

Abstract
Approximately 1% of the global population suffers from epilepsy, a class of disorders characterized by recurrent and unpredictable seizures. Of these cases roughly one-third are refractory to current antiepileptic drugs, which typically target neuronal excitability directly. The events leading to seizure generation and epileptogenesis remain largely unknown, hindering development of new treatments. Some recent experimental models of epilepsy have provided compelling evidence that glial cells, especially astrocytes, could be central to seizure development. One of the proposed mechanisms for astrocyte involvement in seizures is astrocyte swelling, which may promote pathological neuronal firing and synchrony through reduction of the extracellular space and elevated glutamate concentrations. In this review, we discuss the common conditions under which astrocytes swell, the resultant effects on neural excitability, and how seizure development may ultimately be influenced by these effects.
Keywords: Cellular edema, glia, NMDA receptors, extracellular space, VRAC
1. Introduction: Epilepsy models and astrocytes
Epilepsy is one of the most common neurological disorders in the world, affecting roughly 65 million people worldwide (Hirtz et al. 2007; Ngugi et al. 2010) and encompassing over 40 different seizure disorders (Berg et al. 2010). Current antiepileptic drugs (AEDs) are inadequate, as most produce negative side effects on cognition (Aldenkamp 2001; Aldenkamp et al. 2003; Lagae 2006). Moreover, AEDs fail to control seizures in approximately 30% of patients (Brodie et al. 2012; Brodie and Dichter 1996; Kwan and Brodie 2000), increasing mortality (Devinsky 2004a; Laxer et al. 2014; Park et al. 2015) and risk of comorbid neurological disorders (Boylan et al. 2004; Devinsky 2004b; Kwan and Brodie 2001). Research on drug-resistant forms of epilepsy, such as temporal lobe epilepsy (TLE), has historically focused almost exclusively on acute and/or long-term changes in neuronal firing and excitability. This approach, however, does not account for non-neuronal cells, which comprise ~50% of the brain (Azevedo et al. 2009). A growing body of work suggests that astrocytes, microglia and other glial cell types may play essential roles in the pathology of epilepsy.
Many recently developed epilepsy models have been built upon manipulations of glial cells, especially astrocytes. Astrocytes are a particularly attractive target for epilepsy given their control over neuronal excitability (Hubbard et al. 2013; Wetherington et al. 2008). Key changes contributing to hyperexcitability in epileptic tissue can be directly linked to changes in astrocyte function. For example, extracellular glutamate levels are excessively high in epileptic tissue (Cavus et al. 2005; During and Spencer 1993), resulting at least in part from impaired astrocytic glutamate metabolism (Eid et al. 2004) and possibly glutamate transport (Proper et al. 2002). Broberg and colleagues (2008) found that seizures could be reliably induced simply by focal intracerebral injection of the astrocyte metabolic inhibitor fluorocitrate (FC). Similarly, the astrocytic enzyme glutamine synthetase (which recycles glutamate into glutamine for transport back to neurons) has been targeted with methionine sulfoximine (MSO) infusions into the hippocampus, producing acute status epilepticus (SE, a prolonged seizure) and gradual development of spontaneous recurrent seizures (SRS) (Albright et al. 2016; Eid et al. 2008; Wang et al. 2009). Serum albumin, which can sometimes enter the brain through a compromised blood brain barrier (BBB) in epileptic tissue, was found to directly bind astrocytic TGF-β receptors (producing reactive gliosis) and was sufficient to induce seizures in rats (Ivens et al. 2007; Seiffert et al. 2004). On the other hand, genetic deletion of β-integrin from astrocytes, which produces reactive gliosis without BBB disruption (Robel et al. 2009) was also sufficient to produce SRS within the first six weeks of life (Robel et al. 2015), indicating reactive gliosis alone may be sufficient for epileptogenesis. These and other findings suggest that astrocytes may be actively involved in the development of recurrent seizures.
Although there are a plethora of different ions, molecules and even cell types which can be altered to induce epilepsy, most (if not all) share a common factor: they produce (or are capable of producing) astrocyte swelling. In the following sections, we will discuss the evidence pointing to a significant role for astrocyte swelling in the development and maintenance of seizures, including the specific observations of tissue volume in seizures, physiological and pathological factors which affect astrocyte volume, and finally the consequences of astrocyte swelling in the context of hyperexcitable tissue.
2. Tissue swelling and seizures
Space within the brain is both limited and tightly regulated, and neuronal function depends upon the careful balancing of water, ions and neurotransmitter concentrations in the extracellular space (ECS). Accordingly, losing this balance can be a medical emergency. Acute plasma hypoosmolality, often due to low serum Na+ levels (hyponatremia) has been known for nearly a century to cause direct swelling of the brain, muscle spasms, generalized seizures, coma or even death (Andrew 1991; Castilla-Guerra et al. 2006; Rowntree 1926). The effects of plasma hypoosmolality are most directly induced by accidental overhydration, as might occur in psychogenic polydipsia (compulsive water drinking, a symptom occurring in some cases of schizophrenia), or rapid water intake following dehydration. Multiple disorders which can acutely reduce plasma osmolality are also associated with seizures, including syndrome of inappropriate ADH secretion (SIADH), dialysis disequilibrium syndrome, transurethral resection of the prostate (TURP) syndrome and diabetes mellitus (Andrew 1991). Ironically, certain antiepileptic drugs may even result in hyponatremia, although generally asymptomatic (Berghuis et al. 2016; Kim et al. 2014; Lu and Wang 2017; Shepshelovich et al. 2017). Reductions of brain interstitial osmolarity can lead to “cellular” edema (occasionally sub-classified as “osmotic” edema), as water flows into neural cells causing them to swell (Kimelberg 1995; Thrane et al. 2014). This in turn shrinks the ECS (Andrew and MacVicar 1994; Chebabo et al. 1995b; Kilb et al. 2006) which is a critical regulator of neuronal excitability. In addition to increasing effective concentrations of extracellular ions and neurotransmitters, ECS reductions bring neurons closer together and increase nonsynaptic, neuron-to-neuron electrical field (ephaptic) interactions, resulting in more synchronous firing and bursting activity (Andrew et al. 1989; Ballyk et al. 1991; Dudek et al. 1986). Unsurprisingly, neuronal excitability is highly sensitive to extracellular shifts in osmolarity (Azouz et al. 1997; Chebabo et al. 1995a; Huang et al. 1997; Lauderdale et al. 2015), and neurons become more susceptible to seizure in hypoosmolar conditions. Multiple studies have demonstrated that seizures and other epileptiform activity can either be induced by lowering extracellular osmolarity, or abolished by increasing extracellular osmolarity (Dudek et al. 1990; Kilb et al. 2006; Roper et al. 1992; Rosen and Andrew 1990; Saly and Andrew 1993; Traynelis and Dingledine 1989), particularly in regions such as the hippocampal CA1 where the ECS is smaller at baseline (McBain et al. 1990). Similarly, a recent study has found that genetic knockdown of the extracellular matrix glycosaminoglycan Hyaluronan (HA) leads to a 40% ECS reduction within the CA1 stratum pyramidale and results in both ictal (seizure, or seizure-like) and interictal (inter-seizure) epileptiform activity. The epileptiform activity is subsequently abolished in hyperosmolar conditions known to cause cell shrinkage and increase the volume of the ECS (Arranz et al. 2014). Indeed, pre-seizure ECS constriction has been observed in multiple models of epilepsy (Binder et al. 2004b; Broberg et al. 2008; Traynelis and Dingledine 1989). Tissue swelling may therefore represent both an important treatment target, and an important early warning sign, for seizures (Binder and Haut 2013).
2.1. Water channels and brain volume
In general, brain water content is thought to be controlled by astrocytes due to their selective expression of the water channel aquaporin-4 (AQP4) (Nagelhus et al. 2004; Nielsen et al. 1997). AQP4 expression is particularly enriched at astrocytic endfeet adjoining cerebral vasculature, providing tight control of water fluxes both into and out of the brain (Amiry-Moghaddam et al. 2003a; Nagelhus et al. 2004; Nielsen et al. 1997). Consequently, the role of AQP4 in neurological disease reflects the role of water movements in said disease. For example, AQP4-dependent water movement is essential for recovery from vasogenic edema (in which plasma ultrafiltrate leaks into and accumulates in the ECS), since clearance of the edema fluid requires water flow through astrocytes back into capillaries. The same water permeability can exacerbate cellular/cytotoxic (intracellular) edema, making AQP4 a liability in cases of stroke (Katada et al. 2014), liver failure (Rama Rao et al. 2014) or other disorders which cause cell swelling in the brain (Papadopoulos and Verkman 2007). Genetic deletion studies have provided evidence that AQP4 may be more than a simple, passive water channel, and is most likely not the sole mechanism for astrocyte water permeability. In one study, knockout of the anchoring protein α-syntrophin, which removes perivascular AQP4 (Amiry-Moghaddam et al. 2004), significantly inhibited swelling in severe (~33%) hypoosmolar conditions, but had no effect on swelling in mild (~17%) hypoosmolar conditions (Anderova et al. 2014). With a full knockout of AQP4−/−, another study found nearly the opposite effect; little or no change in volume in 30% hACSF, but significant reduction in volume at 20% hACSF (Thrane et al. 2011).
During synaptic transmission, AQP4 may be important for the activity-dependent fluxes of water which occur alongside potassium uptake and buffering in astrocytes. Increases in extracellular potassium, whether activity-induced or bath-applied, cause astrocyte swelling and a reduction in size of the ECS (Andrew and MacVicar 1994; Dietzel et al. 1980; Holthoff and Witte 1996; Risher et al. 2009; Walz and Hinks 1985). Loss or mislocalization of AQP4 channels is reported to impair astrocyte [K+]o clearance and buffering (Amiry-Moghaddam et al. 2003b; Binder et al. 2006; Eid et al. 2005; Haj-Yasein et al. 2015; Strohschein et al. 2011). Suppression of activity-dependent cell swelling has also been reported in AQP4−/− mice (Kitaura et al. 2009), but these data are inconsistent with more recent work in which AQP4 deletion led to an increase in activity-induced tissue swelling (Haj-Yasein et al. 2012). These disparate findings may result from regional astrocyte heterogeneity or methodological differences, but they also suggest that the precise role of AQP4 in activity-driven glial swelling may be more complex than initially anticipated.
In contrast to astrocytes, most CNS neurons do not express any known functional water channels (Papadopoulos and Verkman 2013) and are considered resistant to osmotically-driven volume changes (Andrew et al. 2007; Caspi et al. 2009). This is perhaps due to their functional roles in synaptic transmission as opposed to clearance of neurotransmitters and ions. This does not imply, however, that neuronal volume is static. For example, neuronal somata will rapidly swell in cases of excitotoxic damage, a common occurrence in stroke or traumatic brain injury (Andrew et al. 2007; Choi 1992; Liang et al. 2007; Risher et al. 2009). Excessive entry of Na+ (often as a result of overactive ionotropic glutamate receptors) depolarizes neurons and opens a voltage-gated Cl− channel. Both ions increase intracellular osmolarity, drawing water into and swelling the neuron (Choi 1992; Lee et al. 1999; Rungta et al. 2015). In light of these data, the reported lack of neuronal sensitivity to hypoosmolar conditions in some studies (Andrew et al. 2007; Caspi et al. 2009) is rather curious, since there is little reason to believe that osmotic swelling of neurons would occur exclusively in excitotoxic conditions. In fact, several studies of neurons in culture have reported varying degrees of neuronal swelling in hypoosmolar conditions (Aitken et al. 1998; Borgdorff et al. 2000; Boss et al. 2013; Somjen 1999). Disparate conditions between culture vs. intact tissue settings may in part account for these differences.
2.2. Potassium uptake and glial swelling
Three general mechanisms contribute to potassium uptake and redistribution (“spatial buffering”) by astrocytes: The Na+/K+-ATPase (NKA), the inwardly-rectifying K+ channel 4.1 (Kir4.1), and the Na+/K+/2Cl− cotransporter 1 (NKCC1). The individual contributions of each to basal [K+]o regulation, K+ uptake and activity-dependent astrocyte swelling have been debated for some time (Kofuji and Newman 2004; Larsen et al. 2014; Macaulay and Zeuthen 2012; Walz 2000). Many authors have found evidence that the astrocytic NKA isoforms are partially (if not predominantly) responsible for the rapid clearance of extracellular K+ which enters the extracellular space following neuronal firing (D’Ambrosio et al. 2002; Larsen et al. 2014; Ransom et al. 2000; Xiong and Stringer 2000). NKA is also required to maintain physiologically low [K+]o concentrations (D’Ambrosio et al. 2002; Larsen et al. 2014; Ransom et al. 2000), which is in line with the traditional role NKA plays in establishing transmembrane gradients of Na+ and K+. NKCC1 may supplement K+ uptake under conditions of especially high [K+]o (Larsen et al. 2014; Walz and Hertz 1982; Walz and Hertz 1984). Kir4.1, the most abundant K+ channel expressed by astrocytes, is generally considered essential for the passive K+ conductance in astrocytes (Chever et al. 2010; Meeks and Mennerick 2007; Neusch et al. 2006; Seifert et al. 2009; Tang et al. 2009). Passive movement of K+ through Kir4.1 channels may be particularly important for spatial buffering (Djukic et al. 2007; Haj-Yasein et al. 2011; Kofuji and Newman 2004).
Kir4.1 and the Na+/K+-ATPase have both been shown to physically (and possibly functionally) interact with AQP4 (Illarionova et al. 2010; Nagelhus et al. 2004; Strohschein et al. 2011). Kir4.1 has been particularly well-studied in this context. Initial reports of Kir4.1 colocalizing with AQP4 in astrocytic endfeet provided the first indication of a functional interaction (Connors et al. 2004; Nagelhus et al. 2004). These data have since received support from studies in which AQP4 was either deleted, or mislocalized away from astrocyte endfeet through deletion of the scaffolding protein α-syntrophin (to which AQP4 and Kir4.1 both bind). In either case K+ clearance following synaptic activity was impaired (Amiry-Moghaddam et al. 2003b; Binder et al. 2006; Haj-Yasein et al. 2015; Strohschein et al. 2011). The exact nature of the functional interaction between Kir4.1 and AQP4 remains debated however, as some studies have found no impairment of glial Ba2+-sensitive (Kir4.1) currents and no change in Kir4.1 distribution or expression levels in AQP4−/− mice (Binder et al. 2006; Ruiz-Ederra et al. 2007; Zhang and Verkman 2008). It has been suggested that AQP4 deletion may indirectly alter activity-driven K+ elevations as a consequence of impaired ECS volume regulation (Haj-Yasein et al. 2015; Haj-Yasein et al. 2012; Jin et al. 2013). Additionally, AQP4 deletion may impair the function of NKA. The NKA was recently shown to be capable of forming a macromolecular complex with AQP4 alongside the metabotropic glutamate receptor mGluR5, which may also regulate AQP4 permeability (Illarionova et al. 2010). Group I mGluRs (such as mGluR5) or Kir4.1 channels may indirectly increase or decrease astrocyte water permeability through phosphorylation or dephosphorylation, respectively, of AQP4 channels (Gunnarson et al. 2008; Song and Gunnarson 2012), although these data are controversial (Assentoft et al. 2013; Assentoft et al. 2014). Lack of a direct functional interaction between NKA or Kir4.1 and AQP4 could explain why Larsen and colleagues found neither K+ uptake pathway to be directly responsible for stimulus-induced shrinkage of hippocampal tissue (Larsen et al. 2014).
Similar to Kir4.1 and NKA, NKCC1 has been implicated in [K+]o-induced glial swelling (MacVicar et al. 2002; Su et al. 2002a; Su et al. 2002b). NKCC1 may be capable of transporting water directly rather than through interactions with AQP4 (Hamann et al. 2010). The extent to which NKCC1 directly participates in stimulus-induced [K+]o clearance and glial swelling, however, is debatable and may be overshadowed by NKA (Larsen et al. 2014). NKCC1 activity also increases [Na+]i and [Cl−]i in glial cells, which may be important for processes dependent upon these ions. For example, NKCC1-dependent increases in [Na+]i help offset the acute [Na+]i reductions caused by NKA activity following stimulus-induced [K+]o elevations (Rose and Ransom 1996). This suggests a potential indirect, if not direct, role for NKCC1 in [K+]o clearance and glial swelling.
2.3. Astrocyte swelling in seizures and epilepsy
Evidence for the role of astrocyte swelling in epilepsy has been provided through specific targeting of AQP4 or Kir4.1 in various seizure models. In a series of informative studies, Binder and colleagues used PTZ injections (Binder et al. 2004a; Binder et al. 2004b) or hippocampal electrical stimulation (Binder et al. 2006) to evoke acute seizures in wild-type and AQP4−/− mice. As discussed above, ECS in wild-type animals was observed to shrink just prior to a seizure (Binder et al. 2004b). However, in AQP4−/− animals, baseline ECS was significantly larger, and attenuated ECS shrinkage was observed (Binder et al. 2004b). AQP4−/− animals required a correspondingly higher electrical stimulation or PTZ dose to reach seizure threshold (Binder et al. 2004a; Binder et al. 2006), but also experienced seizures nearly 3 times the duration of wild-type animals (2006). The increased seizure threshold may reflect both the significant increase in baseline ECS, and the reduced tissue swelling observed in AQP4−/− animals (Binder et al. 2004b). Impaired K+ buffering was hypothesized as the cause for prolonged seizure duration, as AQP4−/− mice have noticeably slower K+ decay kinetics (Binder et al. 2006; Strohschein et al. 2011). It is interesting to note that, despite this impaired K+ clearance (or perhaps as a result of it), AQP4−/− astrocytes are coupled to a greater degree through gap junctions than their wild-type counterparts (Strohschein et al. 2011). Loss of astrocyte gap junctional coupling is associated with impaired [K+]o buffering, seizures and neuronal death (Bedner et al. 2015; Wallraff et al. 2006). Conversely, increased gap junctional coupling between AQP4−/− astrocytes may reflect a compensatory measure to offset reduced Kir and/or NKA activity by increasing the K+ redistribution capacity in the syncytium.
A key finding in both human epileptic tissue and animal models of epilepsy is the presence of reactive astrogliosis, a complex array of changes in the morphology, expression patterns and function of astrocytes following neural injury (Hubbard et al. 2013; Sofroniew 2009). Two of these changes are particularly relevant to astrocyte swelling and the development of epilepsy: 1.) Kir4.1 currents in astrocytes are consistently smaller (Hinterkeuser et al. 2000; Kivi et al. 2000), with reduced Kir expression particularly at vascular endfeet (Heuser et al. 2012); and 2.) AQP4 expression increases but polarization at the vascular endfeet is lost, paralleling the loss of Kir4.1 (Alvestad et al. 2013; Eid et al. 2005; Hubbard et al. 2016). Together, these reactive changes impair potassium clearance and spatial buffering (Amiry-Moghaddam et al. 2003b; Bedner et al. 2015; Bordey and Sontheimer 1998; Kivi et al. 2000), and could augment cellular swelling, although this has not yet been directly measured (Hubbard et al. 2013; Wetherington et al. 2008). Several animal models of epilepsy have demonstrated that these reactive changes in astrocytes likely precede development of seizures. For example, in kainic acid models of SRS, AQP4 was severely downregulated by 1 day after SE and mislocalized weeks before the development of seizures (Alvestad et al. 2013; Hubbard et al. 2016; Lee et al. 2012). Using a β1-integrin deletion from astrocytes, Robel and others (2015 (2009) provided evidence that reactive changes in astrocytes (including AQP4 and dystrophin mislocalization, astrocyte hypertrophy and loss of K+ buffering) are sufficient to induce recurrent seizures. In another seizure model, serum albumin directly taken up by astrocytic TGF-β receptors produced astrogliosis a few days before seizures developed (David et al. 2009; Ivens et al. 2007). Interestingly, while reduced K+ buffering efficacy in the albumin model correlated directly with reduced Kir4.1 expression (Ivens et al. 2007), no such reduction in Kir4.1 expression was found in the β1−/− model (Robel et al. 2015). These data suggest that the loss of K+ buffering may not necessarily be a direct consequence of Kir4.1 expression changes, but rather a functional change resulting from the loss of perivascular AQP4 channels (Amiry-Moghaddam et al. 2003b). The changes occurring in reactive astrocytes may create a “perfect storm” of epileptogenic conditions: Impaired K+ buffering increases neuronal excitability, augmenting local glutamate concentrations and further raising extracellular K+ to promote synchronization; loss of Kir4.1 and AQP4 at perivascular endfeet exacerbates astrocyte swelling which can no longer be effectively relieved through water efflux into the bloodstream (Hubbard et al. 2013; Wetherington et al. 2008), further augmenting neurotransmitter and ion concentrations while also increasing ephaptic interactions between neurons.
3. VRAC and volume regulation
Most cell types, including neural cells, exhibit some form of volume regulation. Cells swollen or shrunken by osmotic stress will, over time, return to their original volume even in the continued presence of the osmotic stressor. These two processes are correspondingly termed regulatory volume decrease (RVD) and regulatory volume increase (RVI) (Hoffmann et al. 2009). As described above, astrocyte swelling may be a catalyst for inducing and/or augmenting seizures, so the following discussion will focus specifically on RVD subsequent to cell swelling. RVD has been repeatedly observed in cultured astrocytes swollen by hypoosmolar or high K+ media (Eriksson et al. 1992; Kimelberg and Frangakis 1985; Olson et al. 1995; Vitarella et al. 1994). Disagreement exists as to whether astrocytic RVD occurs in intact tissue, as various groups have reported astrocytic swelling in brain slices without RVD (Andrew et al. 1997; Hirrlinger et al. 2008; Risher et al. 2009). Others have observed swelling with an RVD, but only at certain hypoosmolar doses (Thrane et al. 2011), and even swelling which does not recover following removal of the hypoosmolar stimulus (Anderova et al. 2014). These data (with the possible exception of the latter study) may reflect the ability of intact tissue to maintain a relatively constant volume with more gradual changes in osmolarity, referred to as “isovolumetric” volume regulation or IVR (Franco et al. 2000). The mechanisms involved in IVR are likely similar to those in RVD (Pasantes-Morales and Cruz-Rangel 2010). It is possible that RVD does not occur in some brain slice preparations due to depletion of osmotically-active substances (“osmolytes”) within the slice (Kreisman and Olson 2003). It must also be noted that RVD may depend on the method used to reduce extracellular osmolarity (Andrew et al. 1997). Emerging evidence suggests that astrocyte swelling itself may provide the stimulus necessary for RVD to occur. In Müller glia (an “astrocyte-like” glial cell type in the retina), rapid water influx through AQP4 produces membrane stretch, which then activates nearby stretch-activated TRPV4 channels to permit Ca2+ entry to promote RVD (Iuso and Krizaj 2016; Jo et al. 2015). While the requirement of TRPV4 opening and Ca2+ influx in astrocytic RVD has recently been questioned (Mola et al. 2016), these findings suggest that rapid water influx is an important trigger for subsequent astrocyte volume regulation (Benfenati et al. 2011; Mola et al. 2016).
In general terms, RVD occurs following either active transport or opening of volume-sensitive channels resulting in efflux of K+, Cl−, organic osmolytes and water from the cell (Chamberlin and Strange 1989; Pasantes-Morales et al. 2006). While the exact transporters and channels involved remain unclear, it is generally agreed that the “volume-regulated anion channel” (VRAC) is one of the essential components of astrocytic RVD (Benfenati and Ferroni 2010). Until recently, VRAC were identified pharmacologically, and by their unique swelling-activated currents known as ICl,Swell (Akita and Okada 2014). In 2014 the likely pore-forming subunits comprising VRAC were finally identified as belonging to the leucine-rich repeat containing protein 8 (LRRC8) family, with LRRC8A being required for VRAC currents and other members (B-E) modifying their kinetics (Hyzinski-Garcia et al. 2014; Qiu et al. 2014; Voss et al. 2014). For clarity and consistency with the large body of prior literature (Jentsch et al. 2016), this multimer will be referred to by the term “VRAC” for the remainder of this section.
VRAC are particularly relevant for the study of epilepsy for two main reasons: First and foremost, their permeability to not only small inorganic anions such as F− and Cl−, but also to large anionic excitatory amino acids (EAAs) including taurine, glutamate and aspartate (Akita and Okada 2014), suggests that VRAC are a pathway through which astrocytic EAAs might be released back into the ECS. Indeed, many studies have demonstrated that cultured astrocytes release EAAs primarily through VRAC following swelling in high-K+ or hypoosmolar conditions (Abdullaev et al. 2006; Kimelberg et al. 1990; Kimelberg et al. 1995; Rutledge et al. 1998). In the middle cerebral artery occlusion (MCAO) model of stroke (an ischemia model which produces substantial astrocyte swelling), the VRAC antagonist DCPIB was found to significantly improve behavioral outcome and reduce infarct volume and extracellular glutamate levels (Zhang et al. 2008). These findings suggest that astrocytic swelling in ischemia could lead to glutamate release through VRAC and directly contribute to excitotoxic tissue death (Risher et al. 2009; Zhang et al. 2008). Second, VRAC are not simple, “stretch-activated” channels, despite being activated consistently in cell-swelling conditions. Reductions in intracellular ionic strength are required for their activation (Syeda et al. 2016; Voets et al. 1999), which often occurs during cell swelling (as water influx dilutes intracellular ions). Similarly, VRAC activity can be potentiated independently of cell volume (discussed further below).
Importantly, VRAC in the CNS are probably not limited to astrocytes. Neurons also appear to express VRAC, with similar activation in hypoosmolar or excitotoxic conditions but differing from astrocytic VRAC in specific pharmacology (Inoue et al. 2005; Inoue et al. 2007; Inoue and Okada 2007; Zhang et al. 2011). Differentiating between neuronal vs. astrocytic VRAC is complicated, however, as there are currently no antagonists specific for neuronal VRAC and some of the most commonly-used VRAC antagonists (NPPB, DIDS, etc) have rather broad nonspecific effects (Evanko et al. 2004). For example, DIDS is a broad-spectrum Cl− channel blocker, and was recently found to block the neuronal voltage-gated Cl− channel SLC26A11 which opens following massive cytotoxic Na+ influx such as occurs during stroke (Rungta et al. 2015). The question of whether neurons can swell and/or release glutamate through VRAC will require more specific tools. Regardless of whether neuronal swelling contributes to increased tissue excitability, glutamate release through astrocytic VRAC is a compelling avenue for future study based on the ample evidence for the role of these channels in glutamate release from astrocytes in vitro.
3.1. Astrocytic glutamate and neuronal excitability
There is little doubt that astrocytic glutamate can influence neuronal activity. In quiescent conditions, astrocytes are thought to maintain the so-called “ambient glutamate” concentration in the extracellular space, possibly through release by VRAC or a related channel (Cavelier and Attwell 2005; Le Meur et al. 2007). In hypoosmolar conditions, NMDA receptor-dependent slow inward currents (SICs) have also been observed in hippocampal CA1 neurons even when neuronal firing is blocked and vesicles depleted of neurotransmitter (Fiacco et al. 2007; Lauderdale et al. 2015), suggesting volume-dependent glutamate release from a nonsynaptic source. Astrocytic glutamate released by a Ca2+-dependent process has also been reported to induce SICs. In these studies, stimuli which evoked Ca2+ elevations in astrocytes including direct mechanical stimulation, group 1 metabotropic glutamate receptor (mGluR) agonists, or simply uncaging of Ca2+ within the astrocyte were sufficient to induce SICs in nearby neurons (Angulo et al. 2004; Fellin et al. 2006; Fellin et al. 2004; Tian et al. 2005). These findings seem at odds with hypoosmolar-induced SICs, which persist when vesicles are emptied using bafilomycin and in transgenic mice in which activity-driven astrocyte Ca2+ elevations are significantly depressed (Fiacco et al. 2007). More puzzlingly, one group found that neuronal SICs were produced following astrocytic Ca2+ elevations induced by activation of PAR-1, but not P2Y1, receptors (Shigetomi et al. 2008). Given these conflicting studies, it is quite interesting to note that VRAC, apart from their typical Ca2+-independent mode of activation, can also be potentiated by several mechanisms which elevate intracellular Ca2+, including src kinase activation by peroxynitrite (Haskew et al. 2002), protein kinase C (PKC) activation from B2 bradykinin receptor activity (Akita and Okada 2011; Liu et al. 2009), and P2Y ATP receptor activation (Mongin and Kimelberg 2002; Mongin and Kimelberg 2005; Takano et al. 2005) Takano et al (2005) also found that P2Y receptor activation induced a transient astrocyte volume increase that was necessary for VRAC to open. Taken together, these studies raise the possibility that most or all astrocytic glutamate leading to SICs is released through VRAC opening.
The unique properties of SICs are particularly interesting in the context of epilepsy. SICs evoked by astrocyte Ca2+ increases are synchronized between neurons located within ~100 μm, occurring in up to 12 neurons simultaneously (Angulo et al. 2004; Fellin et al. 2004). This property supports their astrocytic origin, since a single astrocyte within the hippocampal CA1 area can cover nearly 66,000 μm3 of neuropil and ensheathe over 100,000 synapses (Bushong et al. 2002). Astrocytic glutamate release may thus be capable of synchronizing the activity of local neuron populations. It is unclear to what degree these locally-synchronous events can contribute to the much larger, synchronous activity characterizing interictal and ictal discharges. Hypoosmolar conditions augment hippocampal tissue excitability, at least in part, through rapid reduction of the extracellular space accompanied almost simultaneously by SIC generation in CA1 neurons (Fiacco et al. 2007; Lauderdale et al. 2015). SICs have also been observed in multiple slice seizure models, and it has been debated whether SICs themselves are a type of epileptiform activity (Fellin et al. 2006; Tian et al. 2005). Current evidence suggests that SICs are distinct from epileptiform currents, the latter being dependent upon synaptic transmission and synchronous over large distances, while the former are locally synchronous and nonsynaptic (Fellin et al. 2006; Fellin et al. 2004; Wetherington et al. 2008). SICs are more likely to modulate tissue excitability and thereby play a role in the “build up” to seizure activity during the period of cell swelling and reduction of the ECS that immediately precedes seizures. Small groups of neurons synchronized by SIC activity, for example, may gradually fuse into larger “hypersynchronous” groups that characterize interictal or ictal events (Bikson et al. 2003; Jiruska et al. 2010). Release of glutamate through astrocytic VRAC may be sufficient to turn interictal spikes into full ictal discharges, or exacerbate and lengthen ictal bursts in seizure-prone tissue (Ding et al. 2007; Fellin et al. 2006).
4. Conclusions
Astrocytes exert tight control over neuronal excitability through a number of mechanisms including glutamate uptake, [K+]o buffering, and brain water regulation. These same mechanisms, however, may also be actively involved in seizure generation. High astrocyte water permeability facilitates cellular edema in hypoosmolar, high-[K+]o and other pathological conditions. Intracellular glutamate in astrocytes can be released from volume-regulated channels that open during swelling, increasing local glutamate concentrations and potentially binding directly to neuronal glutamate receptors. Extracellular concentrations of glutamate, as well as K+ and other ions, would also be augmented by astrocyte swelling and subsequent reduction of the ECS. These mechanisms combined would elevate neuronal excitability through increased ephaptic interactions, depolarized membrane potentials, and increased excitatory currents (Figure 1). Seizures are preceded by brain parenchyma swelling in both humans and animal models and abolished by increases in extracellular osmolarity, supporting the idea that astrocyte swelling may promote seizure generation. Moreover, reactive astrogliosis is common to epileptic tissue, and includes changes in AQP4 expression and K+ buffering which may directly impact astrocyte volume regulation (Hubbard and Binder 2016).
Figure 1. Hypothesized role of astrocyte swelling in two different models of neuronal hyperexcitability.
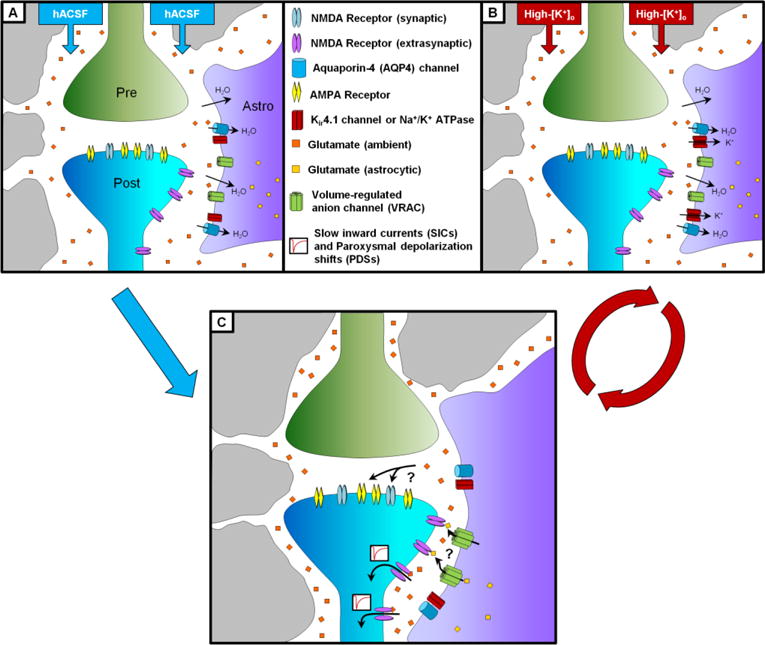
In a hypoosmolar model of neuronal hyperexcitability (A), astrocytes swell through direct water influx. In a high-[K+]o model (B), astrocyte swelling is caused by initial influx of K+ followed by osmotically-obligated water directly across the membrane or through AQP4 channels. In both cases (C), astrocyte swelling leads to a decrease in the extracellular space (which increases local glutamate concentrations and enhances neuronal excitability). In addition, astrocyte swelling may lead to activation of additional glutamate release pathways, such as volume-regulated anion channels (VRAC). The elevated glutamate during astrocyte swelling then results in NMDA receptor-dependent locally-synchronous neuronal depolarization. Neuronal firing results in a positive feedback loop as glutamate and additional K+ are released into the ECS, causing additional astrocyte swelling, opening of VRAC, and further increases in neuronal excitability. As ephaptic interactions cause neuronal firing to become synchronized, this system is driven from locally-synchronous activity (SICs) to global hypersynchrony (PDSs) and finally into ictal discharges. Modified from Lauderdale et al. (2015): “Osmotic edema rapidly increases neuronal excitability through activation of NMDA receptor-dependent slow inward currents in juvenile and adult hippocampus”. ASN Neuro 7(5): 1–21. DOI: 10.1177/1759091415605115. Available from SAGE Publications under a Creative Commons Attribution 3.0 license at: http://asn.sagepub.com/content/7/5/1759091415605115.
5. Future directions
The role of astrocyte/tissue volume changes in seizure generation remains largely unexplored, and many key questions remain to be answered. First and foremost, do astrocytes swell prior to a seizure in vivo? Multiple studies have demonstrated that astrocytes can swell in hyperexcitable environments, and others still have shown that tissue swelling precedes seizure generation. To date, however, astrocyte volume prior to and during preictal, ictal and postictal periods in vivo has not been directly measured. Simultaneous astrocyte imaging and EEG recordings, in multiple seizure models, will be essential to closing this gap. Second, what is the role of AQP4 in astrocyte swelling in vivo? Conventional wisdom holds that AQP4 channels facilitate the majority of water movement across the astrocyte membrane, but is far from the only possible mechanism of astrocyte swelling (Kimelberg 2005). Are different astrocyte swelling mechanisms active under different seizure paradigms (i.e. high [K+]o vs. hypoosmolarity)? Imaging astrocytes from AQP4−/− animals during seizure will be an important first step in answering these questions. Answers to the above questions will guide approaches to a more central one: To what degree does astrocyte swelling facilitate seizure generation? The threshold requirements (such as osmotic pressure) for astrocyte swelling in vivo have not been firmly established, nor has it been established whether the same threshold is capable of eliciting seizures. Moreover, astrocytes may not be the only cells swelling during the preictal period. Volume measurements of neurons and other cell types will be essential, especially if AQP4-independent astrocyte swelling is possible. Lastly, the effect of inhibiting astrocyte swelling on seizures in vivo is currently unknown. The findings from these immediate questions will be invaluable for understanding the role of astrocyte swelling in epilepsy.
Highlights.
Reductions in CNS extracellular space precede and promote seizure activity
Astrocytes regulate neuronal excitability through water, K+, and glutamate uptake
Water and K+ uptake can swell astrocytes and shrink extracellular space
Swollen astrocytes may release glutamate through volume-regulated channels
Astrocyte swelling may promote neuronal hyperexcitability leading to seizure
Acknowledgments
This work was supported by the National Institutes of Health NINDS R01NS082570 to T.A.F. and D.K.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in cultured rat astrocytes. J Physiol. 2006;572:677–89. doi: 10.1113/jphysiol.2005.103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken PG, Borgdorff AJ, Juta AJ, Kiehart DP, Somjen GG, Wadman WJ. Volume changes induced by osmotic stress in freshly isolated rat hippocampal neurons. Pflugers Arch. 1998;436:991–8. doi: 10.1007/s004240050734. [DOI] [PubMed] [Google Scholar]
- Akita T, Okada Y. Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J Physiol. 2011;589:3909–27. doi: 10.1113/jphysiol.2011.208173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita T, Okada Y. Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience. 2014;275:211–31. doi: 10.1016/j.neuroscience.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Albright B, Dhaher R, Wang H, Harb R, Lee TW, Zaveri H, Eid T. Progressive neuronal activation accompanies epileptogenesis caused by hippocampal glutamine synthetase inhibition. Exp Neurol. 2016;288:122–133. doi: 10.1016/j.expneurol.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldenkamp AP. Cognitive and behavioural assessment in clinical trials: when should they be done? Epilepsy Res. 2001;45:155–7. doi: 10.1016/s0920-1211(01)00244-3. discussion 159–61. [DOI] [PubMed] [Google Scholar]
- Aldenkamp AP, De Krom M, Reijs R. Newer antiepileptic drugs and cognitive issues. Epilepsia. 2003;44(Suppl 4):21–9. doi: 10.1046/j.1528-1157.44.s4.3.x. [DOI] [PubMed] [Google Scholar]
- Alvestad S, Hammer J, Hoddevik EH, Skare O, Sonnewald U, Amiry-Moghaddam M, Ottersen OP. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. 2013;105:30–41. doi: 10.1016/j.eplepsyres.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Frydenlund DS, Ottersen OP. Anchoring of aquaporin-4 in brain: molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience. 2004;129:999–1010. doi: 10.1016/j.neuroscience.2004.08.049. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, et al. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A. 2003a;100:2106–11. doi: 10.1073/pnas.0437946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003b;100:13615–20. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderova M, Benesova J, Mikesova M, Dzamba D, Honsa P, Kriska J, Butenko O, Novosadova V, Valihrach L, Kubista M, et al. Altered astrocytic swelling in the cortex of alpha-syntrophin-negative GFAP/EGFP mice. PLoS ONE. 2014;9:e113444. doi: 10.1371/journal.pone.0113444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew RD. Seizure and acute osmotic change: clinical and neurophysiological aspects. J Neurol Sci. 1991;101:7–18. doi: 10.1016/0022-510x(91)90013-w. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Fagan M, Ballyk BA, Rosen AS. Seizure susceptibility and the osmotic state. Brain Res. 1989;498:175–80. doi: 10.1016/0006-8993(89)90417-4. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA. Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex. 2007;17:787–802. doi: 10.1093/cercor/bhk032. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Lobinowich ME, Osehobo EP. Evidence against volume regulation by cortical brain cells during acute osmotic stress. Exp Neurol. 1997;143:300–12. doi: 10.1006/exnr.1996.6375. [DOI] [PubMed] [Google Scholar]
- Andrew RD, MacVicar BA. Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience. 1994;62:371–83. doi: 10.1016/0306-4522(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–7. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz AM, Perkins KL, Irie F, Lewis DP, Hrabe J, Xiao F, Itano N, Kimata K, Hrabetova S, Yamaguchi Y. Hyaluronan deficiency due to Has3 knock-out causes altered neuronal activity and seizures via reduction in brain extracellular space. J Neurosci. 2014;34:6164–76. doi: 10.1523/JNEUROSCI.3458-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assentoft M, Kaptan S, Fenton RA, Hua SZ, de Groot BL, MacAulay N. Phosphorylation of rat aquaporin-4 at Ser(111) is not required for channel gating. Glia. 2013;61:1101–12. doi: 10.1002/glia.22498. [DOI] [PubMed] [Google Scholar]
- Assentoft M, Larsen BR, Olesen ETB, Fenton RA, MacAulay N. AQP4 plasma membrane trafficking or channel gating is not significantly modulated by phosphorylation at COOH-terminal serine residues. Am J Physiol-Cell Ph. 2014;307:C957–C965. doi: 10.1152/ajpcell.00182.2014. [DOI] [PubMed] [Google Scholar]
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, Jacob Filho W, Lent R, Herculano-Houzel S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. 2009;513:532–41. doi: 10.1002/cne.21974. [DOI] [PubMed] [Google Scholar]
- Azouz R, Alroy G, Yaari Y. Modulation of endogenous firing patterns by osmolarity in rat hippocampal neurones. J Physiol. 1997;502(Pt 1):175–87. doi: 10.1111/j.1469-7793.1997.175bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballyk BA, Quackenbush SJ, Andrew RD. Osmotic effects on the CA1 neuronal population in hippocampal slices with special reference to glucose. J Neurophysiol. 1991;65:1055–66. doi: 10.1152/jn.1991.65.5.1055. [DOI] [PubMed] [Google Scholar]
- Bedner P, Dupper A, Huttmann K, Muller J, Herde MK, Dublin P, Deshpande T, Schramm J, Haussler U, Haas CA, et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. 2015;138:1208–22. doi: 10.1093/brain/awv067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci U S A. 2011;108:2563–8. doi: 10.1073/pnas.1012867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati V, Ferroni S. Water transport between CNS compartments: functional and molecular interactions between aquaporins and ion channels. Neuroscience. 2010;168:926–40. doi: 10.1016/j.neuroscience.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- Berghuis B, de Haan GJ, van den Broek MP, Sander JW, Lindhout D, Koeleman BP. Epidemiology, pathophysiology and putative genetic basis of carbamazepine- and oxcarbazepine-induced hyponatremia. European journal of neurology. 2016;23:1393–9. doi: 10.1111/ene.13069. [DOI] [PubMed] [Google Scholar]
- Bikson M, Fox JE, Jefferys JG. Neuronal aggregate formation underlies spatiotemporal dynamics of nonsynaptic seizure initiation. J Neurophysiol. 2003;89:2330–3. doi: 10.1152/jn.00764.2002. [DOI] [PubMed] [Google Scholar]
- Binder DK, Haut SR. Toward new paradigms of seizure detection. Epilepsy Behav. 2013;26:247–52. doi: 10.1016/j.yebeh.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Oshio K, Ma T, Verkman AS, Manley GT. Increased seizure threshold in mice lacking aquaporin-4 water channels. NeuroReport. 2004a;15:259–62. doi: 10.1097/00001756-200402090-00009. [DOI] [PubMed] [Google Scholar]
- Binder DK, Papadopoulos MC, Haggie PM, Verkman AS. In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci. 2004b;24:8049–56. doi: 10.1523/JNEUROSCI.2294-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. GLIA. 2006;53:631–6. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 1998;32:286–303. doi: 10.1016/s0920-1211(98)00059-x. [DOI] [PubMed] [Google Scholar]
- Borgdorff AJ, Somjen GG, Wadman WJ. Two mechanisms that raise free intracellular calcium in rat hippocampal neurons during hypoosmotic and low NaCl treatment. J Neurophysiol. 2000;83:81–9. doi: 10.1152/jn.2000.83.1.81. [DOI] [PubMed] [Google Scholar]
- Boss D, Kuhn J, Jourdain P, Depeursinge C, Magistretti PJ, Marquet P. Measurement of absolute cell volume, osmotic membrane water permeability, and refractive index of transmembrane water and solute flux by digital holographic microscopy. J Biomed Opt. 2013;18:036007. doi: 10.1117/1.JBO.18.3.036007. [DOI] [PubMed] [Google Scholar]
- Boylan LS, Flint LA, Labovitz DL, Jackson SC, Starner K, Devinsky O. Depression but not seizure frequency predicts quality of life in treatment-resistant epilepsy. Neurology. 2004;62:258–61. doi: 10.1212/01.wnl.0000103282.62353.85. [DOI] [PubMed] [Google Scholar]
- Broberg M, Pope KJ, Lewis T, Olsson T, Nilsson M, Willoughby JO. Cell swelling precedes seizures induced by inhibition of astrocytic metabolism. Epilepsy Res. 2008;80:132–41. doi: 10.1016/j.eplepsyres.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Barry SJ, Bamagous GA, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology. 2012;78:1548–54. doi: 10.1212/WNL.0b013e3182563b19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie MJ, Dichter MA. Antiepileptic drugs. N Engl J Med. 1996;334:168–75. doi: 10.1056/NEJM199601183340308. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes inCA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–92. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Benninger F, Yaari Y. KV7/M channels mediate osmotic modulation of intrinsic neuronal excitability. J Neurosci. 2009;29:11098–111. doi: 10.1523/JNEUROSCI.0942-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla-Guerra L, del Carmen Fernandez-Moreno M, Lopez-Chozas JM, Fernandez-Bolanos R. Electrolytes disturbances and seizures. Epilepsia. 2006;47:1990–8. doi: 10.1111/j.1528-1167.2006.00861.x. [DOI] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol. 2005;564:397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, Krystal JH, Spencer DD, Abi-Saab WM. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57:226–35. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- Chamberlin ME, Strange K. Anisosmotic cell volume regulation: a comparative view. Am J Physiol. 1989;257:C159–73. doi: 10.1152/ajpcell.1989.257.2.C159. [DOI] [PubMed] [Google Scholar]
- Chebabo SR, Hester MA, Aitken PG, Somjen GG. Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res. 1995a;695:203–16. doi: 10.1016/0006-8993(95)00778-o. [DOI] [PubMed] [Google Scholar]
- Chebabo SR, Hester MA, Jing J, Aitken PG, Somjen GG. Interstitial space, electrical resistance and ion concentrations during hypotonia of rat hippocampal slices. J Physiol. 1995b;487:685–97. doi: 10.1113/jphysiol.1995.sp020910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chever O, Djukic B, McCarthy KD, Amzica F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J Neurosci. 2010;30:15769–77. doi: 10.1523/JNEUROSCI.2078-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–76. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Connors NC, Adams ME, Froehner SC, Kofuji P. The potassium channel Kir4.1 associates with the dystrophin-glycoprotein complex via alpha-syntrophin in glia. J Biol Chem. 2004;279:28387–92. doi: 10.1074/jbc.M402604200. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009;29:10588–99. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O. Effects of Seizures on Autonomic and Cardiovascular Function. Epilepsy Curr. 2004a;4:43–46. doi: 10.1111/j.1535-7597.2004.42001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O. Therapy for neurobehavioral disorders in epilepsy. Epilepsia. 2004b;45(Suppl 2):34–40. doi: 10.1111/j.0013-9580.2004.452003.x. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Hofmeier G, Lux HD. Transient changes in the size of the extracellular space in the sensorimotor cortex of cats in relation to stimulus-induced changes in potassium concentration. Exp Brain Res. 1980;40:432–9. doi: 10.1007/BF00236151. [DOI] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–84. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–65. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Obenaus A, Tasker JG. Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci Lett. 1990;120:267–70. doi: 10.1016/0304-3940(90)90056-f. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Snow RW, Taylor CP. Role of electrical interactions in synchronization of epileptiform bursts. Adv Neurol. 1986;44:593–617. [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–10. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Ghosh A, Wang Y, Beckstrom H, Zaveri HP, Lee TS, Lai JC, Malthankar-Phatak GH, de Lanerolle NC. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain. 2008;131:2061–70. doi: 10.1093/brain/awn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, Agre P, Ottersen OP, de Lanerolle NC. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci U S A. 2005;102:1193–8. doi: 10.1073/pnas.0409308102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, de Lanerolle NC. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Nilsson M, Wagberg M, Ronnback L, Hansson E. Volume regulation of single astroglial cells in primary culture. Neurosci Lett. 1992;143:195–9. doi: 10.1016/0304-3940(92)90264-8. [DOI] [PubMed] [Google Scholar]
- Evanko DS, Zhang Q, Zorec R, Haydon PG. Defining pathways of loss and secretion of chemical messengers from astrocytes. GLIA. 2004;47:233–40. doi: 10.1002/glia.20050. [DOI] [PubMed] [Google Scholar]
- Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG. Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J Neurosci. 2006;26:9312–22. doi: 10.1523/JNEUROSCI.2836-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–43. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, McCarthy KD. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron. 2007;54:611–26. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Franco R, Quesada O, Pasantes-Morales H. Efflux of osmolyte amino acids during isovolumic regulation in hippocampal slices. J Neurosci Res. 2000;61:701–11. doi: 10.1002/1097-4547(20000915)61:6<701::AID-JNR14>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Gunnarson E, Zelenina M, Axehult G, Song Y, Bondar A, Krieger P, Brismar H, Zelenin S, Aperia A. Identification of a molecular target for glutamate regulation of astrocyte water permeability. GLIA. 2008;56:587–96. doi: 10.1002/glia.20627. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein NN, Bugge CE, Jensen V, Ostby I, Ottersen OP, Hvalby O, Nagelhus EA. Deletion of aquaporin-4 increases extracellular K(+) concentration during synaptic stimulation in mouse hippocampus. Brain Struct Funct. 2015;220:2469–74. doi: 10.1007/s00429-014-0767-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Yasein NN, Jensen V, Ostby I, Omholt SW, Voipio J, Kaila K, Ottersen OP, Hvalby O, Nagelhus EA. Aquaporin-4 regulates extracellular space volume dynamics during high-frequency synaptic stimulation: a gene deletion study in mouse hippocampus. Glia. 2012;60:867–74. doi: 10.1002/glia.22319. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, Hvalby O, Nagelhus EA. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10) Glia. 2011;59:1635–42. doi: 10.1002/glia.21205. [DOI] [PubMed] [Google Scholar]
- Hamann S, Herrera-Perez JJ, Zeuthen T, Alvarez-Leefmans FJ. Cotransport of water by the Na+-K+-2Cl(−) cotransporter NKCC1 in mammalian epithelial cells. J Physiol. 2010;588:4089–101. doi: 10.1113/jphysiol.2010.194738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskew RE, Mongin AA, Kimelberg HK. Peroxynitrite enhances astrocytic volume-sensitive excitatory amino acid release via a src tyrosine kinase-dependent mechanism. J Neurochem. 2002;82:903–12. doi: 10.1046/j.1471-4159.2002.01037.x. [DOI] [PubMed] [Google Scholar]
- Heuser K, Eid T, Lauritzen F, Thoren AE, Vindedal GF, Tauboll E, Gjerstad L, Spencer DD, Ottersen OP, Nagelhus EA, et al. Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J Neuropathol Exp Neurol. 2012;71:814–25. doi: 10.1097/NEN.0b013e318267b5af. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, Schramm J, Steinhauser C. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12:2087–96. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- Hirrlinger PG, Wurm A, Hirrlinger J, Bringmann A, Reichenbach A. Osmotic swelling characteristics of glial cells in the murine hippocampus, cerebellum, and retina in situ. J Neurochem. 2008;105:1405–17. doi: 10.1111/j.1471-4159.2008.05243.x. [DOI] [PubMed] [Google Scholar]
- Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the “common” neurologic disorders? Neurology. 2007;68:326–37. doi: 10.1212/01.wnl.0000252807.38124.a3. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- Holthoff K, Witte OW. Intrinsic optical signals in rat neocortical slices measured with near-infrared dark-field microscopy reveal changes in extracellular space. J Neurosci. 1996;16:2740–9. doi: 10.1523/JNEUROSCI.16-08-02740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Bossut DF, Somjen GG. Enhancement of whole cell synaptic currents by low osmolarity and by low [NaCl] in rat hippocampal slices. J Neurophysiol. 1997;77:2349–59. doi: 10.1152/jn.1997.77.5.2349. [DOI] [PubMed] [Google Scholar]
- Hubbard JA, Binder DK. Astrocytes and epilepsy. Amsterdam, Netherlands: Elsevier Academic Press; 2016. p. xii, 381. pages illustrations (chiefly color), portraits 25 cm. [Google Scholar]
- Hubbard JA, Hsu MS, Fiacco TA, Binder DK. Glial cell changes in epilepsy: overview of the clinical problem and therapeutic opportunities. Neurochem Int. 2013;63:638–51. doi: 10.1016/j.neuint.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Hubbard JA, Szu JI, Yonan JM, Binder DK. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp Neurol. 2016;283:85–96. doi: 10.1016/j.expneurol.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyzinski-Garcia MC, Rudkouskaya A, Mongin AA. LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. J Physiol. 2014;592:4855–62. doi: 10.1113/jphysiol.2014.278887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illarionova NB, Gunnarson E, Li Y, Brismar H, Bondar A, Zelenin S, Aperia A. Functional and molecular interactions between aquaporins and Na, K-ATPase. Neuroscience. 2010;168:915–25. doi: 10.1016/j.neuroscience.2009.11.062. [DOI] [PubMed] [Google Scholar]
- Inoue H, Mori S, Morishima S, Okada Y. Volume-sensitive chloride channels in mouse cortical neurons: characterization and role in volume regulation. Eur J Neurosci. 2005;21:1648–58. doi: 10.1111/j.1460-9568.2005.04006.x. [DOI] [PubMed] [Google Scholar]
- Inoue H, Ohtaki H, Nakamachi T, Shioda S, Okada Y. Anion channel blockers attenuate delayed neuronal cell death induced by transient forebrain ischemia. J Neurosci Res. 2007;85:1427–35. doi: 10.1002/jnr.21279. [DOI] [PubMed] [Google Scholar]
- Inoue H, Okada Y. Roles of volume-sensitive chloride channel in excitotoxic neuronal injury. J Neurosci. 2007;27:1445–55. doi: 10.1523/JNEUROSCI.4694-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iuso A, Krizaj D. TRPV4-AQP4 interactions ‘turbocharge’ astroglial sensitivity to small osmotic gradients. Channels (Austin) 2016:1–3. doi: 10.1080/19336950.2016.1140956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–47. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Lutter D, Planells-Cases R, Ullrich F, Voss FK. VRAC: molecular identification as LRRC8 heteromers with differential functions. Pflugers Arch. 2016;468:385–93. doi: 10.1007/s00424-015-1766-5. [DOI] [PubMed] [Google Scholar]
- Jin BJ, Zhang H, Binder DK, Verkman AS. Aquaporin-4-dependent K(+) and water transport modeled in brain extracellular space following neuroexcitation. J Gen Physiol. 2013;141:119–32. doi: 10.1085/jgp.201210883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiruska P, Csicsvari J, Powell AD, Fox JE, Chang WC, Vreugdenhil M, Li X, Palus M, Bujan AF, Dearden RW, et al. High-frequency network activity, global increase in neuronal activity, and synchrony expansion precede epileptic seizures in vitro. J Neurosci. 2010;30:5690–701. doi: 10.1523/JNEUROSCI.0535-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo AO, Ryskamp DA, Phuong TT, Verkman AS, Yarishkin O, MacAulay N, Krizaj D. TRPV4 and AQP4 Channels Synergistically Regulate Cell Volume and Calcium Homeostasis in Retinal Muller Glia. J Neurosci. 2015;35:13525–37. doi: 10.1523/JNEUROSCI.1987-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada R, Akdemir G, Asavapanumas N, Ratelade J, Zhang H, Verkman AS. Greatly improved survival and neuroprotection in aquaporin-4-knockout mice following global cerebral ischemia. FASEB J. 2014;28:705–14. doi: 10.1096/fj.13-231274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilb W, Dierkes PW, Sykova E, Vargova L, Luhmann HJ. Hypoosmolar conditions reduce extracellular volume fraction and enhance epileptiform activity in the CA3 region of the immature rat hippocampus. J Neurosci Res. 2006;84:119–29. doi: 10.1002/jnr.20871. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim DW, Jung KH, Lee ST, Kang BS, Byun JI, Yeom JS, Chu K, Lee SK. Frequency of and risk factors for oxcarbazepine-induced severe and symptomatic hyponatremia. Seizure. 2014;23:208–12. doi: 10.1016/j.seizure.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. Current concepts of brain edema. Review of laboratory investigations. J Neurosurg. 1995;83:1051–9. doi: 10.3171/jns.1995.83.6.1051. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. GLIA. 2005;50:389–397. doi: 10.1002/glia.20174. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Frangakis MV. Furosemide- and bumetanide-sensitive ion transport and volume control in primary astrocyte cultures from rat brain. Brain Res. 1985;361:125–34. doi: 10.1016/0006-8993(85)91282-x. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci. 1990;10:1583–91. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Rutledge E, Goderie S, Charniga C. Astrocytic swelling due to hypotonic or high K+ medium causes inhibition of glutamate and aspartate uptake and increases their release. J Cereb Blood Flow Metab. 1995;15:409–16. doi: 10.1038/jcbfm.1995.51. [DOI] [PubMed] [Google Scholar]
- Kitaura H, Tsujita M, Huber VJ, Kakita A, Shibuki K, Sakimura K, Kwee IL, Nakada T. Activity-dependent glial swelling is impaired in aquaporin-4 knockout mice. Neurosci Res. 2009;64:208–12. doi: 10.1016/j.neures.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Kivi A, Lehmann TN, Kovacs R, Eilers A, Jauch R, Meencke HJ, von Deimling A, Heinemann U, Gabriel S. Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci. 2000;12:2039–48. doi: 10.1046/j.1460-9568.2000.00103.x. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–56. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisman NR, Olson JE. Taurine enhances volume regulation in hippocampal slices swollen osmotically. Neuroscience. 2003;120:635–42. doi: 10.1016/s0306-4522(03)00359-2. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–9. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Neuropsychological effects of epilepsy and antiepileptic drugs. Lancet. 2001;357:216–22. doi: 10.1016/S0140-6736(00)03600-X. [DOI] [PubMed] [Google Scholar]
- Lagae L. Cognitive side effects of anti-epileptic drugs. The relevance in childhood epilepsy. Seizure. 2006;15:235–41. doi: 10.1016/j.seizure.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N. Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4.1 to hippocampal K(+) clearance and volume responses. GLIA. 2014;62:608–22. doi: 10.1002/glia.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderdale K, Murphy T, Tung T, Davila D, Binder DK, Fiacco TA. Osmotic Edema Rapidly Increases Neuronal Excitability Through Activation of NMDA Receptor-Dependent Slow Inward Currents in Juvenile and Adult Hippocampus. ASN Neuro. 2015;7 doi: 10.1177/1759091415605115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, Resnick T, Benbadis SR. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014;37:59–70. doi: 10.1016/j.yebeh.2014.05.031. [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol. 2007;580:373–83. doi: 10.1113/jphysiol.2006.123570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DJ, Hsu MS, Seldin MM, Arellano JL, Binder DK. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp Neurol. 2012;235:246–55. doi: 10.1016/j.expneurol.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Liang D, Bhatta S, Gerzanich V, Simard JM. Cytotoxic edema: mechanisms of pathological cell swelling. Neurosurg Focus. 2007;22:E2. doi: 10.3171/foc.2007.22.5.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HT, Akita T, Shimizu T, Sabirov RZ, Okada Y. Bradykinin-induced astrocyte-neuron signalling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J Physiol. 2009;587:2197–209. doi: 10.1113/jphysiol.2008.165084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Wang X. Hyponatremia induced by antiepileptic drugs in patients with epilepsy. Expert Opin Drug Saf. 2017;16:77–87. doi: 10.1080/14740338.2017.1248399. [DOI] [PubMed] [Google Scholar]
- Macaulay N, Zeuthen T. Glial K(+) clearance and cell swelling: key roles for cotransporters and pumps. Neurochem Res. 2012;37:2299–309. doi: 10.1007/s11064-012-0731-3. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. GLIA. 2002;37:114–23. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Traynelis SF, Dingledine R. Regional variation of extracellular space in the hippocampus. Science. 1990;249:674–7. doi: 10.1126/science.2382142. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Astrocyte membrane responses and potassium accumulation during neuronal activity. Hippocampus. 2007;17:1100–8. doi: 10.1002/hipo.20344. [DOI] [PubMed] [Google Scholar]
- Mola MG, Sparaneo A, Gargano CD, Spray DC, Svelto M, Frigeri A, Scemes E, Nicchia GP. The speed of swelling kinetics modulates cell volume regulation and calcium signaling in astrocytes: A different point of view on the role of aquaporins. GLIA. 2016;64:139–54. doi: 10.1002/glia.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK. ATP potently modulates anion channel-mediated excitatory amino acid release from cultured astrocytes. Am J Physiol Cell Physiol. 2002;283:C569–78. doi: 10.1152/ajpcell.00438.2001. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK. ATP regulates anion channel-mediated organic osmolyte release from cultured rat astrocytes via multiple Ca2+-sensitive mechanisms. Am J Physiol Cell Physiol. 2005;288:C204–13. doi: 10.1152/ajpcell.00330.2004. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Mathiisen TM, Ottersen OP. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience. 2004;129:905–13. doi: 10.1016/j.neuroscience.2004.08.053. [DOI] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F, et al. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J Neurophysiol. 2006;95:1843–52. doi: 10.1152/jn.00996.2005. [DOI] [PubMed] [Google Scholar]
- Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life-time epilepsy: a meta-analytic approach. Epilepsia. 2010;51:883–90. doi: 10.1111/j.1528-1167.2009.02481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–80. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JE, Alexander C, Feller DA, Clayman ML, Ramnath EM. Hypoosmotic volume regulation of astrocytes in elevated extracellular potassium. J Neurosci Res. 1995;40:333–42. doi: 10.1002/jnr.490400307. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 and brain edema. Pediatr Nephrol. 2007;22:778–84. doi: 10.1007/s00467-006-0411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14:265–77. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Lee HB, Ahn MH, Park S, Choi EJ, Lee HJ, Ryu HU, Kang JK, Hong JP. Identifying clinical correlates for suicide among epilepsy patients in South Korea: A case-control study. Epilepsia. 2015;56:1966–72. doi: 10.1111/epi.13226. [DOI] [PubMed] [Google Scholar]
- Pasantes-Morales H, Cruz-Rangel S. Brain volume regulation: osmolytes and aquaporin perspectives. Neuroscience. 2010;168:871–84. doi: 10.1016/j.neuroscience.2009.11.074. [DOI] [PubMed] [Google Scholar]
- Pasantes-Morales H, Lezama RA, Ramos-Mandujano G, Tuz KL. Mechanisms of cell volume regulation in hypo-osmolality. Am J Med. 2006;119:S4–11. doi: 10.1016/j.amjmed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2002;125:32–43. doi: 10.1093/brain/awf001. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. 2014;157:447–58. doi: 10.1016/j.cell.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rama Rao KV, Jayakumar AR, Norenberg MD. Brain edema in acute liver failure: mechanisms and concepts. Metab Brain Dis. 2014;29:927–36. doi: 10.1007/s11011-014-9502-y. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. The Journal of physiology. 2000;522:427–442. doi: 10.1111/j.1469-7793.2000.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Andrew RD, Kirov SA. Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. GLIA. 2009;57:207–21. doi: 10.1002/glia.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, Sutor B, Sontheimer H. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci. 2015;35:3330–45. doi: 10.1523/JNEUROSCI.1574-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Mori T, Zoubaa S, Schlegel J, Sirko S, Faissner A, Goebbels S, Dimou L, Gotz M. Conditional deletion of beta1-integrin in astroglia causes partial reactive gliosis. Glia. 2009;57:1630–47. doi: 10.1002/glia.20876. [DOI] [PubMed] [Google Scholar]
- Roper SN, Obenaus A, Dudek FE. Osmolality and nonsynaptic epileptiform bursts in rat CA1 and dentate gyrus. Ann Neurol. 1992;31:81–5. doi: 10.1002/ana.410310115. [DOI] [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Intracellular sodium homeostasis in rat hippocampal astrocytes. The Journal of physiology. 1996;491(Pt 2):291–305. doi: 10.1113/jphysiol.1996.sp021216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen AS, Andrew RD. Osmotic effects upon excitability in rat neocortical slices. Neuroscience. 1990;38:579–90. doi: 10.1016/0306-4522(90)90052-6. [DOI] [PubMed] [Google Scholar]
- Rowntree LG. The effects on mammals of the administration of excessive quantities of water. Journal of Pharmacology and Experimental Therapeutics. 1926;29:135–159. [Google Scholar]
- Ruiz-Ederra J, Zhang H, Verkman AS. Evidence against functional interaction between aquaporin-4 water channels and Kir4.1 potassium channels in retinal Muller cells. J Biol Chem. 2007;282:21866–72. doi: 10.1074/jbc.M703236200. [DOI] [PubMed] [Google Scholar]
- Rungta RL, Choi HB, Tyson JR, Malik A, Dissing-Olesen L, Lin PJ, Cain SM, Cullis PR, Snutch TP, MacVicar BA. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell. 2015;161:610–21. doi: 10.1016/j.cell.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Rutledge EM, Aschner M, Kimelberg HK. Pharmacological characterization of swelling-induced D-[3H]aspartate release from primary astrocyte cultures. Am J Physiol. 1998;274:C1511–20. doi: 10.1152/ajpcell.1998.274.6.C1511. [DOI] [PubMed] [Google Scholar]
- Saly V, Andrew RD. CA3 neuron excitation and epileptiform discharge are sensitive to osmolality. J Neurophysiol. 1993;69:2200–8. doi: 10.1152/jn.1993.69.6.2200. [DOI] [PubMed] [Google Scholar]
- Seifert G, Hüttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhäuser C. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. The Journal of neuroscience. 2009;29:7474–88. doi: 10.1523/JNEUROSCI.3790-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–36. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepshelovich D, Schechter A, Calvarysky B, Diker-Cohen T, Rozen-Zvi B, Gafter-Gvili A. Medication-induced SIADH: distribution and characterization according to medication class. Br J Clin Pharmacol. 2017 doi: 10.1111/bcp.13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–63. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends in neurosciences. 2009;32:638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Low external NaCl concentration and low osmolarity enhance voltage-gated Ca currents but depress K currents in freshly isolated rat hippocampal neurons. Brain Res. 1999;851:189–97. doi: 10.1016/s0006-8993(99)02185-x. [DOI] [PubMed] [Google Scholar]
- Song Y, Gunnarson E. Potassium dependent regulation of astrocyte water permeability is mediated by cAMP signaling. PLoS ONE. 2012;7:e34936. doi: 10.1371/journal.pone.0034936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohschein S, Huttmann K, Gabriel S, Binder DK, Heinemann U, Steinhauser C. Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. GLIA. 2011;59:973–80. doi: 10.1002/glia.21169. [DOI] [PubMed] [Google Scholar]
- Su G, Kintner DB, Flagella M, Shull GE, Sun DD. Astrocytes from Na+-K+-Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. American Journal of Physiology-Cell Physiology. 2002a;282:C1147–C1160. doi: 10.1152/ajpcell.00538.2001. [DOI] [PubMed] [Google Scholar]
- Su G, Kintner DB, Sun D. Contribution of Na(+)-K(+)-Cl(−) cotransporter to high-[K(+)](o)- induced swelling and EAA release in astrocytes. Am J Physiol Cell Physiol. 2002b;282:C1136–46. doi: 10.1152/ajpcell.00478.2001. [DOI] [PubMed] [Google Scholar]
- Syeda R, Qiu Z, Dubin AE, Murthy SE, Florendo MN, Mason DE, Mathur J, Cahalan SM, Peters EC, Montal M, et al. LRRC8 Proteins Form Volume-Regulated Anion Channels that Sense Ionic Strength. Cell. 2016;164:499–511. doi: 10.1016/j.cell.2015.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH-C, Yu Y, Li Y, Yang J, Dienel G, Zielke HR, et al. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16466–71. doi: 10.1073/pnas.0506382102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Taniguchi K, Kofuji P. Heterogeneity of Kir4.1 channel expression in glia revealed by mouse transgenesis. GLIA. 2009;57:1706–15. doi: 10.1002/glia.20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrane AS, Rangroo Thrane V, Nedergaard M. Drowning stars: reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014;37:620–8. doi: 10.1016/j.tins.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, Peng W, Wang F, Rangroo Thrane V, Enger R, et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A. 2011;108:846–51. doi: 10.1073/pnas.1015217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G-F, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, et al. An astrocytic basis of epilepsy. Nature medicine. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J Neurophysiol. 1989;61:927–38. doi: 10.1152/jn.1989.61.5.927. [DOI] [PubMed] [Google Scholar]
- Vitarella D, DiRisio DJ, Kimelberg HK, Aschner M. Potassium and taurine release are highly correlated with regulatory volume decrease in neonatal primary rat astrocyte cultures. J Neurochem. 1994;63:1143–9. doi: 10.1046/j.1471-4159.1994.63031143.x. [DOI] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Raskin G, Eggermont J, Nilius B. Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proc Natl Acad Sci U S A. 1999;96:5298–303. doi: 10.1073/pnas.96.9.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. 2014;344:634–8. doi: 10.1126/science.1252826. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–47. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Walz W, Hertz L. Ouabain-sensitive and ouabain-resistant net uptake of potassium into astrocytes and neurons in primary cultures. J Neurochem. 1982;39:70–7. doi: 10.1111/j.1471-4159.1982.tb04702.x. [DOI] [PubMed] [Google Scholar]
- Walz W, Hertz L. Intense furosemide-sensitive potassium accumulation in astrocytes in the presence of pathologically high extracellular potassium levels. J Cereb Blood Flow Metab. 1984;4:301–4. doi: 10.1038/jcbfm.1984.42. [DOI] [PubMed] [Google Scholar]
- Walz W, Hinks EC. Carrier-mediated KCl accumulation accompanied by water movements is involved in the control of physiological K+ levels by astrocytes. Brain Res. 1985;343:44–51. doi: 10.1016/0006-8993(85)91156-4. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zaveri HP, Lee TS, Eid T. The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study. Exp Neurol. 2009;220:293–302. doi: 10.1016/j.expneurol.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington J, Serrano G, Dingledine R. Astrocytes in the Epileptic. Brain Neuron. 2008:168–178. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZQ, Stringer JL. Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol. 2000;83:1443–51. doi: 10.1152/jn.2000.83.3.1443. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cao HJ, Kimelberg HK, Zhou M. Volume regulated anion channel currents of rat hippocampal neurons and their contribution to oxygen-and-glucose deprivation induced neuronal death. PLoS ONE. 2011;6:e16803. doi: 10.1371/journal.pone.0016803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Aquaporin-4 independent Kir4.1 K+ channel function in brain glial cells. Mol Cell Neurosci. 2008;37:1–10. doi: 10.1016/j.mcn.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Feustel PJ, Kimelberg HK. DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol. 2008;210:514–20. doi: 10.1016/j.expneurol.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]