

Abstract
Trichomonas vaginalis, the most common nonviral sexually transmitted parasite, causes ∼283 million trichomoniasis infections annually and is associated with pregnancy complications and increased risk of HIV-1 acquisition. The antimicrobial drug metronidazole is used for treatment, but in a fraction of clinical cases, the parasites can become resistant to this drug. We undertook sequencing of multiple clinical isolates and lab derived lines to identify genetic markers and mechanisms of metronidazole resistance. Reduced representation genome sequencing of ∼100 T. vaginalis clinical isolates identified 3,923 SNP markers and presence of a bipartite population structure. Linkage disequilibrium was found to decay rapidly, suggesting genome-wide recombination and the feasibility of genetic association studies in the parasite. We identified 72 SNPs associated with metronidazole resistance, and a comparison of SNPs within several lab-derived resistant lines revealed an overlap with the clinically resistant isolates. We identified SNPs in genes for which no function has yet been assigned, as well as in functionally-characterized genes relevant to drug resistance (e.g., pyruvate:ferredoxin oxidoreductase). Transcription profiles of resistant strains showed common changes in genes involved in drug activation (e.g., flavin reductase), accumulation (e.g., multidrug resistance pump), and detoxification (e.g., nitroreductase). Finally, we identified convergent genetic changes in lab-derived resistant lines of Tritrichomonas foetus, a distantly related species that causes venereal disease in cattle. Shared genetic changes within and between T. vaginalis and Tr. foetus parasites suggest conservation of the pathways through which adaptation has occurred. These findings extend our knowledge of drug resistance in the parasite, providing a panel of markers that can be used as a diagnostic tool.
Keywords: Trichomonas vaginalis, sexually transmitted infection, comparative genomics, genetic association study, antimicrobial drug resistance
Introduction
Trichomonads are haploid unicellular microaerophilic members of eukaryotic phylum Parabasalia that infect a variety of vertebrates including humans, wildlife, farm animals, and pets (Maritz et al. 2014). Among the several important species in this group is Trichomonas vaginalis, which causes trichomoniasis, the most common nonviral sexually transmitted disease in humans, with 3.7 million cases reported each year in the US (Center for Disease Control and Prevention 2013). T. vaginalis can cause long-term symptomatic infections of vulvar and urethral regions of the genital tract (Petrin et al. 1998). Importantly, T. vaginalis infection is associated with increased risk of HIV infection (McClelland et al. 2007), development of prostate cancer (Sutcliffe et al. 2012; Twu et al. 2014), and complications during pregnancy, such as premature and low-weight births (Cotch et al. 1997). Another trichomonad, Tritrichomonas foetus, is an economically important cattle parasite, and while only distantly related to T. vaginalis, it nevertheless has an analogous site of infection and disease etiology (Yule et al. 1989; Foote 1996; Frey and Muller 2012).
A highly fragmented assembly of the ∼160 Mb T. vaginalis strain G3 genome generated by Sanger sequencing was published in 2007 (Carlton et al. 2007). It consists of ∼17,000 scaffolds containing ∼60,000 predicted protein coding genes, many of them members of high copy number gene families, and ∼40,000 transposable element (TE) genes distributed among ∼59 TE families, members of which are highly similar (average ∼97.5% identity) within a family. Identification of meiosis-specific genes in the genome suggested that the parasite might have a cryptic sexual cycle or may have recently lost the ability to undergo genetic exchange (Carlton et al. 2007; Malik et al. 2008). Genetic markers mined from the genome have been used in population studies of the species (Conrad et al. 2011; Cornelius et al. 2012), revealing significant T. vaginalis genetic diversity that is structured but unrelated to geographical origin (Conrad et al. 2012). Recently, a genome assembly for Tr. foetus strain K has been published, that reveals its genome, too, is also highly repetitive (Benchimol et al. 2017).
Metronidazole (Mz) and tinidazole are the only two United States Food and Drug Administration approved drugs effective against T. vaginalis (de Brum Vieira et al. 2017). However, clinical failure of Mz treatment has been reported since 1959 (Watt and Jennison 1960) and ranges from ∼4% in the US (Kirkcaldy et al. 2012) to 17% in Papua New Guinea (Upcroft et al. 2009). Mz is a prodrug, which must be reduced at its nitro group in order to form the nitroradical anions that are toxic to the parasite. The electrons required for Mz activation are generated by several mechanisms, including the reduction of pyruvate by pyruvate:ferredoxin oxidoreductase (PFO) (Yarlett et al. 1986a,b; Brown et al. 1999; Kulda 1999), an enzyme found in membrane-bound organelles called hydrogenosomes that produce ATP and hydrogen, as well as several pathways hypothesized to exist in the cytosol (Leitsch, et al. 2009, 2010, 2014). Tr. foetus is here again similar to T. vaginalis in displaying both Mz resistance and related hydrogenosomal metabolic pathways (Kulda 1999 and Kulda et al 1984).
Aerobic and anaerobic pathways of Mz resistance have been proposed. Clinical (aerobic) resistance is typically observed only in the presence of oxygen, which decreases Mz toxicity by re-oxidizing nitroradical anions before they can damage the parasite, reverting Mz to an inactive form. Clinically resistant T. vaginalis strains have impaired oxygen scavenging activity and elevated levels of intracellular oxygen. Oxygen stress response genes are thought to be involved in aerobic Mz resistance, with NADH-dependent oxidase and flavin reductase (FR) in particular being implicated as important enzymes involved in oxygen removal (Leitsch et al. 2012, 2014). In contrast, anaerobic resistance has typically been observed only in vitro (Upcroft and Upcroft 2001a,b), with the exception of strain B7268 where anaerobic resistance developed in a patient (Voolmann and Boreham 1993). Anaerobically resistant strains are marked by the absence of certain hydrogenosome metabolic enzymes that would normally activate Mz, including PFO and ferredoxin (Fd) (Yarlett, et al. 1986a; Kulda et al. 1993; Land et al. 2004), as well as cytosolic thioredoxin reductase (TrxR) (Leitsch et al. 2009). Aerobic and anaerobic resistance pathways are proposed to be fundamentally different, although FR, whose primary role is to reduce molecular oxygen to hydrogen peroxide, appears to be involved in both processes (Leitsch et al. 2014). To date, studies of the mechanism of Mz resistance have focused wholly on candidate genes known to be involved in oxygen scavenging or hydrogenosome metabolism, e.g., PFO, Fd, FR, and TrxR (Kulda 1999; Upcroft and Upcroft 2001b; Pal et al. 2009; Leitsch et al. 2010, 2014). However, alterations in these genes do not explain all cases of Mz resistance, nor have other genes been identified as possible candidates. The aim of our study was to address these issues by large-scale genetic comparison of Mz sensitive and resistant strains.
High-throughput methods for large-scale genetic comparison and analysis of mutational phenotypes are particularly difficult to implement in trichomonads. The highly repetitive nature of the T. vaginalis and Tr. foetus genomes makes them refractory to conventional short-read genome sequencing. We therefore employed double digest restriction-site associated DNA sequencing (ddRAD-Seq) to genotype 102 T. vaginalis isolates from eight countries, the largest sampling of T. vaginalis genomes reported to date. We identified single nucleotide polymorphisms (SNPs) in the sequences in order to explore population structure and genome-wide linkage disequilibrium (LD) in the isolates, and provide a basis for a genome-wide association study of Mz genetic indicators. In addition, we compared laboratory-derived Mz resistant isolates with their isogenic parents to identify SNPs that could indicate Mz resistance pathways common to resistant lines. Because there is no efficient high-throughput transfection system for large-scale assay of mutational phenotypes in T. vaginalis, we conducted whole transcriptome analysis of gene regulation in resistant and sensitive isolates, as a proxy for direct functional testing. Finally, to explore the universality of the genetic bases of Mz resistance among trichomonads, we analyzed genetic changes between a parental strain and in vitro-derived Mz resistant lines of Tr. foetus.
Results
We used ddRAD-Seq to partially sequence genomes of 102 T. vaginalis clinical isolates from eight countries (supplementary fig. S1 in supplementary file S1, Supplementary Material online). Sequence reads were filtered, aligned, and single nucleotide polymorphisms (SNPs) were identified (supplementary file S2, Supplementary Material online; see “Materials and Methods” for filtering criteria). Our final set of 3,923 high quality SNPs comprised 1,463 nonsynonymous mutations, 879 silent (synonymous) mutations, 10 nonsense mutations, and 1,571 intergenic mutations.
Population Structure and Fast LD Decay Suggest Recombination in T. vaginalis Genomes
Population structure and linkage disequilibrium (LD) properties play a key role in determining success of mapping genotypes to phenotypes (Mu et al. 2005). The existence of genetic subpopulations and the occurrence of LD decay over short distances are strong indicators of genetic recombination and sexual reproduction in a population (Tibayrenc and Ayala 2002; Halkett et al. 2005), with implications for correlating genotypes to drug resistance. To characterize population structure, we performed principal components analysis (PCA) of SNPs from 102 T. vaginalis isolates (fig. 1). The first two principal components accounted for the highest variation within the sampled isolates and explained 10.1 and 6.1% of the variation, respectively. Similar to published findings, these two PCA axes split the global sample into two subpopulations (Conrad et al. 2012; Bradic et al. 2014), a structure congruent with sexual reproduction. Population structure did not correspond to the geographical origin of the isolates.
Fig. 1.
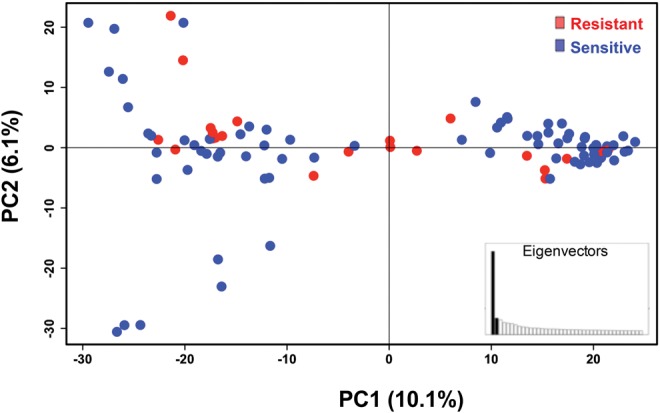
—Population structure in T. vaginalis global samples. Principal components analysis of 102 T. vaginalis isolates using 3,923 genome-wide ddRAD SNPs. The x-axis represents principal component one (PC1) and explains 10.1% of the variation, while the y-axis represents principle component two (PC2), and explains 6.1% of the variation; the inset represents distribution of eigenvalues where each eigenvalue is the variance of the corresponding PC. Colors show variation in 32 Mz resistant (red) and 63 Mz sensitive (blue) isolates as defined by MLC ≥ 100 μg/mL. Individual isolate assignment to each sub-population cluster is summarized in supplementary table S2, Supplementary Material online.
In asexual populations, LD is maintained over long genomic distances or even over the entire genome, while genetic exchange in sexually reproducing populations constrains LD to shorter distances. We plotted LD decay along the 898 genomic contigs containing 2,837 SNPs in one population, and along the 872 contigs containing 2,694 SNPs in the other population, and found decay occurring within 1 kb in both populations (fig. 2), although the rate differed between the two. These results, together with the presence of population structure, provide strong evidence that genetic exchange occurs, or occurred sometime in the recent evolutionary past, of both T. vaginalis populations. The difference in LD decay rates indicates potential differences in recombination, population size, or phenotypic traits (e.g., transmission, drug resistance, virulence) between the two populations.
Fig. 2.

—LD decays over distance in two populations of T. vaginalis. LD decay calculated over 5 kb intervals in 898 contigs (top panel) and 872 contigs (bottom panel) is shown for the two populations of T. vaginalis.
Association Study of T. vaginalis Isolates Identifies Candidate Biomarkers of Mz Resistance
Our findings suggested (1) that population structure should be accounted for in T. vaginalis association studies in order to avoid spurious genotype-to-phenotype association; and that (2) fast LD decay allows for mapping of phenotypic traits in candidate regions. We first tested for association between T. vaginalis population structure and resistance to metronidazole (Mz) as measured using a standard minimum lethal concentration (MLC) protocol under aerobic conditions (supplementary file S2, Supplementary Material online). Of the 95 isolates for which MLC could be determined, 63 were classified as “sensitive” and 32 as “resistant” (MLC ≥ 100 μg/ml), of which 23 were also “highly resistant” (MLC ≥ 400 μg/ml). Neither of the two subpopulations detected by PCA of ddRAD SNPs was significantly associated with Mz resistance (Pearson's chi-squared test for MLC ≥ 100 μg/ml: χ2 = 0.2139, df = NA, P-value = 0.8136; for MLC ≥ 400 μg/ml: χ2 = 0.2291, df = NA, P-value = 0.7271; fig. 1).
We next used Discriminant Analysis of Principal Components (DAPC) (Jombart et al. 2010) to identify SNPs that distinguish Mz resistant from Mz sensitive isolates, and quantified the contribution of each allele to this distinction (fig. 3). Our analysis showed partial overlap of alleles between sensitive and resistant groups (fig. 3A). We identified the SNPs that contribute the most to distinguishing sensitive from resistant phenotypes (fig. 3B). Among these, we identified a set of 72 SNPs associated with moderate or high Mz resistance (MLC ≥ 100 μg/ml or ≥400 μg/ml; supplementary file S3, Supplementary Material online). Along with multiple silent (synonymous) and intergenic SNPs, we identified 16 nonsynonymous SNPs associated with moderate resistance in 13 genes (e.g., TVAG_197160 CAMK family protein kinase, TVAG_158720 BspA-like protein), and two nonsynonymous SNPs in two conserved hypothetical genes (TVAG_124910, TVAG_493120) associated with both moderate (MLC ≥ 100 μg/ml) and high (MLC ≥ 400 μg/ml) resistance. A summary of these genes and their SNPs is shown in supplementary file S3, Supplementary Material online.
Fig. 3.
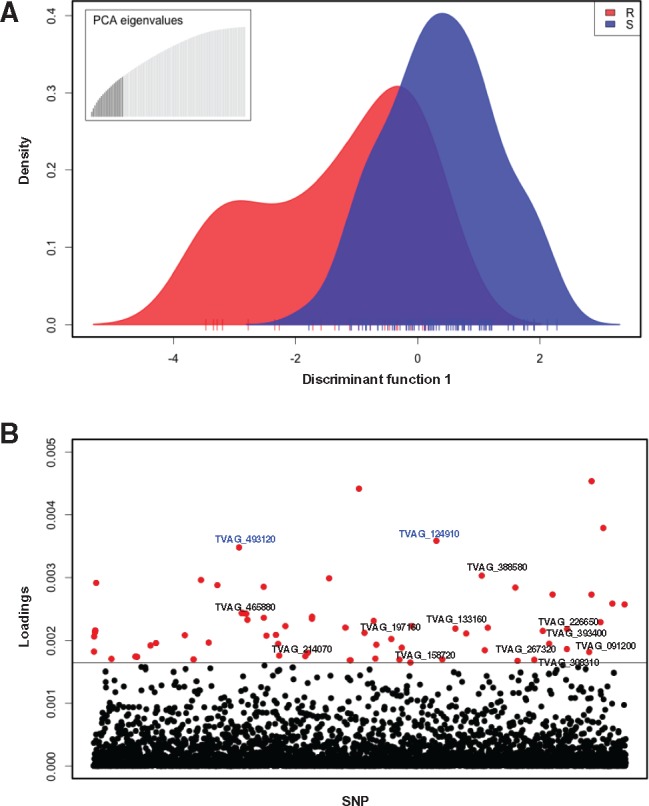
—Association mapping for Mz resistance in T. vaginalis. (A) Discriminant analysis of principal components (DAPC) representing variation and distribution of markers between drug resistant (red) and drug sensitive (blue) isolates. Ticks on the x-axis represent individual isolates. The inset represents the distribution of eigenvalues, with the black histogram showing all the principle components that were used in the DAPC analysis. (B) Loadings plot of the SNPs used for association analysis. The distribution of variances for SNP markers within and between resistant and sensitive isolates is represented as a loading value for each marker. The loading values above the threshold represent the largest between-group variance and the smallest within-group variance and are associated with the resistant phenotype. SNP markers are indicated on the x-axis, and the loadings value for each SNP on the y-axis; a horizontal line indicates the loadings threshold. Red dots represent 72 SNPs identified as significantly contributing to Mz resistance. Text in blue font represents two nonsynonymous SNPs in two conserved hypothetical genes (TVAG_493120 and TVAG_124910) common to both moderate (MLC ≥ 100) and high (MLC ≥ 400 Mz) phenotypes.
SNP Changes in Lab-derived Mz Resistant T. vaginalis Lines Suggest Similar In Vivo and In Vitro Mechanisms of Drug Resistance
While the 72 SNPs we found associated with moderate and high resistance in disparate clinical isolates represent a potentially important set of genetic markers of the resistance phenotype, such association does not demonstrate causation. To identify SNPs that may be more strongly inferred to be consequences of adaptation to drug treatment, we exploited existing pairs of T. vaginalis lines, each consisting of a highly drug-resistant line derived in vitro from a naturally drug-sensitive or moderately resistant parental line by increasing drug selection pressure over time (supplementary file S1 and fig. S2, Supplementary Material online). We analyzed SNPs observed in ddRAD-Seq data from three such pairs of parental and derived lines: (1) Mz-sensitive B7708 and highly resistant B7708-M; (2) Mz-sensitive F1623 and highly resistant F1623-M (Brown et al. 1999); and (3) moderately resistant B7268 and highly resistant B7268-M (Wright et al. 2010). SNPs were identified by comparing the parental and derived line to the reference genome of the Mz-sensitive lab strain G3. For each pair, only bases that were the same in the parental line and the G3 reference but different in the derived strain were considered as SNPs (see “Materials and Methods”). We found 39 nonsynonymous SNPs that were common to all three derived, highly resistant lines, in 36 genes (fig. 4, supplementary file S5, Supplementary Material online). Changes common to two out of three derived lines included seven nonsynonymous mutations in seven genes in B7708-M and F1623-M; 137 nonsynonymous (including one nonsense) SNP in 141 genes in B7708-M and B7268-M; and 170 nonsynonymous and two nonsense SNPs in 155 genes in F1623-M and B7268-M. We also identified multiple synonymous and intergenic SNPs in common between the pairs (data not shown).
Fig. 4.
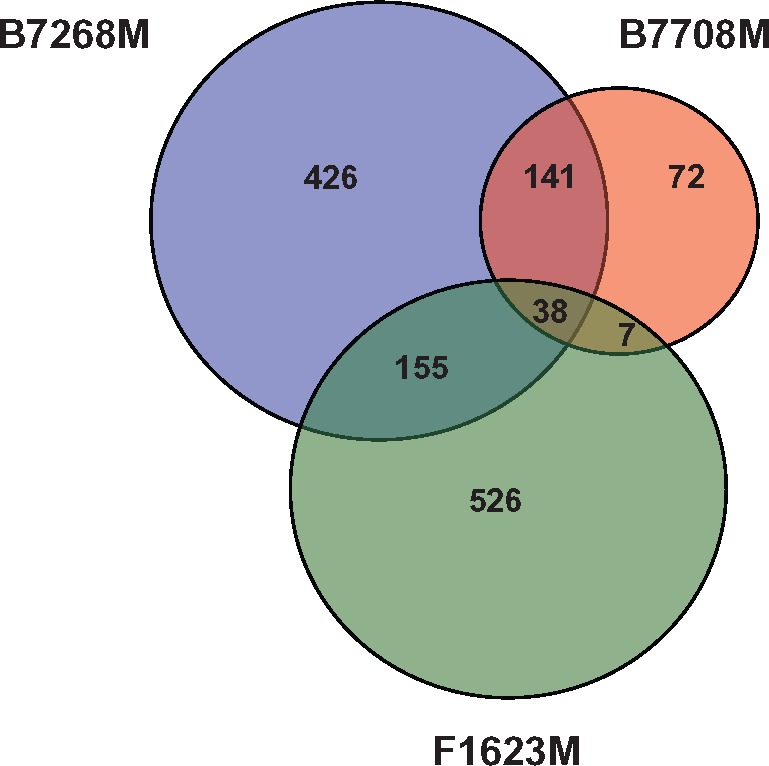
—Common genetic changes in laboratory-derived T. vaginalis lines. Venn diagram circle sizes are based on numbers of genes that have nonsynonymous SNPs. A nonsynonymous SNP was defined as a position where the nucleotide in the ancestral strain was the same as in the reference G3 strain, but different in the derived (more resistant) strain.
Of particular note are five genes (TVAG_124910, TVAG_197160, TVAG_226650, TVAG_393400, and TVAG_465880) containing nonsynonymous SNPs that were (1) present in at least two lab-derived resistant strains, and (2) associated with Mz resistance in our association study of ∼100 clinical isolates described above. TVAG_197160 encodes a predicted CAMK kinase family protein mentioned above, while the rest encode conserved hypothetical proteins, one of which (TVAG_465880) contains multiple SNPs shared between the derived and clinical isolates. These five genes represent novel candidates for roles in both clinical and in vitro-derived Mz resistance. A summary of the genes and their SNPs described in this section is shown in supplementary file S4, Supplementary Material online.
Gene Expression Changes Are Associated with Resistance and SNPs in Clinical and Lab Derived Isolates
We employed whole transcriptome analysis to survey changes in gene expression associated with Mz resistance. RNA-Seq libraries in triplicate were sequenced from nine Mz-sensitive isolates (reference strain G3 and GOR69, NYCA04, NYCB20, NYCD15, NYCE32, NYCF20, NYCG31, and SD2), and three resistant isolates (moderately resistant clinical isolate B7268, its highly resistant in vitro derivative B7268-M, and highly resistant clinical isolate NYCC37). Of the 57,796 predicted protein-coding genes in the T. vaginalis G3 genome (excluding TE genes), a total of 28,919 (∼50%) showed evidence of expression (supplementary file S6, Supplementary Material online), a similar number as reported in a previous transcriptome study of T. vaginalis strain TO16 (Gould et al. 2013). A wide range of gene expression was seen between isolates (PC1, 22.38% and PC2, 14.35% variation explained), with the drug resistant pair B7268 and B7268M clustering together and separately from the other isolates (fig. 5A). It is worth mentioning that a large proportion of gene expression diversity was apparent due to variation in paralogous genes (data not shown).
Fig. 5.
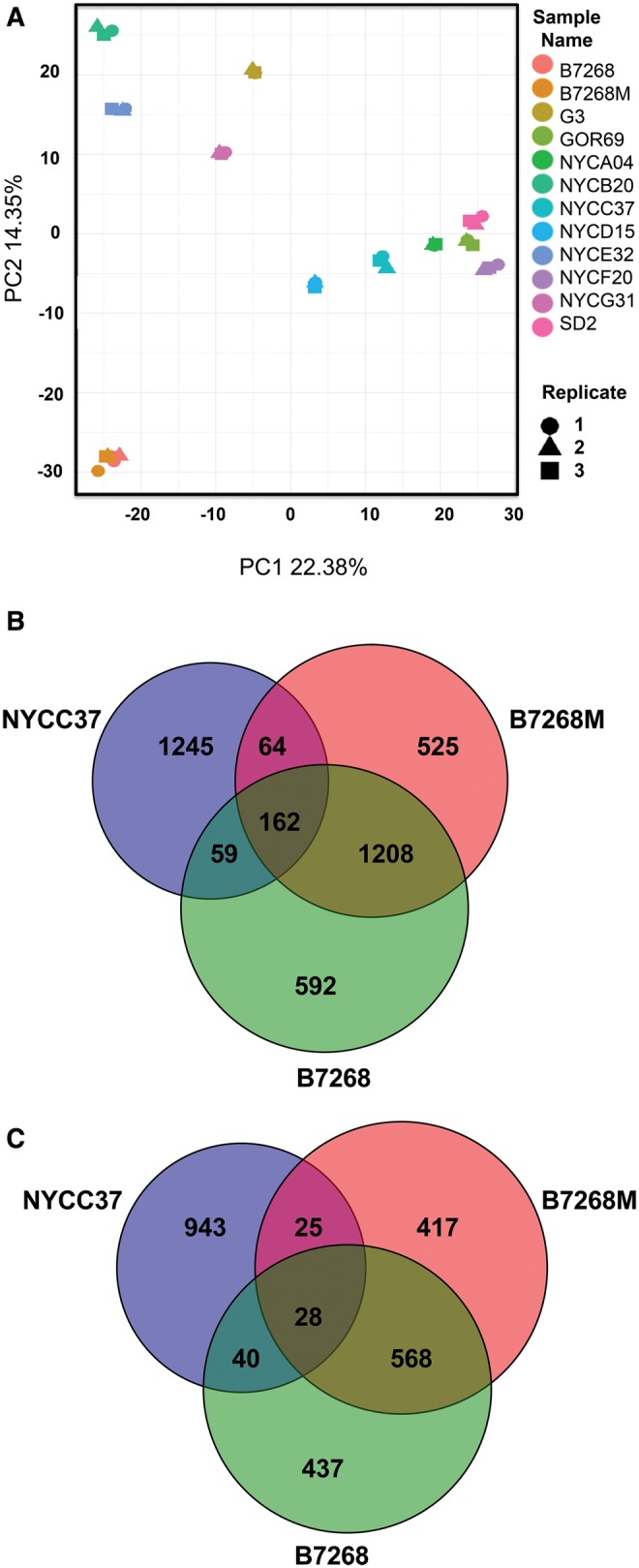
—Differential gene expression in laboratory-derived and clinically resistant T. vaginalis versus sensitive lines. (A) Principal components analysis of 12 isolates and their replicates based on normalized gene read counts. Replicates are indicated as shapes and different isolates as colors. (B) Venn diagram of number of genes in three Mz-resistant strains whose expression is upregulated in comparison with nine Mz-sensitive strains. (C) Venn diagram of number of genes in the same strains whose expression is downregulated in comparison with nine Mz-sensitive strains. Changes in genes where difference in gene expression is significant to P < 0.001 are shown. All the circles are sized based on marker numbers.
To identify genes whose expression is most strongly associated with Mz resistance, we compared each of the three resistant strains with the group of nine sensitive strains. Mz resistant strains B7268 and B7268M shared the highest number of pairwise down- (1208) and up-regulated genes (568) (supplementary file S6, Supplementary Material online), as expected given their common ancestry. All three resistant strains shared 28 upregulated and 162 downregulated genes (FDR = 0.05; fig. 5B and C and supplementary file S6, Supplementary Material online). Comparing these genes to results from our two SNP studies described above, we identified conserved hypothetical gene TVAG_465880 that was upregulated in at least two Mz-resistant lines (B7268M and B7268) and was also among the five novel candidate genes flagged in both of the studies. We also identified nine genes with a nonsynonymous SNP that were either up- or downregulated in the experimentally derived B7268M isolate compared to its B7268 parent. For example, dynein heavy chain protein (TVAG_188120) had a nonsynonymous SNP and expression of the gene was downregulated in B7268M, strongly suggesting its involvement in drug resistance. Another example is a hypothetical gene (TVAG_258020) with one nonsynonymous SNP and downregulation of the gene in both B7268M and clinical strain NYCC37. A summary of the genes and their SNPs described in this section is shown in supplementary file S4, Supplementary Material online.
Mz Resistance Is Associated with Regulation of Genes Involved in Drug Activation, Accumulation, and Detoxification
We first asked if Mz resistance is more likely to occur by drug inactivation or cell repair processes by testing if the number of downregulated genes (fig. 5B) is significantly higher than the number of upregulated genes (fig. 5C). We established that the number of downregulated genes was significantly higher in all the comparisons, suggesting that Mz resistance predominantly occurs through processes that restrict drug activation. Next, we tested for association between the set of 162 downregulated and 28 upregulated genes identified as shared between the three Mz resistant strains and the Mz resistance phenotype by cluster analysis (fig. 6). Not only did the gene expression data cluster by phenotype, but the association was significant (chi-square test, P = 2.76 e−08).
Fig. 6.

—Heatmap and cluster analysis of the association between gene expression and Mz resistance phenotype for commonly up- and down-regulated genes in T. vaginalis. Heatmap is constructed on normalized gene read counts. Cluster analysis and chi-square analysis were performed on genes that had significant expression changes in each resistant strain in comparison to sensitive strains. Drug resistance strains significantly clustered together based on their gene expression profile for 162 downregulated and 28 upregulated genes (chi-square test, P = 2.76e-08). Green color represents downregulated genes, red color represent upregulated genes in resistant strains. Pink cluster represent three resistant strains (Resistant cluster = 9 samples), while blue cluster represents nine sensitive strains (Sensitive cluster = 27 samples) corresponding.
Within our Gene Ontology (GO) enrichment analysis of the 162 downregulated genes, oxidoreductase, catalytic, and acid phosphatase activities were identified as the most enriched molecular processes (P-value < 0.05). These processes are involved in electron transfer and thus likely highly relevant to the activation of Mz (supplementary file S6 and table S6, Supplementary Material online). For example, T. vaginalis oxygen tolerance pathways are important mediators of Mz reduction (Yarlett et al. 1986b; Ellis et al. 1992; Rasoloson et al. 2001). We observed changes in multiple genes associated with oxygen detoxification (supplementary file S6, Supplementary Material online), including in the flavin reductase gene FR1 (TVAG_517010), identified as downregulated in our three Mz resistant strains. It has been suggested that nitrosoimidazol, the reduced form of Mz, can form covalent bonds with cellular proteins such as cytosolic malate dehydrogenase (MDH) and thioredoxin reductase (TrxR) (Leitsch et al. 2009). We observed reduced expression of two variants of MDH (TVAG_354940 in all three resistant isolates, TVAG_412220 in B7268 and B7268M) suggesting their potential role in resistance. As a probable consequence of reduced MDH expression, we also detected an increase in lactate dehydrogenase (TVAG_381310) activity in B7268 and B7268M, which was reported previously (Leitsch et al. 2009). The superoxide dismutase (SOD) gene family is also involved in scavenging oxygen reactive species as part of an oxygen defense mechanism in T. vaginalis (Leitsch et al. 2009). We identified two downregulated paralogs of SOD (TVAG_039980, TVAG_079600) in B7268 and NYCC37 while comparing resistant and sensitive strains. Several other oxidoreductase genes that have not been previously reported were downregulated in the resistant strains (TVAG_414030, TVAG_044820, and TVAG_411220). Other proteins that might contribute to removal of reactive oxygen and help in establishing resistance are the thioredoxin genes (Trx). Paralogs of these genes were upregulated either in B7268 and B7268M (TVAG_277410) or in all three resistant strains (TVAG_253800). Finally, among the eleven Mz activating nitroreductase (ntr) genes, we observed lowered expression of ntr6 (TVAG_354010) in Mz resistant strains B7268 and B7268M, and downregulation of ntr7 (TVAG_474290) (Pal et al. 2009; Paulish-Miller et al. 2014), and two other genes from the ntr family (TVAG_303860, TVAG_473580) in NYCC37. The ntr gene (TVAG_499730) whose protein product is found in the hydrogenosome (Beltran et al. 2013) and is involved in oxygen detoxification, was downregulated in all three resistant strains.
The hydrogenosome is a key site of accumulation and activation of Mz. Of 359 T. vaginalis proteins predicted to be localized to the organelle (Garg et al. 2015) our data revealed down-regulation of several of them in the resistant strains, such as iron-sulfur flavoproteins (TVAG_040030, TVAG_154730, and TVAG_370510) (Smutna et al. 2014), iron hydrogenase (TVAG_182620), alcohol dehydrogenase (TVAG_328940), malic enzymes (TVAG_412220, TVAG_340290, TVAG_238830, and TVAG_416100), and rubrerythrin (TVAG_064490; supplementary file S6, Supplementary Material online). In addition, of the three hydrogenosome-localized nitroimidazole reductase (Nim) paralogs that code for a protein associated with Mz detoxification in the reference strain G3, (TVAG_356810) was upregulated in resistant strains B7268 and B7268M, confirming previous functional studies that showed overexpression of this gene in Eschericia coli inactivates Mz (Pal et al. 2009). However, Nim1 (TVAG_005890) was downregulated in all three resistant strains, making its role less unclear.
Of the 28 upregulated genes found in all three resistant strains compared to the sensitive strains, 14 were enriched for antiporter and drug transmembrane transporter GO functional processes (P < 0.05; supplementary files S4 and S6, Supplementary Material online). Multidrug resistance pump (MDR) (TVAG_291970, TVAG_210540) and metal ABC transporter (TVAG_254060) genes showed overexpression in all three resistance strains, suggesting that T. vaginalis may also use drug efflux to develop resistance. A summary of the genes and their expression differences described in this section is shown in supplementary files S4 and S6, Supplementary Material online.
Similar Genetic Changes Associated with Mz Resistance Occur in Two Distantly Related Trichomonad Species
To investigate whether there are evolutionarily common mechanisms of Mz resistance in trichomonads, we undertook whole genome shotgun sequencing and SNP detection of three strains of Tritrichomonas foetus, a trichomonad distantly related to T. vaginalis that also causes a venereal disease in cattle. We used Mz-sensitive and Mz-resistant lines KV1_M100 (exhibiting in vitro aerobic resistance) and KV1_1MR100 (exhibiting in vitro aerobic and anaerobic Mz resistance) derived by Kulda and colleagues from KV1 (Kulda et al. 1984) (supplementary file S1 and fig. S3, Supplementary Material online). Details of the sequencing are shown in table 1. Sequenced reads of the KV1 genome were assembled into 194,695 contigs (N50 = 2,054 bp) with a size range of 61–19,994 bp. A BLASTX search of the KV1 assembly against the T. vaginalis G3 reference assembly returned 28,632 orthologs of T. vaginalis genes (data not shown). We identified 183 SNPs in the orthologs, 13 of which (in 13 orthologs) were present in both Mz-resistant Tr. foetus lines (supplementary file S7, Supplementary Material online). Of these 13 orthologs two include genes encoding an actin-like protein (TVAG_371880) and TPR domain-containing protein (TVAG_371930), both upregulated in lab derived B7268M resistant strain compared to the sensitive strains.
Table 1.
Characteristics of Whole Genome Shotgun Sequencing Datasets of Three Tr. foetus Strains, KV1, KV1_M100, and KV1_1MR100.
| Tr. foetus KV1 | Tr. foetus KV1_M100 | Tr. foetus KV1_1MR100 | |
|---|---|---|---|
| Library insert size | 350 bp | 350 bp | 350 bp |
| Read length | 100 nc | 100 nc | 100 nc |
| Total no. reads | 67,088,722 | 75,792,262 | 70,337,248 |
| Coverage | 44.27× | 46.23× | 49.48× |
| Avg. GC% | 30 | 30 | 30 |
| Assembly | Velvet | Not done | Not done |
| No. contigs | 194,695 | – | – |
| Contig N50 | 2,054 bp | – | – |
| No. orthologs with T. vaginalis | 28,362 | – | – |
A few Tr. foetus hypothetical proteins with SNP changes in either of the two Tr. foetus strains are also notable for their orthologs in T. vaginalis exhibiting reduced transcription (TVAG_151770, TVAG_451120, and TVAG_400730). Moreover, two of 13 orthologs in the two species (1) CAMK family protein kinase (TVAG_139460), and (2) GP63-like adhesion molecule (TVAG_295740), had SNP mutations and were also downregulated in T. vaginalis (supplementary files S4 and 7, Supplementary Material online). It is also worth noticing that we identified SNP changes in other orthologs that might be important for Mz resistance including tvntr3 (TVAG_35682) and pyruvate:ferredoxin oxidoreductase genes (TVAG_198110, TVAG_242960) that did not show changes in our T. vaginalis transcriptome dataset (supplementary file S4, Supplementary Material online).
Discussion
This study of genetic variation and gene expression related to drug resistance in multiple T. vaginalis isolates is the most comprehensive in this species to date. We successfully used ddRAD-Seq as a method to genotype unique regions of the highly repetitive T. vaginalis genome, demonstrating that it can be applied to protists whose genomes are refractory to cost-effective whole genome sequencing. Our dataset of 3,923 ddRAD-Seq SNP markers also represents a high confidence set of genetic variants, and greatly expands upon the limited number of genetic markers available for this parasite. Using these markers, we explored the genetic diversity of T. vaginalis and present a picture of the parasite’s population structure and linkage disequilibrium (LD) at higher resolution than previous studies (Conrad et al. 2012; Bradic et al. 2014). Our study also offers a panel of biomarkers that can be used to advance personalized treatment and prevention of trichomoniasis.
Genetic variation and exchange through recombination is important to consider in parasites as it influences the spread of drug resistance and virulence genes among them. Our study confirms the existence of two genetically distinct T. vaginalis populations (Conrad et al. 2012; Cornelius et al. 2012; Bradic et al. 2014; Paulish-Miller et al. 2014), congruent with sexually reproducing organisms (Tibayrenc and Ayala 2002). Genome-wide LD, reported from analysis of a small number of genes in previous studies (Conrad et al. 2012; Cornelius et al. 2012), was found to decay rapidly within 1–5 kb in both populations, providing evidence for recent genetic exchange. Differences in LD decay between the two populations suggest that different degrees of inbreeding and rates of transmission might be responsible for maintenance of the two-type population structure. Based on previous work (Conrad et al. 2012; Paulish-Miller et al. 2014), we also expected to observe a difference in average drug susceptibility between the two T. vaginalis populations. Higher mean values of Mz resistance were found in one population compared to the other (98.5 µg/ml vs. 126.2 µg/ml), although the difference was not statistically significant.
A primary goal of generating the SNP markers in this study was to undertake a genome-wide association analysis to identify novel genetic indicators of Mz resistance. From analysis of ∼100 global T. vaginalis Mz resistant and sensitive strains, we identified 72 SNPs associated with moderate or high Mz resistance - a panel of biomarkers that can be used to advance personalized treatment and prevention of trichomoniasis. Currently only a single “reflex test” that identifies a mutation in the T. vaginalis ntr6 gene by real-time PCR (http://www.prnewswire.com/news-releases/medical-diagnostic-laboratories-llc-announces-a-complimentary-reflex-test-to-determine-metronidazole-resistance-in-trichomonas-vaginalis-145736975.html) is available for clinical testing of resistance. Of our 72 biomarkers, several (e.g., those found in TVAG_465880 [conserved hypothetical] and TVAG_308310 [bromodomain containing protein]) were also recurrently identified in other analyses, such as a comparison of three laboratory-derived Mz resistant lines with their isogenic parental strains. While we were unable to undertake a direct functional analysis of these new putative resistance mutations and genes because of the limitations of T. vaginalis transfection, whole transcriptome analysis of three resistant and nine sensitive isolates showed several of the mutations occurred in genes that also showed changes in expression. We also noticed that diversity in gene expression between isolates is primarily due to variation in expression of paralogous genes, as has been noted in previous studies (Pal et al. 2009; Horvathova et al. 2012). For this reason, we suggest that multiple isolates should be used as a control panel in expression studies of T. vaginalis.
Drug resistance in eukaryotic microbes is a complex process and usually involves either drug activation/inactivation, or active drug efflux/reduced drug uptake (Penuliar et al. 2015). A major finding of this study was the many changes found in genes coding for proteins involved in drug reduction, efflux, and inactivation (fig. 7). First, we confirmed that down-regulation of proteins involved in Mz activation such as nitroreductase (ntr) and flavin reductase 1 (FR1) are a primary response of T. vaginalis against Mz. We observed that previously identified mutations in the ntr6 gene (Paulish-Miller et al. 2014) are associated with down-regulation in the resistant lab pair B7268 and B7268M, and that down-regulation of other ntr genes also appears to play an important role (supplementary file S4, Supplementary Material online). Our observation of FR1 reduced expression in resistant isolates is in agreement with Leitsch et al., who first showed reduced activity of this gene in six resistant T. vaginalis strains, and importantly, restoration of Mz sensitivity upon transfection of resistant B7268 with a plasmid bearing a functional FR1 gene (Leitsch et al. 2014). In addition, iron metabolism in T. vaginalis is highly dependent on FR1, and thus resistant parasites must “optimize” their iron-dependent pathways in the absence of this protein (Leitsch et al. 2009). It is possible that the reduced expression of many cytosolic and hydrogenosomal metabolic pathways that we observed, as well as reduced expression of iron-SOD and iron-sulfur flavoproteins and genes coding for proteinases, was as a consequence of that optimization. The effect that iron-induced changes have on the transcriptome and proteome of T. vaginalis hydrogenosomes and proteolytic activity has also been described by others (Horvathova et al. 2012 ; Beltran et al. 2013; Arroyo et al. 2015). As such, these changes in iron-dependent processes represent consequences rather than causes of Mz resistance, and thus may play an important role in parasite fitness.
Fig. 7.
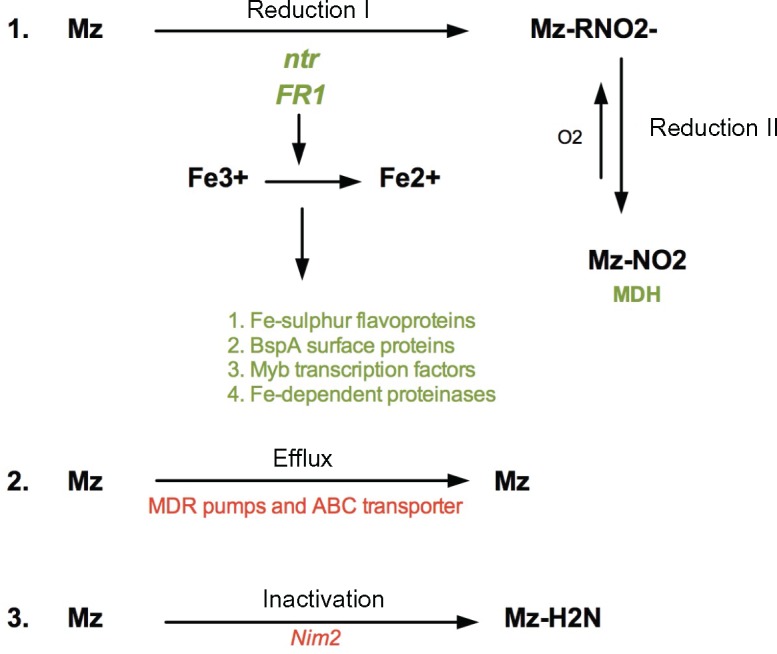
—Summary and proposed model of Mz resistance as three major processes: (1) Drug reduction, (2) Drug Efflux, and (3) Drug inactivation. Red color represents upregulated genes, green color represents downregulated genes.
T. vaginalis contains a large 911 BspA gene family, members of which have been localized to the cell surface and may be involved in pathogenicity (Noel et al. 2010). Interestingly, we detected 14 BspA-like genes as down-regulated in our three resistant strains compared to the sensitive strains (supplementary file S6,Supplementary Material online). This is in contrast to a recent finding by Ansell and colleagues (Ansell et al. 2017) who showed up-regulation of ∼10% of VSP (variant surface protein) genes in drug resistant Giardia strains. Because of the possible modulation of various pathways by iron in T. vaginalis, we consider any association between BspA-like protein regulation and drug resistance to be most likely an epiphenomenon.
We also confirmed a possible role for certain NimA genes in Mz detoxification. Transfection of the nim1 gene in E. coli made the bacterium 20× more resistant to Mz through detoxification of the drug to nontoxic amine (Pal, et al. 2009). While the nim2 paralog in our studies was upregulated in our two anaerobically resistant lab strains suggesting its importance in anerobic resistance to Mz, nim1 was downregulated in all three resistant strains indicating that its function must be furthered studied. Finally, our study identified 14 possible membrane transporter genes that were upregulated in resistant strains, indicating that efflux pumps may play a significant role in the phenotype. Previous work surveyed a single-domain P-glycoprotein-like gene (Pgp) (Johnson et al. 1994), however there was no strong evidence that this protein might be involved in Mz resistance.
While our study is the first to provide a genome-wide scan of genetic variation and transcriptomes in tens of T. vaginalis strains in an attempt to identify novel genes involved in the Mz resistance phenotype, it has limitations. For example, the PFO-ferredoxin pathway in hydrogenosomes and TrxR pathway in the cytosol have been described as potent activators of Mz and linked to anaerobic resistance (Cerkasovova et al. 1984; Leitsch et al. 2009; Rasoloson et al. 2001, 2002). However, our TrxR gene expression results are the opposite to the expression pattern shown in other studies (Leitsch et al. 2009), possibly due to experiments being performed in different strains, or under anaerobic conditions, or through the analysis of gene expression at the RNA level rather than at the protein level. In addition, our use of the “reduced representation” sequencing method means that partial SNP coverage of the highly repetitive, ∼160 Mb genome was obtained, because re-sequencing such a repetitive genome using short read technology would have been too challenging to analyze.
An important question arising from this work is what our analyses reveal about the evolution of Mz resistance in T. vaginalis or other trichomonads. We observed mutations in genes shared between several genetically distinct laboratory-derived T. vaginalis lines and >100 T. vaginalis clinical isolates, as well as with the distantly related Tr. foetus that causes venereal disease in cattle, suggesting strong natural selection on the same sets of genes and the presence of shared pathways of resistance. We propose that one of the first means for developing Mz resistance in trichomonads is the development of tolerance to high oxygen levels, which provides a protective mechanism against the drug (e.g., FR1, ntr).
T. vaginalis already harbors mechanisms to cope with a highly fluctuating oxygen environment (Rasoloson et al. 2001), which may make it “pre-adapted” to Mz treatment. This hypothesis is further supported by the extremely rapid development of aerobic resistance under lab conditions (<50 in vitro passages), and the emergence of clinical Mz resistance within only 2 years of introduction of the drug (Robinson 1962). Other genes may play an important role in developing subsequently higher levels of resistance. For example, several genes in highly resistant isolates show the same mutation recurrently appearing accompanied by transcriptional change; e.g., dynein heavy chain family protein (TVAG_188120), and numerous other hypothetical proteins whose function has yet to be determined (supplementary file S4, Supplementary Material online). Many genes identified in our study have also been associated with Mz resistance in other microaerophilic parasites such as Giardia and Entamoeba (Pal et al. 2009; Penuliar et al. 2015). For example, a recent transcriptomic profile of resistance in Giardia suggests also modification of nitroreductase and oxidoreductase gene families (Ansell et al. 2017). However, in these other species, it has been proposed that laboratory-evolved resistant strains of those parasites accumulate multiple changes not only beneficial for resistance but also those related to decreased fitness. As a consequence, clinical resistance in those parasites has been purported to be rare. It is tempting to speculate in the case of T. vaginalis that due to its “Swiss army knife” of a genome, which shows highly duplicated gene families that may bestow the parasite with extraordinary versatility (Barratt et al. 2016), it is able to establish drug resistant infections because of being able to easily compensate for fitness costs. While the details of these mechanisms and their fitness costs should be further explored, we provide here a first insight into adaptation of the unicellular eukaryote T. vaginalis.
Materials and Methods
Parasite Strains, Culturing and DNA Extraction
We used a set of 102 T. vaginalis isolates originating from eight countries: Australia (13), Italy (2), Mexico (8), Papua New Guinea (18), South Africa (9), Mozambique (1), United States (50), and the United Kingdom (1) (supplementary file S2,Supplementary Material online). Most of the strains were isolated from women undergoing routine examination in sexual health clinics and then adapted to growth in culture. Additional isolates included the reference lab strain G3, and three in vitro-derived Mz resistant lines and their isogenic parents, which were made Mz resistant using the method of Kulda et al. [21] (see Fig. S2 in Supplementary file S1, Supplementary Material online): Mz-sensitive parental strains F1623 and B7708 and their Mz-resistant derivatives F1623-M and B7708-M (MLC ≥ 400 μg/ml, anaerobic) (Brown et al. 1999), and moderately resistant parental strain B7268 (MLC ≥ 200 μg/ml) (Voolmann and Boreham 1993) and its derivative B7268-M (MLC ≥ 400 μg/ml, aerobic and anaerobic) (Wright et al. 2010). All three derived lines are stably resistant in the absence of drug pressure. We also obtained three Tritrichomonas foetus lines derived using a similar selection method [47]: Tr. foetus KV1 is the Mz-sensitive parental strain to two resistant lines: KV1_M100 (exhibits both in vivo and in vitro aerobic resistance) and KV1_1MR100 (exhibits in vitro anaerobic resistance only), respectively (see supplementary fig. S3 in supplementary file S1, Supplementary Material online). T. vaginalis and Tr. foetus isolates were cultured in modified Diamond's media as described (Conrad et al. 2011), pelleted by centrifugation, and parasite DNA extracted using a standard phenol–chloroform procedure (Conrad et al. 2012).
Metronidazole Susceptibility Phenotyping
T. vaginalis susceptibility to Mz was determined using a standard minimum lethal concentration (MLC) protocol under aerobic conditions (Rojas et al. 2004). Each isolate was grown in culture media in 96 well plates in the presence of serial dilutions of Mz ranging from 0.2 to 400 μg/ml for 48 h. The lowest concentration at which no motile parasites were observed by microscopy was recorded as the MLC. Each assay was repeated two times. Control strains [CDC 252, resistant to >400 μg/ml Mz, and Mz sensitive CDC 520 (Crowell et al. 2003)] were obtained from the Centers for Disease Control and Prevention (CDC) and used in each assay. Low-level resistance was defined as aerobic MLC 50–100 µg/ml, moderate-level resistance as 100–400 µg/ml, and high-level resistance as 400 µg/ml or greater, as per previous studies (Lossick et al. 1986; Kirkcaldy et al. 2012).
In Silico Restriction Enzyme Optimization for ddRAD Sequencing
We used ddRAD-Seq to sample unique regions of the highly repetitive T. vaginalis genome. We first evaluated in silico digests of the reference assembly of strain G3 using several common choices of restriction enzymes implemented in GMBX_digest_v1.0.pl script (https://github.com/GenomicsCoreLeuven/GBSX) and the SimRAD package in R (Center for Disease Control and Prevention 2013), starting with digestion with six-base cutting site enzymes (EcoRI, PstI, SdaI, SgrAI, and SbfI) to determine the best enzyme for cutting within unique regions of the genome [defined as regions free of highly repetitive TEs or genes in multicopy families, based on the current genome annotation (www.Trichdb.org)]. EcoRI was found to cut most frequently in the unique regions of the genome (fig. S4A in supplementary file S1, Supplementary Material online). Subsequent testing of EcoRI in combination with other restriction enzymes, both experimentally (data not shown) and in silico, revealed that a double digest of EcoRI and NlaIII produced the optimal 650–850 bp fragment size distribution for Illumina library preparation and sequencing (fig. S4B and C in supplementary file S1, Supplementary Material online).
ddRAD Library Preparation and Illumina Sequencing
ddRAD sequencing libraries were prepared using EcoRI and NlaIII digested DNA followed by the protocol described in (Peterson et al. 2012). Sequencing adapters containing a combinatorial in-line barcode and standard Illumina multiplexing read index were used to individually barcode samples such that the identity of each isolate was kept intact. Libraries were sequenced on an Illumina HiSeq 2000 following standard methods for cluster generation and sequencing, and paired end (PE) reads of 100 bp were generated. Samples were de-multiplexed and individual FASTQ files obtained using Google Documents spreadsheets and database tools in Python (Peterson et al. 2012).
SNP Discovery and Filtering
To estimate the SNP false discovery rate and optimize SNP filtering, the Perl script IRMS.pl (Farrer et al. 2013) was used to introduce 99 random SNPs into the reference G3 genome in silico. SNPs were then detected by aligning ddRAD sequencing G3 data to the G3 reference with introduced SNPs using BWA version 5.09 to align reads (Li and Durbin 2009) and GATK to call variants (DePristo et al. 2011). Multiple filtering steps included removing SNPs based on quality (QUAL > 30), approximate read depth (DP < 5), minimal genotype quality (GQ < 25), quality by depth (QD< 5) and minor allele frequency (MAF) < 0.05. We recovered 82% of introduced SNPs upon alignment with the ddRAD re-sequenced G3 data, thus the same criteria were applied to our actual ddRAD dataset (fig. S5 in supplementary file S1, Supplementary Material online).
Illumina PE reads generated for 102 isolates were aligned to the G3 reference genome in the TrichDB database (Aurrecoechea et al. 2009) using BWA. BAM alignment files of all sequenced isolates were merged and locally realigned to the reference assembly using GATK RealignerTargetCreator in order to minimize the number of mismatching bases across all the reads (DePristo et al. 2011). Genotypes were called from merged BAM files using GATK. In addition to the above-mentioned filtering criteria, SNPs detected from multiple samples were further filtered by removing those with >40% missingness, and those found within TEs. These filtering steps produced a final set of 3,923 high quality filtered SNPs from the 191,671 total SNPs identified from 102 T. vaginalis libraries. Direct SNP comparisons between parental and drug resistant derived pairs were made for each SNPs such that those SNPs found in ancestral pair must match with reference G3 strain (sensitive), and must differ from derived stains with at least three reads for each SNP.
Sanger Sequencing for SNP Validation
Using the reference G3 assembly in the TrichDB database (Aurrecoechea, et al. 2009), primers were designed to 150–300 bp regions flanking six of the 72 SNPs associated with MLC ≥ 100 μg/ml and used to amplify DNA from 20 to 24 T. vaginalis isolates (supplementary table S1 in supplementary file S1, Supplementary Material online). PCRs were performed in 25 µl reactions containing ∼20 ng genomic DNA, 0.2 µM of each primer, 200 µM of each dNTP, 1.5 mM MgCl2, 1 U of Taq DNA polymerase (Sigma) and 1× PCR buffer. Standard PCR amplification programs were used with an initial denaturation for 5 min and 30 cycles, and PCR products visualized on 1.5% agarose gels. Amplicons were cleaned with QIAquick PCR purification kit (Qiagen) and sequenced using standard Sanger sequencing chemistry. The identity of ∼73% (range ∼63% to 88%) of the SNPs identified through ddRAD sequencing were confirmed by Sanger sequencing.
Population Structure
Population structure was assessed from the 3,923 high-quality SNPs representing 100 isolates using Discriminant Analysis of Principal Component (DAPC) implemented in the R adegenet package, using “average” in the snpzip function (Jombart 2008; Jombart et al. 2010). snpzip uses DAPC to calculate the contribution (loadings) of each SNP to each population group or phenotype. Correction of population structure was performed by regressing along the first PCA axis. DAPC with the snpzip function and centroid clustering was also used to identify SNPs that distinguish Mz-resistant and Mz-sensitive isolates. SNP effects from the final data set were predicted using SnpEff (Cingolani et al. 2012), and functional categories were determined using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto 2000; Kanehisa et al. 2014) metabolic enrichment tools in TrichDB (Aurrecoechea et al. 2009).
Linkage Disequilibrium
After filtering out rare variants (MAF < 0.05), which could give spurious LD, we recovered 3,894 SNPs from the 3,923 SNPs used to determine population structure. Filtering this set within each population gave 2,837 SNPs covering 898 genomic contigs in one population, and 2,694 SNPs covering 872 contigs in the other. Linkage disequilibrium decay from SNPs was determined using the function r2 from the toolset PLINK (http://pngu.mgh.harvard.edu/purcell/plink), with pairwise r2 only considered within each contig. The decay of LD over distance was estimated using the Hill and Weir formula (1988), and a nonlinear model was used in order to fit the data to the decay function.
RNA-seq of T. vaginalis Mz Resistant and Sensitive Parasites
Three Mz-resistant T. vaginalis isolates (B7268, B7268-M, and NYCC37), and nine Mz-sensitive isolates (G3, GOR69, NYCA04, NYCB20, NYCD15, NYCE32, NYCF20, NYCG31, and SD2) were grown overnight in 14 ml sealed tubes and total RNA was isolated using the Qiagen® RNeasy Mini Kit, including a column DNase treatment using the Qiagen® RNase-Free DNase kit. Polyadenylated RNA was purified from 5 μg of total RNA using the Dynabeads® mRNA DIRECT™ Purification Kit. All experiments were carried out in triplicate. First strand synthesis was carried out by mixing the entire fraction of isolated polyA+ RNA (8 μl) with 0.5 μl Random Primers (3 μg/μl, Invitrogen), 10 mM DTT and 0.25 μl ANTI-RNase (15–30 U/μl, Ambion) in 1x First-Strand Synthesis Buffer (5×, Invitrogen), with incubation at 65 °C for 3 min to remove RNA secondary structures, then placed on ice. A total of 0.5 μl SuperScript® III enzyme (200 U/μl, Invitrogen) and dNTPs to a final concentration of 0.125 mM were added to the mixture and reverse transcription was carried out using the following incubations: 25 °C for 10 min, 42 °C for 50 min, and 70 °C for 15 min. The resulting cDNA/RNA hybrid was purified from the mix using Agencourt RNAClean XP Beads according to manufacturer's instructions. Second-strand synthesis was carried out by mixing the purified cDNA/RNA hybrid with 1 μl dUTP mix (10 mM, Roche), 0.5 μl Ribonuclease H (2 U/μl, Invitrogen). About 1 μl DNA polymerase I (5–10 U/μl, Invitrogen) in 1 × NEB buffer 2 with 2.5 mM DTT. This mixture was incubated at 16 °C for 2.5 h. The resulting double stranded cDNA was purified using Agencourt AMPure XP beads according to manufactures instructions. The cDNA was end repaired by mixing 5 μl T4 DNA Polymerase (3 U/μl, NEB), 2 μl Klenow DNA Polymerase (3–9 U/μl, Invitrogen), 5 μl T4 Polynucleotide Kinase (10 U/μl, NEB) in 1 × T4 DNA Ligase buffer with 10 mM ATP (NEB) with 0.4 mM dNTPs. The mixture was incubated at room temperature for 30 min. The end-repaired cDNA was purified using Agencourt AMPure XP beads according to manufactures instructions. The purified end-repaired cDNA was then taken through A-tailing, adapter ligation and PCR enrichment using the Illumina TruSeq Stranded mRNA Sample Preparation Kit with different barcodes for each sample. The libraries were pooled and sequenced on the Illumina HiSeq 2500 with 101 cycles, paired-end reads, and multiplexing.
Transcriptome Analyses
Approximately 10–20 million reads were generated per RNA-seq library. The RNA-seq data sets were aligned to the G3 reference genome sequence using BWA version 5.09 (Li and Durbin 2009) with 1300 bp used as a distance between the reads, yielding an overall alignment rate of >95% for all libraries. Counts of reads for each gene were obtained using the program HTseq (Anders et al. 2015). The raw read counts were normalized by rowMeans normalization function based on counts. Quality control of the data was undertaken by comparing the three replicates of each isolate. A statistical test (binomialWaldTest) was used to detect up- and down-log2 fold changes in gene expression. Differential gene expression was explored using the MAplot function in DESeq2 R package, P-values were adjusted using false discovery rate (FDR), and only changes where padj < 0.01 were considered significant. Expression patterns of each of the three Mz resistant strains (B7268, B7268-M, and NYCC37) were compared pair-wise against the group of nine sensitive strains (G3, GOR69, NYCA04, NYCB20, NYCD15, NYCE32, NYCF20, NYCG31, and SD2) using the DESeq2 R package, and FDR = 0.05 and P < 0.05 (Love et al. 2014). Significantly downregulated and upregulated expression cluster groups identified by DESeq2 R were additionally tested for their association with Mz susceptibility using chi-square and a clustering test in the heatmap3 R package (Zhao et al. 2014). Gene Ontology enrichment analyses of genes was performed using TrichDB tools and a P-value cutoff of 0.05 (Aurrecoechea et al. 2009).
Tr. foetus Whole Genome Sequencing
Whole genome sequences of three Tr. foetus samples (1) KV1 (Mz sensitive), (2) KV1_M100 (derived in vitro from KV1 and exhibiting aerobic Mz resistance), and (3) KV1_1MR100 (derived in vitro from KV1 and exhibiting aerobic and anaerobic Mz resistance) as described in Kulda et al. (1984), were generated by standard Illumina sequencing of 350 bp and 500 bp insert libraries and generation of 100 bp paired-end reads. Tr. foetus KV1 Illumina sequencing reads were assembled de novo into 194,695 contigs (N50 = 2,054 bp) with a size range of 61–19,994 bp using Velvet (Zerbino 2010). Sequencing produced between 44.3× coverage and 49.5× coverage of each isolate (Zerbino 2010). Illumina reads from KV1, KV1_M100 and KV1_1MR100 were aligned against the KV1 contigs and SNPs were called using the same GATK pipeline as described above for T. vaginalis SNP detection. We identified 627 high quality SNPs, of which 418 were found in KV1_1MR100, 86 were found in KV1_M100, with 97 were shared between these two resistant lines. To annotate SNPs we used BLASTX (Ye et al. 2006) (with -max_target_seqs 1 -culling_limit 1 -seg no) to search a local T. vaginalis protein database and identify orthologs genes. Only BLASTX hits >50 bp length were considered valid and received a T. vaginalis gene attribute. Candidate SNPs were then assigned to the appropriate T. vaginalis orthologs.
Data Access
This project has been deposited at the Sequence Read Archive under the accession SRP057357 and SRP057311.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Authors' Contributions
MB designed the study, performed experiments, analyzed the data, and drafted the paper. SW made the RNA-seq libraries. GT performed Sanger sequencing validation experiments. PS helped with ddRAD sequencing experimental design. WS provided lab lines and training in drug resistance assays. PH, TC, CL, and PT performed NGS sequencing and raw data processing of three whole genomes. SS undertook data checking, proofreading, and writing the manuscript. JC oversaw all aspects of the research and experimental design, as well as manuscript writing.
Supplementary Material
Acknowledgments
We thank members of the Carlton lab for critical reading of the manuscript and Jonathan Flowers and Swapna Uplekar for helpful discussions. We also thank Peter Augostini at CDC for providing training and protocols for the minimum lethal concentration phenotypic assay. MB is partially supported by National Institutes of Health grant R01 AI097080 to P. Kissinger (Tulane University), S.D.W. by the MacCracken Program in the New York University Graduate School of Arts and Science and a Fleur Strand Fellowship, G.E.T. by a New York University Gallatin Undergraduate Research Fund award, and PT by the Chang Gung Memorial Hospital Research Fund (CMRPD3D0181-3). The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Literature Cited
- Anders S, Pyl PT, Huber W.. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell BR, et al. 2017. Transcriptomics indicates active and passive metronidazole resistance mechanisms in three seminal giardia lines. Front Microbiol. 8:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo R, et al. 2015. Trichomonas vaginalis cysteine proteinases: iron response in gene expression and proteolytic activity. Biomed Res Int. 2015:946787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurrecoechea C, et al. 2009. GiardiaDB and TrichDB: integrated genomic resources for the eukaryotic protist pathogens Giardia lamblia and Trichomonas vaginalis. Nucleic Acids Res. 37:D526–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt J, Gough R, Stark D, Ellis J.. 2016. Bulky trichomonad genomes: encoding a Swiss Army knife. Trends Parasitol. 32:783–797. [DOI] [PubMed] [Google Scholar]
- Beltran NC, et al. 2013. Iron-induced changes in the proteome of Trichomonas vaginalis hydrogenosomes. PLoS One 8:e65148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchimol M, et al. 2017. Draft genome sequence of Tritrichomonas foetus strain K. Genome Announc. 5: e00195-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradic M, Warring SD, Low V, Carlton JM.. 2014. The Tc1/mariner transposable element family shapes genetic variation and gene expression in the protist Trichomonas vaginalis. Mobile DNA 5: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Upcroft JA, Dodd HN, Chen N, Upcroft P.. 1999. Alternative 2-keto acid oxidoreductase activities in Trichomonas vaginalis. Mol Biochem Parasitol. 98:203–214. [DOI] [PubMed] [Google Scholar]
- Carlton JM, et al. 2007. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 315:207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerkasovova A, Cerkasov J, Kulda J.. 1984. Metabolic differences between metronidazole resistant and susceptible strains of Tritrichomonas foetus. Mol Biochem Parasitol 11:105–118. [DOI] [PubMed] [Google Scholar]
- Cingolani P, et al. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, et al. 2011. Microsatellite polymorphism in the sexually transmitted human pathogen Trichomonas vaginalis indicates a genetically diverse parasite. Mol Biochem Parasitol. 175:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad MD, et al. 2012. Extensive genetic diversity, unique population structure and evidence of genetic exchange in the sexually transmitted parasite Trichomonas vaginalis. PLoS Negl Trop Dis. 6: e1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Disease Control and Prevention. 2013. Incidence, Prevalence, and Cost of Sexually Transmitted Infections in the United States. In. CDC Fact Sheet.
- Cornelius DC, et al. 2012. Genetic characterization of Trichomonas vaginalis isolates by use of multilocus sequence typing. J Clin Microbiol. 50:3293–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotch MF, et al. 1997. Trichomonas vaginalis associated with low birth weight and preterm delivery. The Vaginal Infections and Prematurity Study Group. Sex Transm Dis. 24:353–360. [DOI] [PubMed] [Google Scholar]
- Crowell AL, Sanders-Lewis KA, Secor WE.. 2003. In vitro metronidazole and tinidazole activities against metronidazole-resistant strains of Trichomonas vaginalis. Antimicrob Agents Chemother. 47:1407–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brum Vieira P, Tasca T, Secor WE.. 2017. Challenges and persistent questions in the treatment of Trichomoniasis. Curr Top Med Chem. 17:1249–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43:491.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JE, Cole D, Lloyd D.. 1992. Influence of oxygen on the fermentative metabolism of metronidazole-sensitive and resistant strains of Trichomonas vaginalis. Mol Biochem Parasitol. 56:79–88. [DOI] [PubMed] [Google Scholar]
- Farrer RA, Henk DA, MacLean D, Studholme DJ, Fisher MC.. 2013. Using false discovery rates to benchmark SNP-callers in next-generation sequencing projects. Sci Rep. 3: 1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote RH. 1996. Review: dairy cattle reproductive physiology research and management–past progress and future prospects. J Dairy Sci. 79:980–990. [DOI] [PubMed] [Google Scholar]
- Frey CF, Muller N.. 2012. Tritrichomonas—systematics of an enigmatic genus. Mol Cell Probes 26:132–136. [DOI] [PubMed] [Google Scholar]
- Garg S, et al. 2015. Conservation of transit peptide-independent protein import into the mitochondrial and hydrogenosomal matrix. Genome Biol Evol. 7:2716–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould SB, et al. 2013. Deep sequencing of Trichomonas vaginalis during the early infection of vaginal epithelial cells and amoeboid transition. Int J Parasitol. 43:707–719. [DOI] [PubMed] [Google Scholar]
- Halkett F, Simon JC, Balloux F.. 2005. Tackling the population genetics of clonal and partially clonal organisms. Trends Ecol Evol. 20:194–201. [DOI] [PubMed] [Google Scholar]
- Hill WG, Weir BS.. 1988. Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol. 33:54–78. [DOI] [PubMed] [Google Scholar]
- Horvathova L, et al. 2012. Transcriptomic identification of iron-regulated and iron-independent gene copies within the heavily duplicated Trichomonas vaginalis genome. Genome Biol. Evol. 4:1017–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PJ, Schuck BL, Delgadillo MG.. 1994. Analysis of a single-domain P-glycoprotein-like gene in the early-diverging protist Trichomonas vaginalis. Mol Biochem Parasitol. 66:127–137. [DOI] [PubMed] [Google Scholar]
- Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. [DOI] [PubMed] [Google Scholar]
- Jombart T, Devillard S, Balloux F.. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11:94.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S.. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, et al. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42:D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkcaldy RD, et al. 2012. Trichomonas vaginalis antimicrobial drug resistance in 6 US cities, STD Surveillance Network, 2009-2010. Emerg Infect Dis. 18:939–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulda J. 1999. Trichomonads, hydrogenosomes and drug resistance. Int J Parasitol. 29:199–212. [DOI] [PubMed] [Google Scholar]
- Kulda J, Cerkasov J, Demes P, Cerkasovova A.. 1984. Tritrichomonas foetus: stable anaerobic resistance to metronidazole in vitro. Exp Parasitol. 57:93–103. [DOI] [PubMed] [Google Scholar]
- Kulda J, Tachezy J, Cerkasovova A.. 1993. In vitro induced anaerobic resistance to metronidazole in Trichomonas vaginalis. J Eukaryot Microbiol. 40:262–269. [DOI] [PubMed] [Google Scholar]
- Land KM, et al. 2004. Targeted gene replacement of a ferredoxin gene in Trichomonas vaginalis does not lead to metronidazole resistance. Mol Microbiol. 51:115–122. [DOI] [PubMed] [Google Scholar]
- Leitsch D, Drinic M, Kolarich D, Duchene M.. 2012. Down-regulation of flavin reductase and alcohol dehydrogenase-1 (ADH1) in metronidazole-resistant isolates of Trichomonas vaginalis. Mol Biochem Parasitol. 183:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitsch D, Janssen BD, Kolarich D, Johnson PJ, Duchene M.. 2014. Trichomonas vaginalis flavin reductase 1 and its role in metronidazole resistance. Mol Microbiol. 91:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitsch D, et al. 2009. Trichomonas vaginalis: metronidazole and other nitroimidazole drugs are reduced by the flavin enzyme thioredoxin reductase and disrupt the cellular redox system. Implications for nitroimidazole toxicity and resistance. Mol Microbiol. 72:518–536. [DOI] [PubMed] [Google Scholar]
- Leitsch D, Kolarich D, Duchene M.. 2010. The flavin inhibitor diphenyleneiodonium renders Trichomonas vaginalis resistant to metronidazole, inhibits thioredoxin reductase and flavin reductase, and shuts off hydrogenosomal enzymatic pathways. Mol Biochem Parasitol. 171:17–24. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossick JG, Muller M, Gorrell TE.. 1986. In vitro drug susceptibility and doses of metronidazole required for cure in cases of refractory vaginal trichomoniasis. J Infect Dis. 153:948–955. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik SB, Pightling AW, Stefaniak LM, Schurko AM, Logsdon JM. Jr.. 2008. An expanded inventory of conserved meiotic genes provides evidence for sex in Trichomonas vaginalis. PLoS One 3:e2879.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritz JM, Land KM, Carlton JM, Hirt RP.. 2014. What is the importance of zoonotic trichomonads for human health? Trends Parasitol. 30:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland RS, et al. 2007. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis. 195:698–702. [DOI] [PubMed] [Google Scholar]
- Mu J, et al. 2005. Recombination hotspots and population structure in Plasmodium falciparum. PLoS Biol. 3:e335.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel CJ, et al. 2010. Trichomonas vaginalis vast BspA-like gene family: evidence for functional diversity from structural organisation and transcriptomics. BMC Genomics 11:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, et al. 2009. Giardia, Entamoeba, and Trichomonas enzymes activate metronidazole (nitroreductases) and inactivate metronidazole (nitroimidazole reductases). Antimicrob Agents Chemother. 53:458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulish-Miller TE, et al. 2014. Trichomonas vaginalis metronidazole resistance is associated with single nucleotide polymorphisms in the nitroreductase genes ntr4(Tv) and ntr6(Tv). Antimicrob Agents Chemother. 58:2938–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penuliar GM, Nakada-Tsukui K, Nozaki T.. 2015. Phenotypic and transcriptional profiling in Entamoeba histolytica reveal costs to fitness and adaptive responses associated with metronidazole resistance. Front Microbiol. 6:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE.. 2012. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7:e37135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrin D, Delgaty K, Bhatt R, Garber G.. 1998. Clinical and microbiological aspects of Trichomonas vaginalis. Clin Microbiol Rev. 11:300–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasoloson D, Tomkova E, Cammack R, Kulda J, Tachezy J.. 2001. Metronidazole-resistant strains of Trichomonas vaginalis display increased susceptibility to oxygen. Parasitology 123:45–56. [DOI] [PubMed] [Google Scholar]
- Rasoloson D, et al. 2002. Mechanisms of in vitro development of resistance to metronidazole in Trichomonas vaginalis. Microbiology-Sgm 148:2467–2477. [DOI] [PubMed] [Google Scholar]
- Robinson SC. 1962. Trichomonal vaginitis resistant to metronidazole. Can. Med. Assoc. J. 86:665. [PMC free article] [PubMed] [Google Scholar]
- Rojas L, Fraga J, Sariego I.. 2004. Genetic variability between Trichomonas vaginalis isolates and correlation with clinical presentation. Infect Genet Evol. 4:53–58. [DOI] [PubMed] [Google Scholar]
- Smutna T, Pilarova K, Tarabek J, Tachezy J, Hrdy I.. 2014. Novel functions of an iron-sulfur flavoprotein from Trichomonas vaginalis hydrogenosomes. Antimicrob Agents Chemother. 58:3224–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe S, Neace C, Magnuson NS, Reeves R, Alderete JF.. 2012. Trichomonosis, a common curable STI, and prostate carcinogenesis—a proposed molecular mechanism. PLoS Pathog. 8:e1002801.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M, Ayala FJ.. 2002. The clonal theory of parasitic protozoa: 12 years on. Trends Parasitol. 18:405–410. [DOI] [PubMed] [Google Scholar]
- Twu O, et al. 2014. Trichomonas vaginalis homolog of macrophage migration inhibitory factor induces prostate cell growth, invasiveness, and inflammatory responses. Proc Natl Acad Sci U S A. 111:8179–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upcroft JA, et al. 2009. Metronidazole resistance in Trichomonas vaginalis from highland women in Papua New Guinea. Sex Health 6:334–338. [DOI] [PubMed] [Google Scholar]
- Upcroft JA, Upcroft P.. 2001a. Drug susceptibility testing of anaerobic protozoa. Antimicrob Agents Chemother. 45:1810–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upcroft P, Upcroft JA.. 2001b. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin Microbiol Rev. 14:150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voolmann T, Boreham P.. 1993. Metronidazole resistant Trichomonas vaginalis in Brisbane. Med J Aust. 159:490.. [DOI] [PubMed] [Google Scholar]
- Watt L, Jennison RF.. 1960. Clinical evaluation of metronidazole. A new systemic trichomonacide. Br Med J. 2:902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JM, et al. 2010. Susceptibility in vitro of clinically metronidazole-resistant Trichomonas vaginalis to nitazoxanide, toyocamycin, and 2-fluoro-2'-deoxyadenosine. Parasitol Res. 107:847–853. [DOI] [PubMed] [Google Scholar]
- Yarlett N, Yarlett NC, Lloyd D.. 1986a. Ferredoxin-dependent reduction of nitroimidazole derivatives in drug-resistant and susceptible strains of Trichomonas vaginalis. Biochem Pharmacol. 35:1703–1708. [DOI] [PubMed] [Google Scholar]
- Yarlett N, Yarlett NC, Lloyd D.. 1986b. Metronidazole-resistant clinical isolates of Trichomonas vaginalis have lowered oxygen affinities. Mol Biochem Parasitol. 19:111–116. [DOI] [PubMed] [Google Scholar]
- Ye J, McGinnis S, Madden TL.. 2006. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34:W6–W9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yule A, Skirrow SZ, Bonduran RH.. 1989. Bovine trichomoniasis. Parasitol Today 5:373–377. [DOI] [PubMed] [Google Scholar]
- Zerbino DR. 2010. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics Chapter 11: Unit 11 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Guo Y, Sheng Q, Shyr Y.. 2014. Advanced heat map and clustering analysis using heatmap3. Biomed Res Int. 2014:986048.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.