2022. [Google Scholar]
- 78.Dubcenco E, Jeejeebhoy KN, Petroniene R, et al. Capsule endoscopy findings in patients with established and suspected small-bowel Crohn’s disease: correlation with radiologic, endoscopic, and histologic findings. Gastrointest Endosc. 2005;62(4):538–544. doi: 10.1016/j.gie.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 79.Courtice FC, Simmonds WJ, Steinbeck AW. Some investigations of lymph from a thoracic duct fistula in man. Aust J Exp Biol Med Sci. 1951;29:201–210. doi: 10.1038/icb.1951.24. [DOI] [PubMed] [Google Scholar]
- 80.Fanous MY, Phillips AJ, Windsor JA. Mesenteric lymph: the bridge to future management of critical illness. JOP. 2007;8(4):374–399. [PubMed] [Google Scholar]
- 81.Bill A. Plasma protein dynamics: albumin and IgG capillary permeability, extravascular movement and regional blood flow in unanesthetized rabbits. Acta Physiol Scand. 1977;101(1):28–42. doi: 10.1111/j.1748-1716.1977.tb05980.x. [DOI] [PubMed] [Google Scholar]
- 82.Kingham JG, Loehry CA. Selectivity of small intestinal exudate in celiac disease and Crohn’s disease. Am J Dig Dis. 1978;23(1):33–38. doi: 10.1007/BF01072572. [DOI] [PubMed] [Google Scholar]
- 83.Olaison G, Leandersson P, Sjodahl R, Tagesson C. Intestinal permeability to polyethyleneglycol 600 in Crohn’s disease. Peroperative determination in a defined segment of the small intestine. Gut. 1988;29(2):196–199. doi: 10.1136/gut.29.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ainsworth M, Eriksen J, Rasmussen JW, Schaffalitzky de Muckadell OB. Intestinal permeability of 51Cr-labeled ethylenediaminetetraacetic acid in patients with Crohn’s disease and their healthy relatives. Scand J Gastroenterol. 1989;24(8):993–998. doi: 10.3109/00365528909089246. [DOI] [PubMed] [Google Scholar]
- 85.Miki K, Moore DJ, Butler RN, Southcott E, Couper RT, Davidson GP. The sugar permeability test reflects disease activity in children and adolescents with inflammatory bowel disease. J Pediatr. 1998;133(6):750–754. doi: 10.1016/s0022-3476(98)70145-3. [DOI] [PubMed] [Google Scholar]
- 86.Murphy MS, Eastham EJ, Nelson R, Pearson AD, Laker MF. Intestinal permeability in Crohn’s disease. Arch Dis Child. 1989;64(3):321–325. doi: 10.1136/adc.64.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alexander JS, Chaitanya GV, Grisham MB, Boktor M. Emerging roles of lymphatics in inflammatory bowel disease. Ann N Y Acad Sci. 2010;1207(Suppl 1):E75–E85. doi: 10.1111/j.1749-6632.2010.05757.x. [DOI] [PubMed] [Google Scholar]
- 88.Van Kruiningen HJ, Colombel JF. The forgotten role of lymphangitis in Crohn’s disease. Gut. 2008;57(1):1–4. doi: 10.1136/gut.2007.123166. [DOI] [PubMed] [Google Scholar]
- 89.von der Weid PY, Rehal S, Ferraz JG. Role of the lymphatic system in the pathogenesis of Crohn’s disease. Curr Opin Gastroenterol. 2011;27(4):335–341. doi: 10.1097/MOG.0b013e3283476e8f. [DOI] [PubMed] [Google Scholar]
- 90.Garcia de Tena J, Manzano L, Leal JC, San Antonio E, Sualdea V, Alvarez-Mon M. Active Crohn’s disease patients show a distinctive expansion of circulating memory CD4+ CD45RO + CD28null T cells. J Clin Immunol. 2004;24(2):185–196. doi: 10.1023/B:JOCI.0000019784.20191.7f. [DOI] [PubMed] [Google Scholar]
- 91.Kelly CP. Celiac Disease. In: Feldman M, Friedman LS, Brandt LJ, editors. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Vol. 2. Philadelphia, PA: Saunders; 2016. pp. 1849–1872. [Google Scholar]
- 92.Stenhammar L, Falth-Magnusson K, Jansson G, Magnusson KE, Sundqvist T. Intestinal permeability to inert sugars and different-sized polyethyleneglycols in children with celiac disease. J Pediatr Gastroenterol Nutr. 1989;9(3):281–289. doi: 10.1097/00005176-198910000-00004. [DOI] [PubMed] [Google Scholar]
- 93.Laster L, Waldmann TA, Fenster LF, Singleton JW. Albumin metabolism in patients with Whipple’s disease. J Clin Invest. 1966;45(5):637–644. doi: 10.1172/JCI105379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maiwald M, Herbay AV. Whipple’s Disease. In: Feldman M, Friedman LS, Brandt LJ, editors. Sleisenger and Fordtran Gastrointestinal and Liver Disease. 10th ed. Vol. 2. Philadelphia, PA: Saunders; 2016. pp. 1886–1895. [Google Scholar]
- 95.Benner KG, Montanaro A. Protein-losing enteropathy in systemic lupus erythematosus. Diagnosis and monitoring immunosuppressive therapy by alpha-1-antitrypsin clearance in stool. Dig Dis Sci. 1989;34(1):132–135. doi: 10.1007/BF01536168. [DOI] [PubMed] [Google Scholar]
- 96.Zheng WJ, Tian XP, Li L, et al. Protein-losing enteropathy in systemic lupus erythematosus: analysis of the clinical features of fifteen patients. J Clin Rheumatol. 2007;13(6):313–316. doi: 10.1097/RHU.0b013e31815bf9c6. [DOI] [PubMed] [Google Scholar]
- 97.Moos V, Schmidt C, Geelhaar A, et al. Impaired immune functions of monocytes and macrophages in Whipple’s disease. Gastroenterology. 2010;138(1):210–220. doi: 10.1053/j.gastro.2009.07.066. [DOI] [PubMed] [Google Scholar]
- 98.Marth T, Roux M, von Herbay A, Meuer SC, Feurle GE. Persistent reduction of complement receptor 3 alpha-chain expressing mononuclear blood cells and transient inhibitory serum factors in Whipple’s disease. Clin Immunol Immunopathol. 1994;72(2):217–226. doi: 10.1006/clin.1994.1134. [DOI] [PubMed] [Google Scholar]
- 99.Habibagahi M, Habibagahi Z, Jaberipour M, Aghdashi A. Quantification of regulatory T cells in peripheral blood of patients with systemic lupus erythematosus. Rheumatol Int. 2011;31(9):1219–1225. doi: 10.1007/s00296-010-1427-0. [DOI] [PubMed] [Google Scholar]
- 100.Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84(3):282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 101.Schulzke JD, Bentzel CJ, Schulzke I, Riecken EO, Fromm M. Epithelial tight junction structure in the jejunum of children with acute and treated celiac sprue. Pediatr Res. 1998;43(4 Pt 1):435–441. doi: 10.1203/00006450-199804000-00001. [DOI] [PubMed] [Google Scholar]
- 102.Madara JL, Trier JS. Structural abnormalities of jejunal epithelial cell membranes in celiac sprue. Lab Invest. 1980;43(3):254–261. [PubMed] [Google Scholar]
- 103.Citrin Y, Sterling K, Halsted JA. The mechanism of hypoproteinemia associated with giant hypertrophy of the gastric mucosa. N Engl J Med. 1957;257(19):906–912. doi: 10.1056/NEJM195711072571902. [DOI] [PubMed] [Google Scholar]
- 104.Azer M, Sultan A, Zalata K, Abd El-Haleem I, Hassan A, El-Ebeidy G. A case of Menetrier’s disease without Helicobacter pylori or hypoalbuminemia. Int J Surg Case Rep. 2015;17:58–60. doi: 10.1016/j.ijscr.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Meuwissen SG, Ridwan BU, Hasper HJ, Innemee G. Hypertrophic protein-losing gastropathy. A retrospective analysis of 40 cases in The Netherlands. The Dutch Menetrier Study Group. Scand J Gastroenterol Suppl. 1992;194:1–7. [PubMed] [Google Scholar]
- 106.Xiong LS, Gong YY. Natural history of adult-onset Menetrier’s disease: report of a case with 9-year follow-up. Exp Ther Med. 2016;11(6):2462–2466. doi: 10.3892/etm.2016.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burdick JS, Chung E, Tanner G, et al. Treatment of Menetrier’s disease with a monoclonal antibody against the epidermal growth factor receptor. N Engl J Med. 2000;343(23):1697–1701. doi: 10.1056/NEJM200012073432305. [DOI] [PubMed] [Google Scholar]
- 108.Fiske WH, Tanksley J, Nam KT, et al. Efficacy of cetuximab in the treatment of Menetrier’s disease. Sci Transl Med. 2009;1(8):8ra18. doi: 10.1126/scitranslmed.3000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Williamson K, Park HK, Schacht R, Kaistha A. A case of menetrier disease in a child. Pediatr Emerg Care. 2012;28(3):277–279. doi: 10.1097/PEC.0b013e31824957b8. [DOI] [PubMed] [Google Scholar]
- 110.Jarnum S, Jensen KB. Plasma protein turnover (albumin, transfer-rin, IgG, IgM) in Menetrier’s disease (giant hypertrophic gastritis): evidence of non-selective protein loss. Gut. 1972;13(2):128–137. doi: 10.1136/gut.13.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reinhart WH, Weigand K, Kappeler M, Roesler H, Halter F. Comparison of gastrointestinal loss of alpha-1-antitrypsin and chromium-51-albumin in Menetrier’s disease and the influence of ranitidine. Digestion. 1983;26(4):192–196. doi: 10.1159/000198889. [DOI] [PubMed] [Google Scholar]
- 112.Huh WJ, Coffey RJ, Washington MK. Menetrier’s disease: its mimickers and pathogenesis. J Pathol Transl Med. 2016;50(1):10–16. doi: 10.4132/jptm.2015.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takagi H, Jhappan C, Sharp R, Merlino G. Hypertrophic gastropathy resembling Menetrier’s disease in transgenic mice overexpressing transforming growth factor alpha in the stomach. J Clin Invest. 1992;90(3):1161–1167. doi: 10.1172/JCI115936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Miura S, Asakura H, Tsuchiya M. Lymphatic abnormalities in protein-losing gastropathy, especially in Menetrier’s disease. Angiology. 1981;32(5):345–354. doi: 10.1177/000331978103200508. [DOI] [PubMed] [Google Scholar]
- 115.Oderda G, Cinti S, Cangiotti AM, Forni M, Ansaldi N. Increased tight junction width in two children with Menetrier’s disease. J Pediatr Gastroenterol Nutr. 1990;11(1):123–127. doi: 10.1097/00005176-199007000-00024. [DOI] [PubMed] [Google Scholar]
- 116.Kelly DG, Miller LJ, Malagelada JR, Huizenga KA, Markowitz H. Giant hypertrophic gastropathy (Menetrier’s disease): pharmacologic effects on protein leakage and mucosal ultrastructure. Gastroenterology. 1982;83(3):581–589. [PubMed] [Google Scholar]
- 117.Becheur H, Pauwels A, Mostefa-Kara N, et al. Gastric protein loss and alcoholic cirrhosis. Study by measurement of gastric clearance of alpha 1-antitrypsin. Gastroenterol Clin Biol. 1996;20(8–9):669–673. [PubMed] [Google Scholar]
- 118.Donnelly JP, Rosenthal A, Castle VP, Holmes RD. Reversal of protein-losing enteropathy with heparin therapy in three patients with univentricular hearts and Fontan palliation. J Pediatr. 1997;130(3):474–478. doi: 10.1016/s0022-3476(97)70214-2. [DOI] [PubMed] [Google Scholar]
- 119.Ang YS, Mahmud N, White B, et al. Randomized comparison of unfractionated heparin with corticosteroids in severe active inflammatory bowel disease. Aliment Pharmacol Ther. 2000;14(8):1015–1022. doi: 10.1046/j.1365-2036.2000.00802.x. [DOI] [PubMed] [Google Scholar]
- 120.Folwaczny C, Wiebecke B, Loeschke K. Unfractioned heparin in the therapy of patients with highly active inflammatory bowel disease. Am J Gastroenterol. 1999;94(6):1551–1555. doi: 10.1111/j.1572-0241.1999.01143.x. [DOI] [PubMed] [Google Scholar]
- 121.Chande N, MacDonald JK, Wang JJ, McDonald JW. Unfractionated or low molecular weight heparin for induction of remission in ulcerative colitis: a Cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm Bowel Dis. 2011;17(9):1979–1986. doi: 10.1002/ibd.21776. [DOI] [PubMed] [Google Scholar]
- 122.Bode L, Salvestrini C, Park PW, et al. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest. 2008;118(1):229–238. doi: 10.1172/JCI32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Murch SH, Winyard PJ, Koletzko S, et al. Congenital enterocyte heparan sulphate deficiency with massive albumin loss, secretory diarrhea, and malnutrition. Lancet. 1996;347(9011):1299–1301. doi: 10.1016/s0140-6736(96)90941-1. [DOI] [PubMed] [Google Scholar]
- 124.Wang Z, Li R, Tan J, et al. Syndecan-1 acts in synergy with tight junction through stat3 signaling to maintain intestinal mucosal barrier and prevent bacterial translocation. Inflamm Bowel Dis. 2015;21(8):1894–1907. doi: 10.1097/MIB.0000000000000421. [DOI] [PubMed] [Google Scholar]
- 125.Muller T, Hess MW, Schiefermeier N, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40(10):1163–1165. doi: 10.1038/ng.225. [DOI] [PubMed] [Google Scholar]
- 126.Thoeni C, Amir A, Guo C, et al. A novel nonsense mutation in the EpCAM gene in a patient with congenital tufting enteropathy. J Pediatr Gastroenterol Nutr. 2014;58(1):18–21. doi: 10.1097/MPG.0000000000000106. [DOI] [PubMed] [Google Scholar]
- 127.Stephen J, Vilboux T, Haberman Y, et al. Congenital protein losing enteropathy: an inborn error of lipid metabolism due to DGAT1 mutations. Eur J Hum Genet. 2016;24(9):1268–1273. doi: 10.1038/ejhg.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Elkadri A, Thoeni C, Deharvengt SJ, et al. Mutations in plasmalemma vesicle associated protein result in sieving protein-losing enteropathy characterized by hypoproteinemia, hypoalbuminemia, and hypertriglyceridemia. Cell Mol Gastroenterol Hepatol. 2015;1(4):381–394 e387. doi: 10.1016/j.jcmgh.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hoffmann JC, Pawlowski NN, Kuhl AA, Hohne W, Zeitz M. Animal models of inflammatory bowel disease: an overview. Pathobiology. 2002;70(3):121–130. doi: 10.1159/000068143. [DOI] [PubMed] [Google Scholar]
- 130.Matricon J, Meleine M, Gelot A, et al. Review article: Associations between immune activation, intestinal permeability and the irritable bowel syndrome. Aliment Pharmacol Ther. 2012;36(11–12):1009–1031. doi: 10.1111/apt.12080. [DOI] [PubMed] [Google Scholar]
- 131.Fischbach W, Becker W, Mossner J, Koch W, Reiners C. Fecal alpha-1-antitrypsin and excretion of 111indium granulocytes in assessment of disease activity in chronic inflammatory bowel diseases. Gut. 1987;28(4):386–393. doi: 10.1136/gut.28.4.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Biancone L, Fantini M, Tosti C, Bozzi R, Vavassori P, Pallone F. Fecal alpha 1-antitrypsin clearance as a marker of clinical relapse in patients with Crohn’s disease of the distal ileum. Eur J Gastroenterol Hepatol. 2003;15(3):261–266. doi: 10.1097/00042737-200303000-00009. [DOI] [PubMed] [Google Scholar]
- 133.Klotz U, Teml A, Schwab M. Clinical pharmacokinetics and use of infliximab. Clin Pharmacokinet. 2007;46(8):645–660. doi: 10.2165/00003088-200746080-00002. [DOI] [PubMed] [Google Scholar]
- 134.Brandse JF, van den Brink GR, Wildenberg ME, et al. Loss of infliximab into feces is associated with lack of response to therapy in patients with severe ulcerative colitis. Gastroenterology. 2015;149(2):350–355 e352. doi: 10.1053/j.gastro.2015.04.016. [DOI] [PubMed] [Google Scholar]