

Abstract
Background
Avelumab, a fully human IgG1 immune checkpoint inhibitor targeting programmed death ligand 1 (PD-L1), has shown antitumour activity and an acceptable safety profile in patients with advanced solid tumours. Here, we assessed avelumab treatment in patients with advanced, platinum-treated non-small cell lung cancer (NSCLC).
Methods
In this dose-expansion cohort of a multicentre, open-label, phase 1 study, patients with progressive or platinum-resistant metastatic or recurrent NSCLC were enrolled at 58 sites in the United States. Eligible patients had confirmed stage IIIB or IV NSCLC with squamous or nonsquamous histology, measurable disease by Response Evaluation Criteria In Solid Tumors version 1.1 (RECIST v1.1), tumour biopsy or archival sample for biomarker assessment, and Eastern Cooperative Oncology Group performance status 0 or 1, among other criteria. Patient selection was not based on PD-L1 expression or other biomarkers, including EGFR or KRAS mutation or ALK translocation status. Patients received avelumab monotherapy 10 mg/kg every 2 weeks until disease progression or toxicity. The primary endpoint was safety and tolerability. Secondary endpoints included best overall response, progression-free survival, overall survival, and clinical activity associated with PD-L1 expression. Responses were evaluated using RECIST v1.1, and analyses of antitumour activity and safety were performed in all patients who received at least one dose of avelumab. This trial is registered with ClinicalTrials.gov (NCT01772004); enrolment in this cohort is closed and the trial is ongoing.
Findings
Between 10 September 2013 and 24 June 2014, 184 patients were enrolled and initiated treatment with avelumab. Median follow-up duration was 8·8 months (interquartile range [first and third quartiles], 7·2–11·9 months). The most common treatment-related adverse events of any grade were fatigue (n=46; 25%), infusion-related reaction (n=38; 21%), and nausea (n=23; 13%). Grade ≥3 treatment-related adverse events occurred in 23 of 184 patients (13%); the most common were infusion-related reaction (n=4; 2%), elevated lipase (n=3; 2%), constipation (n=2; 1%), and dyspnoea (n=2; 1%). 16 of 184 patients (9%) had a serious adverse event related to treatment with avelumab, with infusion-related reaction (4 [2%]) and dyspnoea (2 [1%]) occurring in more than one patient. Immune-related treatment-related events occurred in 22 patients (12%). The confirmed objective response rate, regardless of PD-L1 status, was 12% (95% CI, 8–18), including one complete response and 21 partial responses. Seventy patients had stable disease, for an overall disease-control rate of 50%.
Interpretation
Avelumab showed an acceptable safety profile and antitumour activity in patients with progressive or resistant NSCLC, providing a rationale for additional studies of avelumab in this disease setting.
Funding
Merck KGaA, Darmstadt, Germany and Pfizer, Inc, New York, USA.
Introduction
Lung cancer is the leading cause of cancer death worldwide.1 Most patients present with stage IV disease, which has a median overall survival of 8–10 months and a 5-year relative survival rate of approximately 4%.2–4 First-line treatment for patients with non-small cell lung cancer (NSCLC) without any actionable mutation is generally based on platinum doublet chemotherapy. Until recently, eligible patients with progressive disease following first-line therapy typically received chemotherapy with docetaxel or pemetrexed, which has been associated with a 1-year survival rate of approximately 30%.5 In eligible subsets of patients with specific tumour biomarkers, such as epidermal growth factor receptor (EGFR) gene mutation or anaplastic lymphoma kinase (ALK) or protein kinase ROS1 rearrangements, targeted therapy with tyrosine kinase inhibitors has shown clinical efficacy, but resistance eventually develops.6,7
NSCLC tumours have been found to evade immune activity through multiple mechanisms, including the expression of molecules (immune checkpoints) that inhibit T-cell activation. In particular, PD-L1 expression is often upregulated in immunogenic tumours, including NSCLC,8,9 and binding of PD-L1 to its receptor on T cells, PD-1, inhibits tumour immunity by suppressing T-cell activation,8,10,11 enabling tumours to escape T-cell surveillance. PD-L1/PD-1 pathway blockade may stimulate a patient’s antitumour immune response by promoting T-cell reactivity against tumour neoantigens.12 Recently, PD-L1/PD-1–targeted immune checkpoint inhibitors have been shown to increase overall survival vs docetaxel in patients with previously treated advanced NSCLC, leading to regulatory approval of three anti–PD-L1/PD-1 therapies in this setting.13–16 Clinical benefits of immune checkpoint inhibition may be influenced by NSCLC histology, mutational load, molecular drivers of disease, and expression of PD-L1 by tumours, although responses have been achieved independent of these factors.9,17,18
Avelumab (MSB0010718C) is a fully human anti–PD-L1 IgG1 antibody that inhibits PD-L1/PD-1 interactions but leaves the PD-L2/PD-1 pathway intact. In contrast to other PD-L1/PD-1 drugs assessed in clinical trials to date, avelumab binding to the surface of tumour cells via PD-L1 has the potential to induce natural killer cell-mediated antibody-dependent cellular cytotoxicity (ADCC) of tumour cells, as shown by preclinical models, which may contribute to the clinical activity of avelumab.19,20 A large, multicohort, phase 1 dose-escalation and dose-expansion trial is being conducted to assess the safety and activity of avelumab in patients with a range of advanced solid tumours. In the phase 1a dose-escalation part of the study, avelumab showed preliminary evidence of antitumour activity, including durable responses and stable disease.21 Avelumab was safely administered by intravenous infusion every 2 weeks and had a predictable pharmacokinetic profile at doses of up to 20 mg/kg.22 The 10-mg/kg dose, which has a half-life of approximately 4 days, was selected for further study in dose-expansion cohorts in a range of tumour types. Here we present phase 1 results from this study in a cohort of patients with advanced NSCLC progressing after platinum-based chemotherapy and unselected for PD-L1 expression.
Methods
Study design and participants
JAVELIN Solid Tumour is an ongoing, international, multicentre, phase 1, open-label trial that includes several expansion cohorts. This trial included a dose-escalation part (phase 1a), the results of which are reported separately, and a dose-expansion part (phase 1b) comprised of 16 different cohorts. In the dose-expansion cohort reported here, eligible patients had histologically or cytologically confirmed stage IIIB or IV NSCLC, with squamous or non-squamous histology, which had progressed after treatment with platinum-based doublet chemotherapy for metastatic disease. Eligible patients were aged ≥18 years and had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, a life expectancy of ≥3 months, no active or history of CNS metastases, and adequate haematologic, hepatic, and renal function (defined by the following laboratory values: white blood cell count of ≥3 × 109 cells per L with an absolute neutrophil count ≥1·5 × 109 cells per L, lymphocyte count ≥0·5 × 109 cells per L, platelet count ≥100 × 109 platelets per L, haemoglobin ≥9 g/dL, total bilirubin concentration of ≤1·5 × the upper limit of normal [ULN] range, aspartate aminotransferase and alanine aminotransferase concentrations of ≤2·5 × ULN), and an estimated creatinine clearance >50 mL/min according to the Cockcroft-Gault formula). Patients were required to have measurable disease by CT or MRI scan and Response Evaluation Criteria In Solid Tumors (RECIST), version 1.123 and available fresh biopsy or tumour archival material for biomarker analyses (see appendix pages 16–18 for full eligibility criteria). Patient selection was not based on PD-L1 expression or other biomarkers, including EGFR or KRAS mutation or ALK translocation status. Patients were enrolled in accordance with an approved protocol, international standards of good clinical practice, and institutional safety monitoring, and written informed consent was provided by patients or their representatives.
Procedures
Avelumab (EMD Serono, Research & Development Institute, Billerica, MD, USA, a business of Merck KGaA, Darmstadt, Germany) was supplied as a 10 mg/mL solution. Patients received avelumab 10 mg/kg by 1-hour intravenous infusion once every 2 weeks until confirmed disease progression, unacceptable toxicity, or any other criterion for withdrawal occurred. Permanent treatment discontinuation occurred for any grade 3 or worse adverse event (with the exception of transient [≤6 hours] influenza-like symptoms or pyrexia controlled with medical management; fatigue, local infusion-related reaction, headache, nausea, or emesis that resolved to grade ≤1 within 24 hours; single laboratory values out of the normal range that were unrelated to study treatment and without clinical correlate [except for elevation in liver enzyme concentrations] that resolved to grade ≤1 within 7 days; and tumour flare, defined as local pain, irritation, or rash localised at sites of known or suspected malignant tissue) or recurring grade 2 treatment-related adverse events. Grade 2 adverse drug reactions were managed by dose modifications (changes in the infusion rate) and dose delays, and those that did not resolve to grade 1 or less by the end of the next cycle led to permanent discontinuation of avelumab. Dose modifications were not recommended, however, interruptions in delivering the planned dose that resulted in an actual dose that was less than 90% of the planned dose were defined as dose reductions. Detailed guidelines were provided for delaying or discontinuing treatment following specified adverse events of different grades (appendix pages 18–19).
Safety was assessed at each biweekly trial visit and included performance status evaluation, physical examination, clinical laboratory tests (haematology, serum chemistry, and hepatic panels), and documentation of concurrent medications and adverse events. Adverse events and laboratory abnormalities were classified and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Adverse events that had an immune-related aetiology were identified using a prespecified list of Medical Dictionary for Regulatory Activities (MedDRA) terms. Infusion-related reaction was classified as an adverse event of special interest, and signs and symptoms such as fever, chills, or rigors reported on the same day or next day following treatment were queried with investigators to ascertain whether an adverse event of infusion-related reaction should be recorded. A premedication regimen of diphenhydramine and paracetamol, implemented as mandatory on 29 January 2014, was required 30–60 minutes before all infusions of avelumab. Patients enrolled prior to this date may not have received premedication.
Clinical activity was assessed by investigators using RECIST version 1.1 to determine best overall response, defined as the best response obtained among all tumour assessments after the start of treatment with avelumab until documented disease progression, and duration of response. Radiographic tumour evaluations were performed at baseline and every 6 weeks. Modified immune-related response criteria, derived from RECIST version 1.1, were used to evaluate response patterns related to immunotherapeutic agents that may not have been adequately captured by RECIST or modified WHO criteria (appendix page 20).24
Levels of PD-L1 protein expressed by tumour cells and immune cells within the tumour microenvironment were measured in a central laboratory by immunohistochemistry staining of formalin-fixed, paraffin-embedded blocks (preferred) or slides of the most recent suitable biopsy or surgical specimen with a proprietary assay (Dako, Carpinteria, CA, USA) based on an anti–PD-L1 rabbit monoclonal antibody clone (73–10) under license from Merck KGgA. PD-L1 positivity in tumour cells was scored based on the proportion of tumour cells showing membranous PD-L1 staining. Three thresholds were prospectively defined, based on preliminary assessments of PD-L1 expression and tumour response: 1% and 5% tumour cells PD-L1 positive with any staining intensity, and 25% of tumour cells positive with moderate-to-high staining intensity (2+ to 3+). PD-L1 positivity in tumour-associated immune cells (identified as non-malignant cells based on conventional morphological features) was determined using a prospectively defined threshold of 10% of immune cells showing PD-L1 staining of any intensity within “hotspots” (dense aggregates of tumour-associated immune cells adjacent to tumour cells showing PD-L1 staining in immune cells). Tumour assessment for EGFR or KRAS mutation or ALK translocation was performed at individual centres based on local protocols.
Outcomes
The primary endpoint of the trial was occurrence of dose-limiting toxicities during the first 3 weeks of treatment in the dose-escalation part of the study; these data are reported elsewhere.21 Secondary endpoints included best overall response (defined as complete response, partial response, stable disease, or progressive disease), duration of response (defined as the time from first documented complete or partial response until documented progressive disease or death, whichever occurred first), and progression-free survival (defined as time from the first administration of avelumab until documented progressive disease or death, whichever occurred first) by RECIST v1.1 and by modified immune-related response criteria per investigator assessment; overall survival (defined as the time from first administration of avelumab until the date of death); safety; and activity according to PD-L1 expression on tumour and tumour-associated immune cells. Other secondary endpoints (pharmacokinetic and pharmacodynamic profile and immunogenicity of avelumab) will be analysed across multiple cohorts of this phase 1 study and will be reported elsewhere. Subgroup analyses based on patient and disease characteristics at baseline were conducted post hoc.
Statistical analyses
Enrolment of 150 patients was planned in this cohort, although this target was exceeded due to acceleration of recruitment by investigators at the end of the enrolment period. The sample size was chosen to explore safety and antitumour activity of avelumab in the overall cohort in addition to subgroups defined by prespecified PD-L1 tumour expression status and to provide data to aid in future study design. Safety and activity were analysed in all patients who received at least one dose of avelumab. Patients with no post-baseline assessments due to discontinuation or death within the first 6 weeks were not evaluable for a confirmed best overall response. The specified time frame for the primary analysis was 6 months after the date of the first dose in the last patient enrolled. Objective response rate, determined as the proportion of patients with a confirmed best overall response of complete or partial response, was calculated with corresponding Clopper-Pearson CI; an objective response rate of ≥10% (95% CI, 6–16) was considered indicative of clinical benefit (ie, 15 patients with a response out of 150 patients planned for enrolment). Time-to-event endpoints were estimated using Kaplan-Meier methodology; median values were calculated with corresponding CI using Brookmeyer-Crowley methodology. SAS version 9.2 was used for the statistical analysis, and R software package version 2.15.0 was used for the sample-size calculations (SAS Institute, Inc, Cary, NC, USA). This trial is registered with ClinicalTrials.gov (NCT01772004).
Role of the funding source
The sponsor, Merck KGaA, Darmstadt, Germany, provided the study drug and worked with investigators on the trial design and plan, collection and analyses of data, and interpretation of results. Data sets were reviewed by the authors, and all authors participated fully in developing and reviewing the manuscript for publication (J.L.G., A.R., D.R.S., N.I., J.C., D.J.L.W., J.L., W.J.E, D.W., H.J.G, A.vH., K.C., J-M.C., K.K.). Funding for a professional medical writer with access to the data was provided by the sponsor and Pfizer, Inc. for initial drafts of the manuscript. The corresponding author had full access to all of the data and the final responsibility to submit for publication.
Results
From 10 September 2013 to 24 June 2014, of 288 patients screened, 184 patients with locally advanced or metastatic measurable disease that had relapsed following treatment with a platinum-based doublet therapy were enrolled at 58 sites in the United States (appendix page 6) and received avelumab (figure 1). Due to interest in the study, planned enrolment was exceeded by 34 patients. Median age was 65·0 years (interquartile range [IQR, defined as first and third quartiles], 58·0–69·5) and 54% (100 of 184) were male (table 1). Of 184 treated patients, most had an ECOG performance status of 1 (n=128; 70%), had stage IV disease (n=170; 92%), were current or former smokers (n=159; 86%), and had received only one prior line of chemotherapy for metastatic disease (n=122; 66%). Tumours had squamous cell histology in 53 of 184 patients (29%). EGFR mutational status was evaluated in 110 of 184 patients (60%), and a mutation was found in 9 (5%). ALK translocation status was determined in 104 of 184 patients (57%), and one tumour (1%) was positive. PD-L1 expression was evaluable in 142 of 184 patients (77%); 122 of 142 (86%) had PD-L1+ tumours based on a 1% threshold. Additional patient demographic and disease characteristics and prior anticancer therapies are provided in the appendix (pages 8–9).
Figure 1.

Trial profile
Table 1.
Select baseline characteristics
| Characteristics | (N=184) | |
|---|---|---|
|
| ||
| Median age (IQR), years | 65·0 (58·0–69·5) | |
|
| ||
| Sex, n (%) | ||
| Male | 100 (54) | |
| Female | 84 (46) | |
|
| ||
| ECOG PS, n (%) | ||
| 0 | 55 (30) | |
| 1 | 128 (70) | |
| 2 | 0 (0) | |
| 3* | 1 (1) | |
|
| ||
| Disease stage, n (%)† | ||
| Stage IIIB | 13 (7) | |
| Stage IV | 170 (92) | |
|
| ||
| Median time since first diagnosis (IQR), months | 11·5 (6·8–21·7) | |
|
| ||
| Median time since diagnosis of metastatic disease (IQR), months | 8·5 (5·5–16·2) | |
|
| ||
| Histology, n (%) | ||
| Adenocarcinoma | 114 (62) | |
| Large cell | 5 (3) | |
| Squamous cell carcinoma | 53 (29) | |
| Other§ | 12 (7) | |
|
| ||
| Smoking history, n (%) | ||
| Never smoked | 24 (13) | |
| Ever smoked | 159 (86) | |
| Unknown | 1 (1) | |
|
| ||
| EGFR mutation status, n (%) | ||
| Wildtype | 101 (55) | |
| Mutant | 9 (5) | |
| Unknown | 74 (40) | |
|
| ||
| KRAS mutation status, n (%) | ||
| Wildtype | 38 (21) | |
| Mutant | 21 (11) | |
| Unknown | 125 (68) | |
|
| ||
| ALK translocation status, n (%) | ||
| Negative | 103 (56) | |
| Positive | 1 (1) | |
| Unknown | 80 (44) | |
|
| ||
| No. of prior anticancer lines for advanced disease (%) | ||
| 0§ | 1 (1) | |
| 1 | 122 (66) | |
| 2 | 44 (24) | |
| ≥3 | 17 (9) | |
| No. of prior anticancer lines for advanced disease, median (IQR) | 1·0 (1–2) | |
|
| ||
| PD-L1 expression status, n (%)¶ | PD-L1+ | PD-L1− |
| ≥1% tumour cells | 122 (86) | 20 (14) |
| ≥5% tumour cells | 84 (59) | 58 (41) |
| ≥25% tumour cells | 53 (37) | 89 (63) |
| ≥10% tumour-associated immune cells in hotspots | 27 (19) | 115 (81) |
ECOG PS, Eastern Cooperative Oncology Group performance status; IQR, interquartile range, defined as first and third quartiles. Some percentages do not sum to 100 because of rounding.
The patient had an ECOG PS of 1 at screening, which had increased to 3 at start of treatment.
Data on disease stage was missing for one patient.
Other category includes poorly differentiated carcinoma (n=3), poorly differentiated adenocarcinoma (n=2), poorly differentiated non-squamous (n=1), poorly differentiated sarcomatoid carcinoma (n=1), moderately differentiated adenocarcinoma (n=1), adenosquamous (n=2), neuroendocrine (n=1), and carcinoma not further defined (n=1).
One patient was enrolled without having received prior systemic therapy for advanced disease (protocol violation).
PD-L1 expression was evaluable in 142 of 184 patients (77%). Nonevaluable specimens (n=42) included those that were missing (n=8), of poor quality or quantity (insufficient tissue on slide [n=8] or insufficient tumour sample [n=9]), or otherwise not available to provide results (n=17). Thresholds for positive PD-L1 status were based on any intensity of staining except for ≥25% tumour cells, for which moderate-to-high intensity staining was required.
Patients received a median of six doses of avelumab (IQR, 3–15) administered every 2 weeks for a median of 12·2 weeks (IQR, 6·1–30·0). At data cut-off on 15 January 2015, median follow-up was 8·8 months (IQR, 7·2–11·9; minimum, 6 months), and 41 of 184 patients (22%) were still on treatment. Among 143 patients who discontinued avelumab, the most common reason was disease progression (93 patients [65%]). Dosing was modified (planned dose not administered in full) in nine of 184 patients (5%) due to infusion-related reaction in seven patients (five grade 1–2, one grade 3, and one grade 4), treatment-related grade 2 chest discomfort in one patient, and unrelated grade 1 erythema in one patient. Dosing was delayed for 3–6 days in 34 patients (19%) and for ≥7 days in 42 patients (23%); delays were due to an adverse event (related or unrelated to treatment) in 28 of 184 patients (15%), most commonly diarrhoea and upper respiratory tract infection (n=3 [2%] each).
Of 184 treated patients, 182 (99%) had an adverse event of any grade (appendix pages 10–13); 142 (77%) had a treatment-related adverse event (table 2), of which fatigue (25%), infusion-related reaction (21%), and nausea (13%) were most common. Grade ≥3 treatment-related adverse events occurred in 23 patients (13%), of which only infusion-related reaction (n=4 [2%]) and increased lipase level (n=3 [2%]) occurred in more than two patients. One patient developed grade 4 pneumonitis (table 2). Avelumab was permanently discontinued because of a treatment-related adverse event in 17 patients (9%): infusion-related reaction (8 patients), increased lipase level (2), dyspnoea (2), syncope (1), increased gamma-glutamyltransferase (1), autoimmune neutropenia (1), adrenal insufficiency (1), stomatitis (1), anaphylactic reaction (1), and radiation pneumonitis (1).
Table 2.
Treatment-related adverse events (worst grade per patient)
| Adverse event (N=184) | Any grade | Grade 1–2 | Grade 3 | Grade 4 | Grade 5 |
|---|---|---|---|---|---|
| Treatment-related adverse events [n, (%)] occurring at any grade in ≥5% patients or any grade ≥3 | |||||
| Any event | 142 (77) | 119 (65) | 16 (9) | 6 (3) | 1 (1) |
| Fatigue | 46 (25) | 46 (25) | 0 | 0 | 0 |
| Infusion-related reaction* | 38 (21) | 34 (19) | 2 (1) | 2 (1) | 0 |
| Nausea | 23 (13) | 23 (13) | 0 | 0 | 0 |
| Decreased appetite | 13 (7) | 13 (7) | 0 | 0 | 0 |
| Diarrhoea | 13 (7) | 13 (7) | 0 | 0 | 0 |
| Chills | 12 (7) | 12 (7) | 0 | 0 | 0 |
| Hypothyroidism | 11 (6) | 11 (6) | 0 | 0 | 0 |
| Arthralgia | 9 (5) | 9 (5) | 0 | 0 | 0 |
| Vomiting | 9 (5) | 9 (5) | 0 | 0 | 0 |
| Anaemia | 7 (4) | 6 (3) | 1 (1) | 0 | 0 |
| Dyspnoea | 6 (3) | 4 (2) | 1 (1) | 1 (1) | 0 |
| Constipation | 5 (3) | 3 (2) | 2 (1) | 0 | 0 |
| Elevated lipase | 4 (2) | 1 (1) | 3 (2) | 0 | 0 |
| Elevated amylase | 3 (2) | 2 (1) | 0 | 1 (1) | 0 |
| Elevated gamma-glutamyltransferase | 2 (1) | 1 (1) | 1 (1) | 0 | 0 |
| Radiation pneumonitis† | 2 (1) | 1 (1) | 0 | 0 | 1 (1) |
| Anaphylactic reaction | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Autoimmune neutropenia | 1 (1) | 0 | 0 | 1 (1) | 0 |
| Chronic obstructive pulmonary disease | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Elevated transaminases | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Embolic stroke | 1 (1) | 0 | 0 | 1 (1) | 0 |
| Fall | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Hypertension | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Hyponatraemia | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Hypovolaemia | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Hypoxia | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Lung abscess | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Monoplegia | 1 (1) | 0 | 0 | 1 (1) | 0 |
| Musculoskeletal pain | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Pneumonitis | 1 (1) | 0 | 0 | 1 (1) | 0 |
| Syncope | 1 (1) | 0 | 0 | 1 (1) | 0 |
| Systemic inflammatory response syndrome | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Urosepsis | 1 (1) | 0 | 1 (1) | 0 | 0 |
| Treatment-related adverse events [n, (%)] classified as immune-related occurring at any frequency | |||||
| Any event | 22 (12) | 18 (10) | 1 (1) | 2 (1) | 1 (1) |
| Hypothyroidism | 11 (6) | 11 (6) | 0 | 0 | |
| Adrenal insufficiency | 2 (1) | 2 (1) | 0 | 0 | |
| Radiation pneumonitis† | 2 (1) | 1 (1) | 0 | 0 | 1 (1) |
| Arthritis | 1 (1) | 1 (1) | 0 | 0 | 0 |
| Autoimmune neutropenia | 1 (1) | 0 | 0 | 1 (1) | 0 |
| Dry eye | 1 (1) | 1 (1) | 0 | 0 | 0 |
| Iritis | 1 (1) | 1 (1) | 0 | 0 | 0 |
| Pneumonitis | 1 (1) | 0 | 0 | 1 (1) | 0 |
| Psoriasis | 1 (1) | 1 (1) | 0 | 0 | 0 |
| Rheumatoid factor increased | 1 (1) | 1 (1) | 0 | 0 | 0 |
| Systemic inflammatory response syndrome | 1 (1) | 0 | 1 (1) | 0 | 0 |
Signs and symptoms of a potential infusion-related reaction (e.g., fever, chills, rigors) reported on the day of infusion were queried with investigators to ascertain whether an adverse event of “infusion-related reaction” should be recorded.
Grade 5 radiation pneumonitis was reported in one patient, which was later regraded to a grade 3 event of radiation pneumonitis that had not resolved at the time of death. The primary cause of death in this patient was attributed to disease progression.
The incidence of infusion-related reaction was analysed in more detail using a composite definition including three MedDRA preferred terms (infusion-related reaction, drug hypersensitivity, and anaphylactic reaction). Of 39 patients who had an event using this definition, 34 (87%) were grade 1 or 2, and 5 (13%) were grade 3 or 4. Most infusion-related reactions (35 of 39 cases; 90%) occurred during the first or second administration of avelumab (appendix page 2). Of 166 patients who received premedication before at least one dose of avelumab, 26 (16%) had an infusion-related reaction, which reached grade ≥3 in two patients (1%).
Immune-related adverse events of any grade occurred in 36 of 184 patients (20%) and were considered treatment-related by the investigator in 22 patients (12%), of which hypothyroidism (11 [6%]), adrenal insufficiency (2 [1%]), and radiation pneumonitis (2 [1%]) were most common. Four patients (2%) had a grade ≥3 immune-related treatment-related adverse event. Three patients (2%) permanently discontinued avelumab treatment following immune-related events, all of which were considered treatment-related.
Serious adverse events regardless of cause occurred in 80 of 184 patients (44%). Those occurring in more than five patients (≥3%) were dyspnoea (10 [5%]), pneumonia (9 [5%]), and chronic obstructive pulmonary disease (6 [3%]). 16 of 184 patients (9%) had a serious adverse event related to treatment with avelumab, with infusion-related reaction (4 [2%]) and dyspnoea (2 [1%]) occurring in more than one patient, and abdominal pain, anaphylactic reaction, autoimmune neutropenia, chronic obstructive pulmonary disease, embolic stroke, hyponatraemia, hypovolaemia, monoplegia, pneumonitis, pleural effusion, radiation pneumonitis, generalised rash, syncope, systemic inflammatory response syndrome, and urosepsis occurring in one patient each (1%). Of these treatment-related events deemed serious, grade 3 systemic inflammatory response syndrome, grade 4 autoimmune neutropenia, grade 4 pneumonitis, and grade 5 radiation pneumonitis (n=1 patient each) were classified as immune-related. There was one death (1%) that was initially assessed by the investigator as being related to trial treatment and attributed to radiation pneumonitis. The patient had a history of dyspnoea and had received radiation therapy to the chest and right lung 4 months before treatment with avelumab. After further evaluation, the event of radiation pneumonitis was reclassified as a grade 3 event that had not resolved at the time of death, and disease progression was recorded as the primary cause of death.
Of 184 patients, 22 had a confirmed objective response, including one complete response and 21 partial responses, resulting in a confirmed objective response rate of 12% (95% CI, 8–18) (table 3). The unconfirmed objective response rate was 14% (95% CI, 9–20), including one complete response and 25 partial responses. The objective response rate by immune-related response criteria was 12% (95% CI, 8–18). Twenty-six of 184 patients (14%) had a reduction in size of target lesions by ≥30% from baseline (appendix page 3), including four patients not classified as achieving a confirmed response because of lack of response confirmation or new lesion developing. The disease control rate, based on patients who had a confirmed response or stable disease as best overall response (n=92), was 50%. Figure 2 shows change in target lesions over time in evaluable patients. Based on confirmed or unconfirmed responses, 10 of 26 responding patients (39%) had responded by the first assessment at 6 weeks, and 19 of 26 (73%) had responded by 12 weeks (figure 3); median duration of response was not reached, with response durations ranging from 0·1 to ongoing at 54·1 weeks (figure 3). Among 22 patients with a confirmed response, response was maintained for 24 weeks or longer in 83% of cases (95% CI, 54–94) by Kaplan-Meier estimates. Median progression-free survival was 11·6 weeks (95% CI, 8·4–13·7), and progression-free survival rates at 24 weeks and 48 weeks were 26% (95% CI, 20–33) and 18% (95% CI, 12–26), respectively. At the time of analysis, 139 of 184 patients (76%) had an event of progressive disease (116 [63%]) or death (23 [13%]). Based on immune-related response criteria, median progression-free survival was 17·6 weeks (95% CI, 12·1–22·9), and rates at 24 and 48 weeks were 39% (95% CI, 31–46) and 33% (95% CI, 25–42), respectively. Median overall survival was 8·4 months (95% CI, 7·3–10·6), and the survival rate at 12 months was 36% (95% CI, 26–46) (table 3), based on 90 of 184 patients (49%) with an event. Thirty-eight patients (21%) received anticancer therapy after discontinuing avelumab, including drug therapy in 33 patients (18%) and radiotherapy in 16 patients (9%); drugs administered comprised cytotoxic chemotherapy in 28 patients (15%) and targeted therapy in 13 patients (7%).
Table 3.
Clinical activity of avelumab
| (N=184) | |
|---|---|
| Complete response, n (%) | 1 (1) |
| Partial response, n (%) | 21 (11) |
| Stable disease, n (%) | 70 (38) |
| Progressive disease, n (%) | 69 (38) |
| Nonevaluable, n (%)* | 23 (13) |
| Objective response rate (95% CI), % | 12 (8–18) |
| Disease control rate, % | 50 |
| Median progression-free survival (95% CI), weeks | 11·6 (8·4–13·7) |
| Progression-free survival rate at 24 weeks (95% CI), % | 26 (20–33) |
| Progression-free survival rate at 48 weeks (95% CI), % | 18 (12–26) |
| Median overall survival (95% CI), months | 8·4 (7·3–10·6) |
| Overall survival rate at 12 months (95% CI), % | 36 (26–46) |
Response rates are based on confirmed responses.
Patients with missing and/or no assessable information included 19 patients without post-baseline tumour assessments (12 patients died within 6 weeks, one patient had an unevaluable post-baseline target lesion, four patients withdrew consent, and two patients discontinued due to disease progression) and four patients with stable disease who did not meet minimum duration requirement and in which there were no further tumour assessments during available follow-up.
Figure 2. Percent change from baseline in target lesions over time.
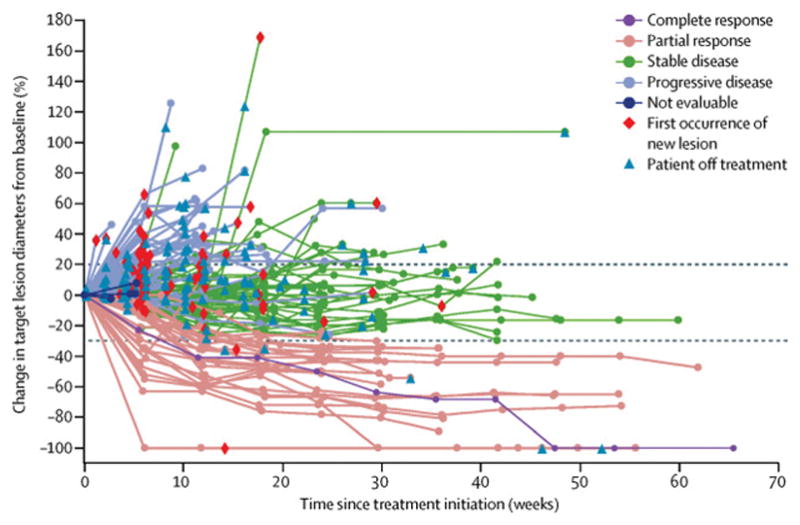
Spider plot of the change in sum of target lesions diameters from baseline over time for all evaluable patients (n=158), defined as those with baseline tumour assessments and at least one post-baseline assessment. Lines are color-coded based on BOR (confirmed and unconfirmed). Horizontal dashed lines represent RECIST v1.1 guideline for partial response (≥30% decrease in target lesion) and progressive disease (≥20% increase in target lesion).
Figure 3. Time to and duration of response in patients with complete response or partial response.
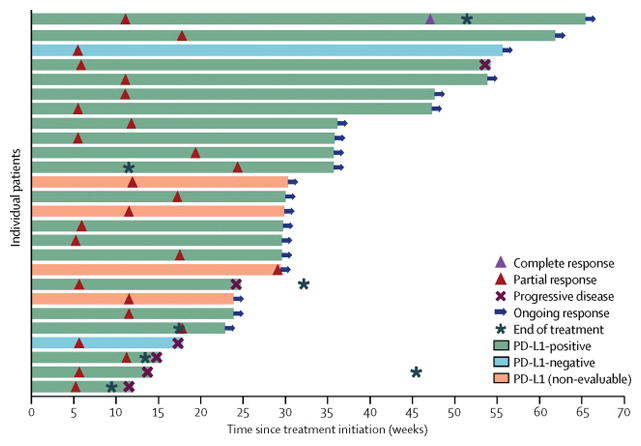
Bars represent individual patients with partial response or complete response (confirmed and unconfirmed) and are color-coded based on PD-L1 status at the ≥1% tumour cell staining cut-off. Responses were ongoing in 20 patients at the time of data cut-off. Two PD-L1–, 20 PD-L1+, and four patients not evaluable for PD-L1 staining achieved a response. NE, not evaluable.
Antitumour activity was seen in tumours defined as PD-L1+ and PD-L1− using prespecified PD-L1 expression levels on tumour cells (1%, 5%, and 25%) and tumour-associated immune cells (table 4). In patients with PD-L1+ vs PD-L1− tumours, there was no significant difference in response rates or OS (table 4), whereas PFS was longer in those patients with PD-L1+ tumours compared with PD-L1− tumours in an analysis based on a 1% cut-off for tumour cell staining (hazard ratio for progressive disease, 0·45 [95% CI, 0·27–0·75]) (figure 4, table 4, appendix pages 4–5).
Table 4.
Confirmed responses, progression-free survival, and overall survival associated with PD-L1 expression
| PD-L1 expression (n=142) | PD-L1+ | PD-L1− | P value* | Hazard ratio (95% CI) |
|---|---|---|---|---|
| ≥1% tumour cells, any intensity | ||||
| Prevalence, n | 122 | 20 | — | — |
| Objective response rate, n/N1 (% [95% CI]) | 17/122 (14 [8–21]) | 2/20 (10 [1–32]) | >0·99 | — |
| Median progression-free survival (95% CI), weeks | 12·0 (10·4–17·7) | 5·9 (5·6–7·1) | — | — |
| Progression-free survival rate at 48 weeks (95% CI), % | 21 (13–30) | 5 (0–21) | — | 0·45 (0·27–0·75) |
| Median overall survival (95% CI), months | 8·9 (8·0–NE) | 4·6 (2·8–NE) | — | — |
| Overall survival rate at 12 months (95% CI), % | 39 (26–52) | 36 (14–58) | — | 0·64 (0·34–1·20) |
| ≥5% tumour cells, any intensity | ||||
| Prevalence, n | 84 | 58 | — | — |
| Objective response rate, n/N1 (% [95% CI]) | 12/84 (14 [8–24]) | 7/58 (12 [5–23]) | 0·81 | — |
| Median progression-free survival (95% CI), weeks | 11·9 (6·7–18·3) | 7·8 (6·0–12·0) | — | — |
| Progression-free survival rate at 48 weeks (95% CI), % | 25 (16–36) | 10 (3–21) | — | 0·70 (0·48–1·02) |
| Median overall survival (95% CI), months | 10·6 (7·9–NE) | 8·4 (5·6–NE) | — | — |
| Overall survival rate at 12 months (95% CI), % | 39 (22–55) | 37 (22–52) | — | 0·79 (0·48–1·28) |
| ≥25% tumour cells, moderate-to-high intensity | ||||
| Prevalence, n | 53 | 89 | — | — |
| Objective response rate, n/N1 (% [95% CI]) | 9/53 (17 [8–30]) | 10/89 (11 [6–20]) | 0·45 | — |
| Median progression-free survival (95% CI), weeks | 11·9 (6·1–22·9) | 10·79 (6·0–13·7) | — | — |
| Progression-free survival rate at 48 weeks (95% CI), % | 28 (16–41) | 15 (8–24) | — | 0·79 (0·53–1·18) |
| Median overall survival (95% CI), months | 8·44 (6·0–11·1) | 8·57 (7·16–NE) | — | — |
| Overall survival rate at 12 months (95% CI), % | 28 (12–47) | 48 (35–60) | — | 1·14 (0·70–1·85) |
| ≥10% tumour-associated immune cells in hotspots, any intensity | ||||
| Prevalence, n | 27 | 115 | — | — |
| Objective response rate, n/N1 (% [95% CI]) | 4/27 (15 [4–34]) | 15/115 (13 [8–21]) | 0·76 | — |
| Median progression-free survival (95% CI), weeks | 8·4 (5·7–15·1) | 11·3 (6·7–15·3) | — | — |
| Progression-free survival rate at 48 weeks (95% CI), % | NE | 19 (11–28) | — | 1·19 (0·74–1·92) |
| Median overall survival (95% CI), months | 8·5 (3·9–NE) | 8·9 (7·9–NE) | — | — |
| Overall survival rate at 12 months (95% CI), % | 30 (10–53) | 41 (28–54) | — | 1·20 (0·68–2·14) |
N1, number of patients with tumours evaluable for PD-L1 expression; NE, not estimable; PD-L1, programmed death ligand 1.
Fisher exact test; P values were calculated based on the binary comparison of positive vs negative.
Figure 4.
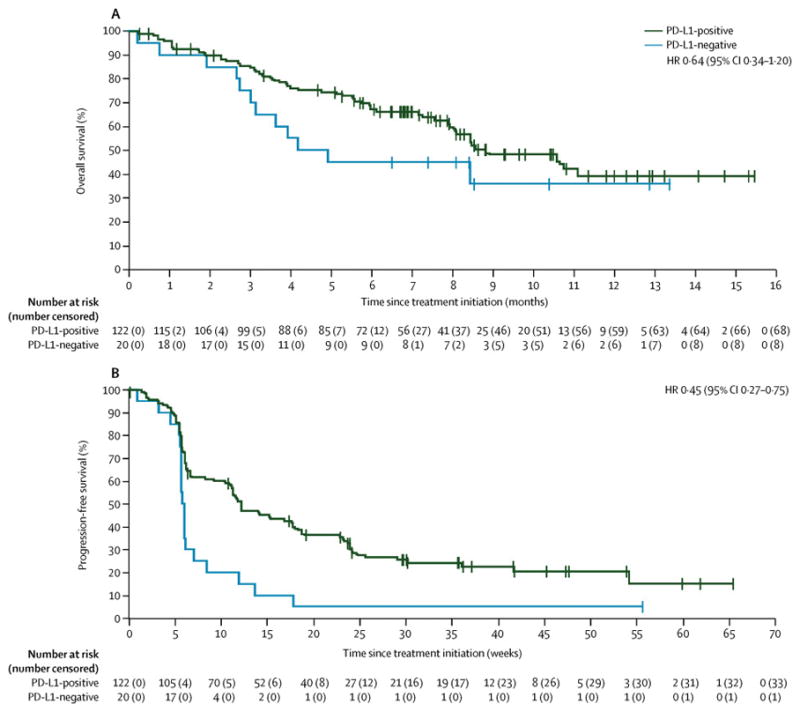
Kaplan-Meier estimates of progression-free survival (A) and overall survival (B) by PD-L1 expression status (≥1% tumour cell cut-off)
Responses were observed independent of patient and disease characteristics, including tumour histology, prior lines of therapy, and smoking status. There were no responses among the few patients with known EGFR-mutant (n=9) or ALK translocation–positive (n=1) tumours, and one responder among 21 patients with known KRAS-mutant tumours (table 5, appendix pages 14–15).
Table 5.
Responses, progression-free survival, and overall survival in select subgroups
| (N=184) | Confirmed response by RECIST v1·1, n (% [95% CI]) | Median PFS (95% CI), weeks | Median OS (95% CI), months |
|---|---|---|---|
|
| |||
| Age categorization, years | |||
| <65 (n=90) | 12 (13 [7–22]) | 11·9 (11·1–15·3) | 8·1 (6·0–NE) |
| ≥65 (n=94) | 10 (11 [5–19]) | 8·7 (6·1–14·3) | 8·4 (6·4–10·7) |
|
| |||
| Sex | |||
| Male (n=100) | 8 (8 [4–15]) | 11·7 (6·4–17·6) | 8·4 (6·0–10·7) |
| Female (n=84) | 14 (17 [9–26]) | 11·6 (6·1–15·1) | 8·4 (7·3–ne) |
|
| |||
| ECOG PS | |||
| 0 (n=55) | 7 (13 [5–25]) | 11·3 (6·1–16·9) | 11·1 (8·5–NE) |
| ≥1 (n=129) | 15 (12 [7–19]) | 11·6 (8·4–14·3) | 7·6 (5·7–8·4) |
|
| |||
| Histology | |||
| Adenocarcinoma (n=114) | 12 (11 [6–18]) | 10·9 (6·1–12·0) | 8·5 (7·6–NE) |
| Squamous cell carcinoma (n=53) | 7 (13 [6–25]) | 12·0 (6·4–23·9) | 7·3 (5·6–NE) |
| Other (n=17) | 3 (18 [4–43]) | 15·3 (5·6–NE) | 8·4 (5·0–NE) |
|
| |||
| Smoking history | |||
| Never smoked (n=24) | 3 (13 [3–32]) | 12·1 (5·9–21·9) | 8·1 (4·0–NE) |
| Ever smoked (n=159) | 19 (12 [7–18]) | 11·6 (7·1–12·1) | 8·4 (7·6–10·7) |
|
| |||
| EGFR mutation status | |||
| Wildtype (n=101) | 10 (10 [5–18]) | 11·7 (6·3–14·3) | 8·6 (7·6–NE) |
| Mutant (n=9) | 0 (0 [0–34]) | 5·4 (1·9–24·0) | 3·0 (1·1–NE) |
| Unknown (n=74) | 12 (16 [9–27]) | 11·9 (6·7–18·1) | 8·1 (6·5–10·6) |
|
| |||
| KRAS mutation status | |||
| Wildtype (n=38) | 3 (8 [2–21]) | 11·5 (6·0–18·0) | NE (5·6–NE) |
| Mutant (n=21) | 1 (5 [0–24]) | 6·1 (5·4–12·1) | 8·1 (3·7–10·7) |
| Unknown (n=125) | 18 (14 [9–22]) | 11·9 (10·4–17·6) | 8·1 (6·5–10·6) |
|
| |||
| ALK translocation status | |||
| Negative (n=103) | 10 (10 [5–17]) | 11·3 (6·1–12·0) | 8·4 (6·0–NE) |
| Positive (n=1) | 0 (0 [0–98]) | 17·6 (NE–NE) | NE (NE–NE) |
| Unknown (n=80) | 12 (15 [8–25]) | 12·0 (9·1–18·9) | 8·4 (7·3–10·7) |
|
| |||
| No. of prior anticancer lines for advanced disease | |||
| ≤1 (n=123) | 14 (11 [6–18]) | 11·9 (8·4–14·3) | 8·4 (7·3–11·1) |
| 2 (n=44) | 6 (14 [5–27]) | 11·3 (6·0–18·1) | 8·6 (4·2–NE) |
| ≥3 (n=17) | 2 (12 [2–36]) | 11·6 (4·1–54·1) | 8·1 (1·1–NE) |
NE, not estimable; OS, overall survival; PFS, progression-free survival; RECIST, Response Evaluation Criteria In Solid Tumors.
Discussion
In this large cohort of patients with metastatic NSCLC that had progressed following chemotherapy with a platinum-containing doublet, avelumab monotherapy showed antitumour activity and an acceptable safety profile. 22 of 184 patients (12%) achieved confirmed responses, which generally occurred early and were durable, and an additional 70 patients (38%) had stable disease. The time to response observed with avelumab (responses were seen by first assessment at 6 weeks in 39% of responders and by 12 weeks in 73% of responders) is comparable to median time to response reported for chemotherapy administered in the second-line setting5; assessment of early responses was enabled by the prespecified schedule for radiologic tumour assessments. Subgroup analyses conducted post hoc were generally consistent with overall findings and included responses in tumours with squamous and non-squamous histology. Recently, response rates of 15%–21% have been reported in the literature for patients treated with other anti–PD-L1/PD-1 monotherapy in the second-line setting, although data cannot be compared directly because of differences in study designs and eligibility criteria, including reporting of unconfirmed responses and differences in eligibility based on prior treatment (agents administered and number of prior lines) or PD-L1 status using different assays.16,25–28 In addition, head-to-head comparisons of the various anti–PD-L1/PD-1 antibodies in randomised trials have not been conducted. Thus, the data reported here do not support more detailed conclusions about the efficacy of avelumab compared with other agents in the same therapeutic class. One-year progression-free and overall survival rates with avelumab were 18% and 36%, respectively. Based on immune-related response criteria, the 1-year progression-free survival rate with avelumab was 33%. These data support previous suggestions that the standard endpoints measured by RECIST may underestimate the benefit of treatment with anti–PD-L1/PD-1 antibodies.16,29
In some tumour types, a trend of better response rates and improved progression-free and overall survival with anti–PD-L1/PD-1 treatment has been associated with PD-L1+ tumour cells or tumour-associated lymphocytes.15,16,25,30. In our study, patients with PD-L1+ tumour cells using the 1% cut-off had longer progression-free survival with avelumab than patients with PD-L1− tumours, based on tumour classification using the novel antibody clone (73–10), although this trend should be interpreted with caution. Work is ongoing to further investigate the use of PD-L1 as a potential biomarker for avelumab in NSCLC and other tumour types. Direct comparisons of clinical activity based on PD-L1 status with different agents is hampered by differences in PD-L1 assays in terms of sensitivity and staining properties between antibody clones, criteria for evaluating PD-L1 expression on tumour cell membranes, stroma, and/or immune cells within the tumour microenvironment, testing platforms, and scoring algorithms. PD-L1 positivity was defined in the current study at three prespecified levels for frequency and intensity of staining on tumour cell membranes and one prespecified level on tumour-associated immune cells. At the staining cut-offs defined for this study, the proportion of tumours expressing PD-L1 was higher than levels reported by other investigators using different assays.15,16,25 However, preliminary data using commercially procured NSCLC samples suggest that our assay is highly sensitive across a broad dynamic range. These data will be reported elsewhere. Efforts to develop standardised approaches to PD-L1 expression diagnostics are underway, including the BLUEPRINT proposal initiated by the Food and Drug Administration, American Association for Cancer Research, American Society of Clinical Oncology, and the International Association for the Study of Lung Cancer.31,32 No data have been reported to date comparing the novel PD-L1 assay used in this study with other assays, although studies are ongoing and will be published in the future. Clinical data on other potential predictive biomarkers of response to PD-L1/PD-1 antibodies have also been reported, including an association between longer overall survival with atezolizumab (anti–PD-L1), and pre-existing immunity based on high T-effector-interferon-γ-associated gene expression.16 Exploratory analyses of potential correlates of response to avelumab are ongoing, including analyses of cytokines, CD8+ T cells in tumour specimens, pharmacokinetic parameters, and expanded scoring algorithms for PD-L1 expression.
Rates of adverse events related to avelumab treatment were low and generally consistent with those reported for other anti–PD-L1/PD-1 agents.9,13–18,25,26 One patient (1%) experienced treatment-related pneumonitis (grade 4); in studies of other anti–PD-L1/PD-1 antibodies administered as monotherapy in patients with metastatic NSCLC, rates of treatment-related pneumonitis were 1%–5% for all grades and 1%–3% for grade ≥3 events.13,14,25,26 Immune-related adverse events occurred in 12% of patients, were mostly grade 1 or 2, and led to treatment discontinuation in 2% of patients. In this study, infusion-related reaction was monitored as an adverse event of special interest and occurred in 21% of patients. However, the vast majority of infusion-related reactions with avelumab were mild to moderate in severity, occurred after the first or second infusion, and did not lead to treatment discontinuation. The frequency and management of infusion-related reactions will be further characterised in ongoing clinical trials of avelumab.
Enrolment in this cohort exceeded our prespecified target of 150 patients because of an acceleration in investigator recruitment towards the end of the enrolment process. As a result, a relatively large number of patients were screened and signed informed consent forms within a short period. We decided to include all patients who met eligibility criteria rather than exclude these patients once the planned total had been exceeded.
Preclinical studies suggest that avelumab can mediate tumour lysis through ADCC, which is in contrast to other anti–PD-L1/PD-1 antibodies that are based on a nonactive IgG subtype (IgG4) or contain engineered mutations in the Fc region designed to exclude ADCC.19,33 Although ADCC could theoretically provide a secondary mechanism of action for avelumab in addition to PD-L1/PD-1 blockade, there are currently no clinical data to demonstrate whether ADCC contributes to the clinical activity of avelumab. However, clinical studies have shown that avelumab treatment does not lead to any decrease in the frequency of various circulating immune cell subsets, suggesting that avelumab does not have ADCC activity against PD-L1+ immune cells.21,33
In conclusion, the results of this study show that avelumab has promising efficacy and safety in patients with NSCLC that progressed after platinum doublet therapy. Although interpretation of these results is limited by the early-phase and single-arm study design, our findings provided the rationale for an ongoing phase 3 head-to-head trial of avelumab vs docetaxel in patients with recurrent NSCLC, in which patients are stratified according to PD-L1 expression status and NSCLC histology (NCT02395172). Studies of avelumab monotherapy in first-line NSCLC are also ongoing, including an expansion cohort in the JAVELIN Solid Tumor phase 1 trial and a randomised phase 3 trial comparing avelumab with platinum-based doublet therapy in patients with squamous and non-squamous PD-L1+ NSCLC (NCT02576574).
Supplementary Material
Research in Context.
Evidence before this study
Because the field of immunotherapy with anti–PD-L1/PD-1 immune checkpoint inhibitors has expanded rapidly in recent years, we conducted a 5-year systematic search of the literature using both PubMed and selected annual congresses (American Society of Clinical Oncology, European Society for Medical Oncology, European Cancer Congress, and World Conference on Lung Cancer). Search dates were 1 January 2011 to 22 August 2016, and terms included “PD-1” and “PD-L1,” in addition to relevant generic and investigational drug names of immune checkpoint inhibitors (“nivolumab,” “BMS-936558,” “pembrolizumab,” “lambrolizumab,” “MK-3475,” “atezolizumab,” “MPDL3280A,” “durvalumab,” “MEDI4736,” “avelumab,” and “MSB0010718A”). Most clinical studies were published after 2013. Additional search terms were used to identify literature on patients with advanced non-small cell lung cancer (NSCLC). Patients with recurrent or metastatic NSCLC have limited therapeutic options, and recent literature reporting the clinical activity and acceptable safety profile of anti–PD-L1/PD-1 monoclonal antibodies supports the potential of these agents to address a significant unmet need. In addition, correlative and translational studies in NSCLC and other tumour types suggest that patient selection and stratification, based on predictive or prognostic biomarkers, tumour histology, and PD-L1 expression, are important characteristics to consider in clinical study design and prespecified subgroup analyses. Recent evidence from phase 2 and phase 3 trials indicates that anti–PD-L1/PD-1 antibodies have efficacy and are well tolerated in patients with squamous and non-squamous histological subtypes. Tumour PD-L1 expression may have predictive value for efficacy of anti–PD-1 antibodies in non-squamous NSCLC. Avelumab is a fully human anti–PD-L1 IgG1 antibody that has shown promising antitumour activity and a manageable safety profile in multiple tumour types. Unlike other anti–PD-L1/PD-1 antibodies approved or in advanced clinical development, which are based on an IgG4 subtype or contain engineered mutations in their Fc region, avelumab has been shown to mediate antibody-dependent cellular cytotoxicity in vitro. Based on safety, pharmacokinetic, target occupancy, and preliminary efficacy data obtained during a dose-escalation study of avelumab, the dose of 10 mg/kg administered as a 1-hour intravenous infusion every 2 weeks was selected for phase 1 dose expansion in various tumours.
Added value of this study
We report antitumour activity of avelumab monotherapy in a large phase 1 cohort of patients with progressive or platinum-resistant metastatic or recurrent NSCLC. Eligible patients in this cohort were not preselected based on NSCLC histology, PD-L1 expression status, or EGFR or ALK status. Clinical activity was recorded independent of PD-L1 expression, or squamous vs non-squamous histology. This is the largest cohort of patients studied in a phase 1 trial of avelumab and further characterises its clinical attributes.
Implications of all the available evidence
Avelumab had promising clinical activity and a manageable safety profile in a population of patients with progressive, platinum-treated, metastatic or recurrent NSCLC. Responses occurred irrespective of PD-L1 expression status and in squamous and non-squamous tumours. These findings support the therapeutic benefit of anti–PD-L1 antibodies in patients with previously treated NSCLC. These results with avelumab have provided the rationale for an ongoing phase 3 trial in the second-line NSCLC population and underscore the potential benefits of immunotherapy for patients with this difficult-to-treat disease.
Acknowledgments
The authors would like to thank the patients and their families, the investigators, co-investigators, and the study teams at each of the participating centres and at Merck KGaA, Darmstadt, Germany, EMD Serono Research & Development Institute, Billerica, MA, USA (a business of Merck KGaA, Darmstadt, Germany), and Quintiles, Inc., Durham, NC, USA. This trial was sponsored by Merck KGaA, Darmstadt, Germany and is part of an alliance between Merck KGaA and Pfizer, Inc (New York, NY). Medical writing support was provided by ClinicalThinking, Inc, Hamilton, NJ and funded by Merck KGaA, Darmstadt, Germany and Pfizer, Inc.
Footnotes
Contributions to the manuscript:
J.L.G., A.R., N.I., D.W., H.J.G., A.vH., K.C., and J-M.C. conceived and designed the study. J.L.G., A.R., D.R.S. N.I., J.C., D.J.L.W., J.L., W.J.E., D.W., H.J.G., A.vH., K.C., J-M.C., and K.K. collected and assembled the data. All authors analysed and interpreted the data, wrote the manuscript, and approved the final version of the manuscript.
Conflict of Interest Statement:
D.J.L.W. reports research grant funding from Merck Serono. J.L. is an employee at PRA Health Sciences. J.L. reports institutional research funding from EMD Serono, relevant to the submitted work. J.L. reports institutional research funding from AstraZeneca, Genentech, and Clovis, outside the submitted work. W.J.E. reports participation in Speaker’s Bureau’s for Astellas and Novartis, outside the submitted work. H.J.G. and A.vH. are employees at Merck KGaA. A.vH. is a stockholder at Merck KGaA. K.C. is an employee at EMD Serono. J-M.C. was an employee of Merck KGaA. J-M.C. is an employee and stockholder at Angenus Bio. All other authors declare no competing interests.
References
- 1.Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Dicker D, et al. The global burden of cancer 2013. JAMA Oncol. 2015;1:505–27. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dela Cruz CS, Tanoue LT, Matthay RA. Lung cancer: Epidemiology, etiology, and prevention. Clin Chest Med. 2011;32:605–44. doi: 10.1016/j.ccm.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–8. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 4.Fossella F, Pereira JR, von Pawel J, et al. Randomized, multinational, phase III study of docetaxel plus platinum combinations versus vinorelbine plus cisplatin for advanced non-small-cell lung cancer: the TAX 326 study group. J Clin Oncol. 2003;21:3016–24. doi: 10.1200/JCO.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 5.Hanna N, Shepherd FA, Fossella FV, et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol. 2004;22:1589–97. doi: 10.1200/JCO.2004.08.163. [DOI] [PubMed] [Google Scholar]
- 6.Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357–60. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isozaki H, Takigawa N, Kiura K. Mechanisms of acquired resistance to ALK inhibitors and the rationale for treating ALK-positive lung cancer. Cancers (Basel) 2015;7:763–83. doi: 10.3390/cancers7020763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 9.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirano F, Kaneko K, Tamura H, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–96. [PubMed] [Google Scholar]
- 12.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–50. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 16.Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–46. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 17.Gettinger SN, Horn L, Gandhi L, et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2015;33:2004–12. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antonia S, Goldberg SB, Balmanoukian A, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17:299–308. doi: 10.1016/S1470-2045(15)00544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyerinas B, Jochems C, Fantini M, et al. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res. 2015;3:1148–57. doi: 10.1158/2326-6066.CIR-15-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujii R, Friedman ER, Richards J, et al. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget. 2016;7:33498–511. doi: 10.18632/oncotarget.9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heery CR, O’Sullivan Coyne G, Madan RA, et al. Phase I open-label, multiple ascending dose trial of MSB0010718C, an anti-PD-L1 monoclonal antibody, in advanced solid malignancies. Proc Am Soc Clin Oncol. 2014;32(suppl 5S) abstr 3064. [Google Scholar]
- 22.Heery CR, O’Sullivan Coyne G, Marte JL, et al. Pharmacokinetic profile and receptor occupancy of avelumab (MSB00110718C), an anti-PD-L1 monoclonal antibody, in a phase I, open-label, dose escalation trial in patients with advanced solid tumors. Proc Am Soc Clin Oncol. 2015;33(suppl) abstr 3055. [Google Scholar]
- 23.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 24.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 25.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 26.Rizvi NA, Mazieres J, Planchard D, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16:257–65. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rizvi NA, Brahmer JR, Ou S-I, et al. Safety and clinical activity of MEDI4736, an anti-programmed cell death-ligand (PD-L1) antibody, in patients with non-small cell lung cancer (NSCLC) Proc Am Soc Clin Oncol. 2015;33(suppl 5S) abstr 8032. [Google Scholar]
- 28.Horn L, Spigel DR, Gettinger SN, et al. Clinical activity, safety, and predictive biomarkers of the engineered antibody MPDL3280A (anti-PDL1) in non-small cell lung cancer (NSCLC): update from a phase Ia study. Proc Am Soc Clin Oncol. 2015;33(suppl 5S) abstr 8029. [Google Scholar]
- 29.Pilotto S, Carbognin L, Karachaliou N, et al. Moving towards a customized approach for drug development: Lessons from clinical trials with immune checkpoint inhibitors in lung cancer. Transl Lung Cancer Res. 2015;4:704–12. doi: 10.3978/j.issn.2218-6751.2015.10.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirsch FR, Suda K, Wiens J, Bunn PA., Jr New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet. 2016;388:1012–24. doi: 10.1016/S0140-6736(16)31473-8. [DOI] [PubMed] [Google Scholar]
- 32.Scheel AH, Dietel M, Heukamp LC, et al. Harmonized PD-L1 immunohistochemistry for pulmonary squamous-cell and adenocarcinomas. Mod Pathol. 2016;29:1165–72. doi: 10.1038/modpathol.2016.117. [DOI] [PubMed] [Google Scholar]
- 33.Grenga I, Donahue RN, Lepone LM, Richards J, Schlom J. A fully human IgG1 anti-PD-L1 MAb in an in vitro assay enhances antigen-specific T-cell responses. Clin Transl Immunolog. 2016;5:e83. doi: 10.1038/cti.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.