

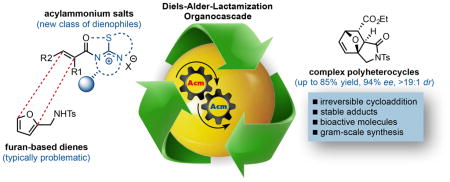
Abstract
α,β-Unsaturated acylammonium salts are useful dienophiles enabling highly enantioselective and stereodivergent Diels-Alder-initiated organocascades with furan-based dienes. Complex polycyclic systems can thus be obtained from readily prepared dienes, commodity acid chlorides, and a chiral isothiourea organocatalyst under mild conditions. We describe the use of a furan-based diene bearing pendant sulfonamides leading to the generation of oxa-bridged, trans-fused tricyclic γ-lactams. This process constitutes the first highly enantio- and diastereoselective, organocatalytic Diels-Alder cycloadditions with these typically problematic dienes due to their reversibility. Computational studies suggest that high diastereoselectivity with these furan dienes may be due to a reversible Diels-Alder cycloaddition for the endo adducts. In addition, the utility of this methodology is demonstrated through a concise approach to a core structure with similarity to the natural product isatisine A and a nonpeptidyl ghrelin-receptor inverse agonist.
Graphical Abstract
A long-standing problem for enantioselective, organocatalytic Diels-Alder (DA) cycloadditions is the use of furan as dienes. Due to their aromaticity, furans are often unreactive and undergo reversible DA cycloadditions, thus strong Lewis acids or high pressure are typically required to promote these cycloadditions.1 Furthermore, the potential for facile cycloreversion even at lower temperatures,2 and the sensitivity to acidic conditions of both the furanyl dienes and cycloadducts typically necessitates immediate post-reaction modification and limit reaction conditions including use of strong Lewis acids. Only two examples of effective catalytic asymmetric DA reactions of furans (67–94% yield, 97–99% ee, 4–7.3:1 endo/exo) were accomplished by Evans3,4 and Corey5,6 utilizing chiral bis(oxazoline)Cu(II) and oxazaborolidine Lewis acids 5 and 6, respectively (Figure 1, 1 → 7). The former reaction must be conducted at low temperature (−78 °C), due to rapid equilibration at higher temperatures (−20 °C) with concomitant erosion of both endo/exo diastereoselectivity and enantioselectivity, while the latter method is limited to the use of 2,2,2-trifluoroethyl acrylate as the only dienophile providing practical yields. Kotsuki attempted the first and only organocatalytic DA reaction of furans catalyzed by 50 mol% D-proline under high-pressure (0.8 GPa), however this resulted in low yields and selectivities (26% yield, 20% ee, 1.4:1 exo/endo).7 Building on our previous studies of Diels-Alder-initiated organocascades with chiral α,β-unsaturated acylammonium salts (Figure 1, e.g. 2),8,9) we reasoned that the termination step of a Diels-Alder-lactamization (DAL) organocascade (Figure 1, e.g. 3→7) could provide a solution to the reversibility of these cycloadditions with furanyl dienes by careful choice of a suitable pendant, terminating nucleophile. Herein we describe the successful implementation of this strategy leading to the first highly enantioselective DA cycloadditions with a furanyl diene through a Diels-Alder lactamization cascade employing organocatalysis. Enantioselectivity and diastereoselectivity were rationalized by computational studies and applications of this methodology are presented.
Figure 1.
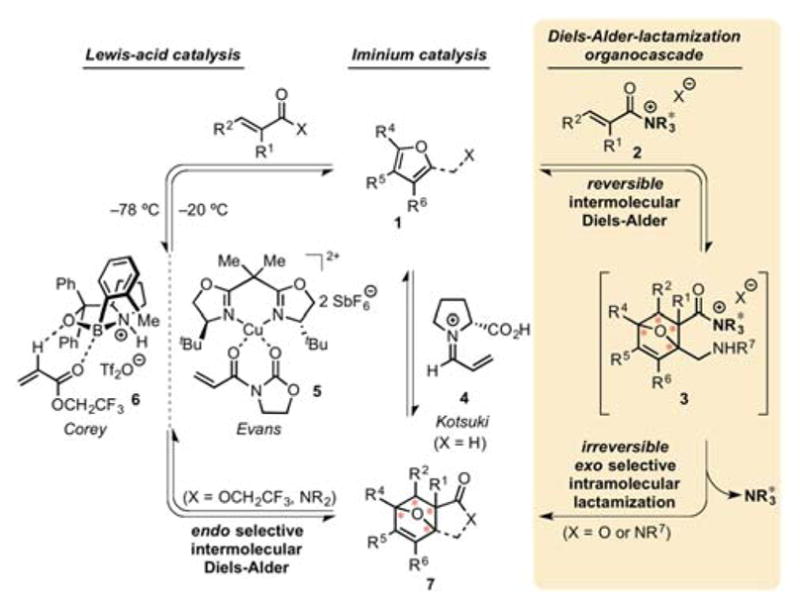
Comparison of enantioselective Diels-Alder cycloadditions with furans as dienes. Lewis-acid catalyzed, iminium-based, and the current asymmetric, α,β-unsaturated acylammonium salt-based, Diels-Alder-lactamization organocascade.
We began our studies with furans bearing pendant alcohols but discovered that these gave nearly racemic cycloadducts. Presumably this is due to reversible DA cycloaddition following the initial enantioselective DAL organocascade due to a slow lactonization step or a subsequent non-enantioselective intramolecular DA cycloaddition following ester formation.10,11 Further evidence for a racemization pathway involving retro-DA followed by intramolecular DA came from monitoring the enantiopurity of cycloadducts as a function of time (see ESI, Table S1) showing a decrease in enantiopurity with prolonged reaction times.
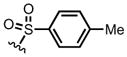
On the other hand, use of a furan bearing a secondary pendant amine, namely furanyl p-tolylsulfonamide 8a, in the DA lactamization organocascade led to greater success (Scheme 1). Employing our standard conditions developed previously for other dienes8,9 but employing less expensive levamisole hydrochloride (10 mol%) as Lewis base promoter, gave the oxa-bridged, trans-fused bicyclic γ-lactam 10 in 76% yield and 91% ee as a single exo-diastereomer (>19:1, 1H NMR 500 MHz). Importantly, this bicyclic γ-lactam could be stored at ambient temperature (23 °C) for an extended period without racemization pointing to much greater stability compared to analogous lactone adducts. Thus, it appears in this case that the conversion of the cycloadducts to lactams precludes cycloreversion and racemization through intramolecular DA cycloaddition and enables a successful enantioselective, organocascade process.
Scheme 1.
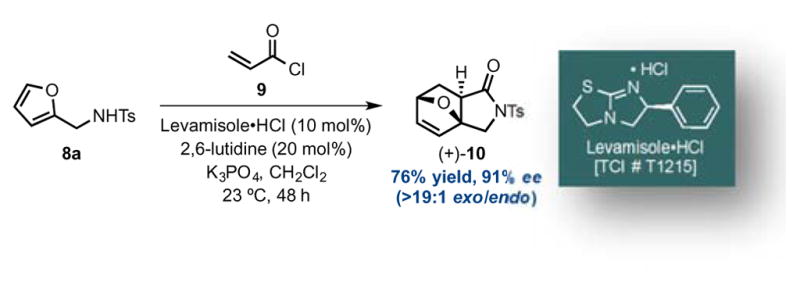
Diels-Alder-lactamization of furan-based sulfonamide 8a and acryloyl chloride (9) mediated by (R)-(−)-levamisole hydrochloride providing bridged cycloadduct (+)-10.
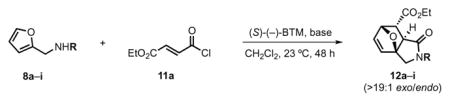
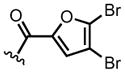
To gain further insights into this cycloaddition, in particular the requirements of the pendant nucleophile, we studied various substituents with varying electronic and steric properties on the pendant amine, namely dienes 8a–i (Table 1). For this study, we employed ethyl fumaroyl chloride (11a) and (S)-(−)-BTM as Lewis base promoter. Predictably, attempted DAL with furan 8b, containing a sterically demanding trityl group failed, likely due to a slow lactamization step (Table 1, entry 1). Similarly, furanyl dienes 8c–e possessing tert-butoxycarbonyl (Boc), benzoyl (Bz), and 4,5-dibromofuranyl groups did not afford the corresponding lactams 12b–d (Table 1, entries 2–4), likely due to the reduced nucleophilicity of these amine derivatives retarding the rate of lactamization. On the other hand, the nucleophilic benzylamine-containing furan 8f generated the desired cycloadduct 12f in 92% yield (Table 1, entry 5), however as a racemate, suggesting initial N-acylation followed by a non-enantioselective intramolecular DA cycloaddition revealing a possible non-selective background pathway for these DAL processes.
Table 1.
Varying the N-substituent on amino furans 8 for the enantioselective DAL organocascade with acid chloride 11a providing lactams 12.a
| ||||
|---|---|---|---|---|
| entry | cycloadducts R(12) | catalyst loading (mol%) | base | ee (yield†) % |
| 1 | CPh3 (12b) | 100 | 2,6-lutidine | n.r. |
| 2 | Boc (12c) | 100 | 2,6-lutidine | n.r. |
| 3 | Bz (12d) | 100 | 2,6-lutidine | n.r. |
| 4 |
 (12e) (12e) |
100 | 2,6-lutidine | n.r. |
| 5 | Bn (12f) | 100 | 2,6-lutidine | 3 (92) |
| 6 |
 (12g) (12g) |
0 | 2,6-lutidine | −(28) |
| 7 |
|
100 | 2,6-lutidine | 40 (46) |
| 8 |
 (12h) (12h) |
100 | 2,6-lutidine | 51 (40) |
| 9 |
 (Ts, 12a) (Ts, 12a) |
100 | 2,6-lutidine | 70 (75) |
| 10 |
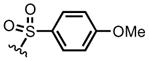 (12i) (12i) |
100 | 2,6-lutidine | 75 (82) |
| 11 | Ts | 20 | 2,6-lutidine | 42 (86) |
| 12 | Ts | 20 | pyridine | 83 (88) |
| 13‡ | Ts | 20 | pyridine | 94 (85) |
DAL reactions were performed with dienes 8 (1.0 equiv), ethyl fumaroyl chloride (11a, 1.2 equiv), (S)-(−)-BTM (20–100 mol%) and Brønsted base (1.0 equiv) in CH2Cl2 (0.1 M).
All yields refer to isolated, purified yields of cycloadducts. Diastereomeric (endo/exo) ratios were determined by 1H NMR (500 MHz) analysis of the crude reaction mixtures. Enantiomeric excess (ee) was determined by chiral phase HPLC.
Fumaroyl chloride 11a was added as a solution in CH2Cl2 by syringe pump over 5 h.
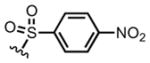
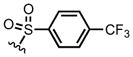
We next screened several para-substituted sulfonamides 8g–i to study the electronic effects of the N-substituent on the lactamization step in comparison to the N-Ts substituted diene 8a. The effect on enantioselectivity due to para-substituents of the N-sulfonamides was investigated and found to follow the order: NO2 < CF3 < Me < OMe (Table 1, entries 7–10). This trend is consistent with increasing acidity of the corresponding sulfonamides.12 In the case of p-NO2 sulfonamide furan 8g, the greater acidity may lead to initial N-acylation and a competitive, racemic intramolecular DA pathway. As anticipated, in the absence of a chiral Lewis base, a substantial background DAL was found, with the p-NO2 sulfonamide furan 8g affording a single exo-diastereomer of racemic bicyclic γ-lactam (±)-12g albeit in modest 28% yield (Table 1, entry 6). Thus, in this series the p-OMe sulfonamide diene 8i provided the best results employing 1.0 equiv of (S)-(−)-BTM leading to the cycloadduct 12i in 82% ee (75% yield). However, the differential cost of tosyl chloride (TsCl ~$0.06/g) compared to methoxy benzene sulfonyl chloride (MbsCl ~$6/g) led us to continue optimization with furanyl p-toluene sulfonamide 8a. Lowering the catalyst loading to 20 mol% delivered the bicyclic γ-lactam 12a (Table 1, entry 11) with comparable yield (86%) but diminished enantiocontrol (42% ee). Use of pyridine, improved the enantioselectivity dramatically to 83% ee (88% yield, Table 1, entry 12) and may be be due to rapid deprotonation with this base compared to the more sterically encumbered base, 2,6-lutidine, which reduces opportunity for reversible DAL. Extending the addition times of acid chloride 11 (Table 1, entry 13) by syringe pump addition led to high enantioselectivity (85% yield, 92% ee) presumably by enabling the asymmetric DAL process to compete effectively with the non-selective background pathway.
In addition to acryloyl and ethyl fumaroyl chloride, we investigated other dienophiles including crotonyl, α-methyl, and β-phenyl acryloyl chloride which, not unexpectedly, did not yield detectable amounts of cycloadduct under the optimized conditions due to the low reactivity of furanyl dienes. However, the electron deficient trifluorocrotonyl chloride 11b participated under identical conditions to provide the corresponding fluorinated cycloadduct 13 in 54% yield and 85% ee (Scheme 2).
Scheme 2.
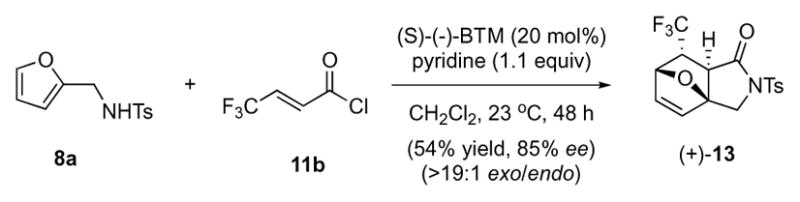
Diels-Alder-lactamization of furan 8a and trifluorocrotonyl chloride (11b) delivering bridged cycloadduct (+)-13.
The possibility of a facile retro-DA reaction between these furan-derived cycloadducts and the isothiourea-bound α,β-unsaturated acylammonium salt in conjunction a potentially terminating lactamization step led us to study the energetics of this organocascade computationally. We optimized transition state structures (TSSs), reactants, and products for the DA step with diene 8a, fumaroyl chloride (11), and (−)-(S)-BTM and their computed relative free energies are shown in Figure 2a. All structures were optimized with Gaussian 0916 using the M06-2X17,18 functional and the 6–31G(d)19,20 basis set. Solvent effects were modelled using the SMD continuum model with implicit dichloromethane.21 The identities of TSSs were verified by the presence of one imaginary frequency and confirmed with intrinsic reaction coordinate (IRC)22–24 calculations.25
Figure 2.

Calculated TSSs for the DA step (optimized with SMD(DCM)-M06-2X/6-31G(d)). Free energies (ΔG, normal text) and enthalpies (ΔH, bold, italic) are shown relative to separated reactant species, furan 8a, acryloyl chloride (9), and (S)-(−)-BTM (energies in kcal/mol).
The computed free energy barriers for the retro-DA cycloaddition were found to be approximately 19 – 23 kcal/mol (ΔG), consistent with reversibility at 23 °C (Figure 2). The DA reactions were found to be concerted, but highly asynchronous, with one C–C bond formation preceding the other (e.g. TS1exo and TS1endo in Figure 2). For the endo pathway, a structure resembling an intermediate for a stepwise process could be optimized to a minimum (e.g. INT1endo in Figure 2), but this structure is higher in energy than the flanking TSSs (TS1endo and TS2endo) when zero-point energy corrections and/or entropy is taken into account. In other words, conversion of structures resembling intermediates in stepwise cycloaddition pathways are predicted to collapse to cycloadducts without a barrier.
Complete pathways for conversion of the cycloadducts to lactam products were not modelled due to various issues with solvent participation, proton transfers, etc.; however, lactam products were optimized to a minimum and their free energies were calculated relative to separated reactant species (Figure 3). The free energies are 2.9 and −12.4 kcal/mol for the endo (10′) and exo (10) lactams, respectively, pointing to a potentially reversible lactamization process (optimized geometries and coordinates available in the ESI) and in particular for the endo adduct 10′. We expect this reversibility comes from the addition of the BTM catalyst to the lactam to reform cycloadducts followed by retro-DA. While the exo (+)-10 lactam is thermodynamically favoured, due to reduced angle strain and the gauche effect, we predict the endo 10′ diastereomer, for which formation is endergonic, will reverse quickly to the corresponding cycloadduct, providing a relief in ring strain (Figure 2b).26,27 For the exo lactam, the nitrogen and bridged oxygen are gauche, which allows for a favourable σC-H↔σ*C-O interaction. However, the N and O are anti in the endo lactam, resulting in a less favourable interaction between the σN-C orbital, a worse donor, and the σ*C-O. The reversibility of the endo-DA adduct could not be tested experimentally since it was never isolated, however we verified that the exo-adduct does not undergo reversible DA since no erosion of enantioselectivity was observed upon resubjection to the reaction conditions. Alternatively, lactamization of the endo cycloadduct may have too high a barrier to compete with lactamization to the exo lactam. This would lead to a Curtin-Hammet situation in which the endo/exo DA adducts can equilibrate but the differential ΔG≠ for the lactamization leads to exclusive formation of the exo adduct.
Figure 3.
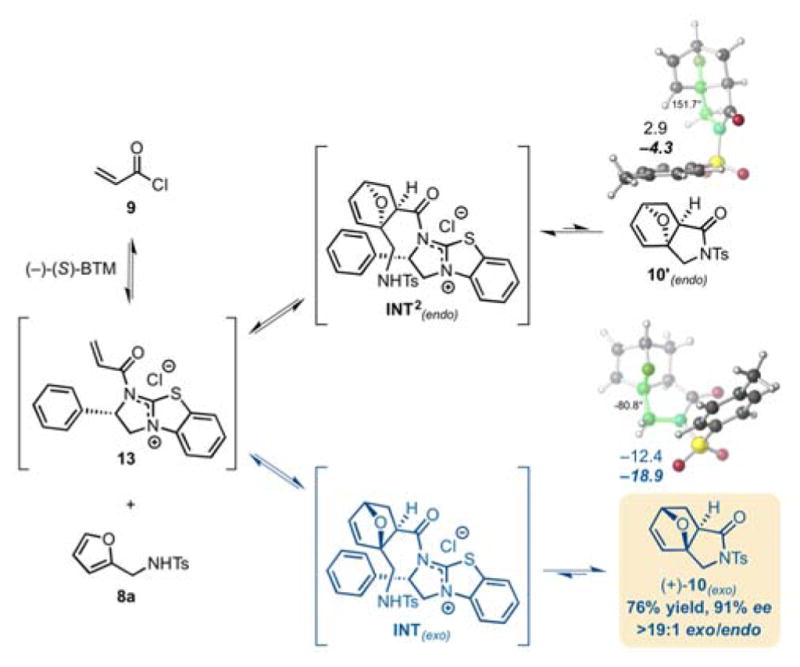
Potential funneling of endo 10′ diastereomer to the thermodynamically favored exo (+)-10 diastereomer involving a retro-Diels-Alder/Diels-Alder-lactamization sequence. Optimized geometries of endo 10′ and exo (+)-10 lactams calculated using SMD(DCM)-M06-2X/6-31G(d). Select dihedral angles (O-C-C-N, green) are shown.
Toward demonstrating the utility of the described DAL organocascade process, we targeted further functionalization of cycloadducts and applications to the core structures of two bioactive molecules, an isatasine A derivative and a non-peptidyl grehlin-receptor inverse antagonist (Figure 4). The acetonide derivative of the natural product (−)-isatisine A was an artifact obtained during isolation found to exhibit cytotoxic activity against the leukemic C8166 cell line and also anti-HIV activity.28 Lactam (+)-12a was converted in a three-step, single-pot process to a fully substituted tetrahydrofuran (−)-15 with correspondence to the core structure of (−)-isatisine A. The process involved ozonolytic cleavage of the olefin followed by in situ double Wittig olefination to deliver diester (−)-15. The cycloadduct 12a, bearing an oxa-bridged tricyclic γ-lactam, also bears resemblance to the nonpeptidyl ghrelin-receptor inverse agonists that were recently disclosed by 7TM Pharma.29 In addition, epoxidation of the tricyclic γ-lactam (+)-12a furnished a fully substituted cyclohexane bearing four fused rings with six contiguous stereogenic centers, (+)-14, as needle-like crystals. This enabled unambiguous assignment of the absolute and relative configuration of cycloadduct (+)-12a (Figure 4, inset; ESI, Figure S1).
Figure 4.
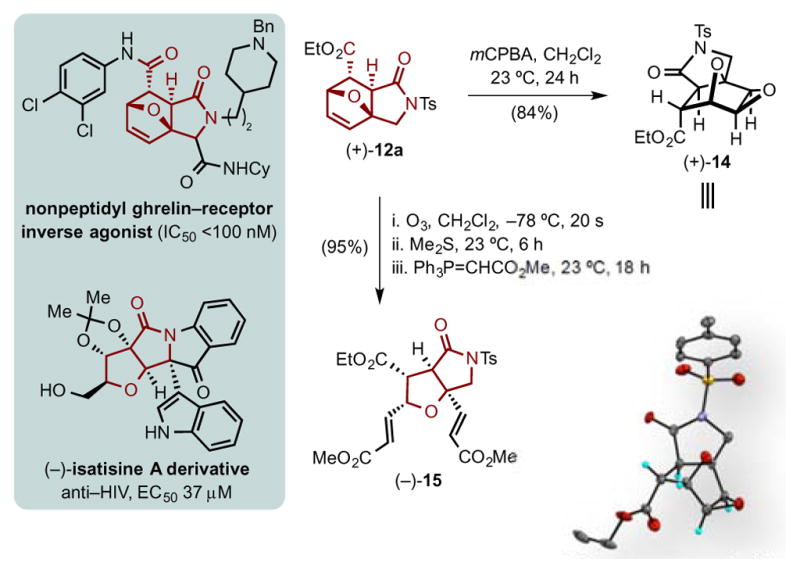
Epoxidation of tricyclic γ-lactam (+)-12a to a fully substituted oxa-bridged cyclohexane (+)-14 with six contiguous stereocenters. Transformation of (+)-12a to a fully substituted tetrahydrofuran (−)-15 with correspondence to the natural product, isatisine A. (Inset: single crystal X-ray structure in ORTEP format of (+)-14; 50% probability, see ESI Figure S1).
In summary, the first highly enantio- and diastereoselective organocatalytic DA cycloaddition of a furanyl diene is described through a Diels-Alder-lactamization organocascade employing chiral, α,β-unsaturated acylammonium salts as dienophiles. The use of a furan with a pendant sulfonamide led to the generation of oxa-bridged trans-fused tricyclic γ-lactams. Computational evidence was gathered to support the notion that these DAL organocascades are susceptible to thermodynamic control through potential reversible lactamization and Diels-Alder steps. While the scope of this furan DAL is limited at this time to highly electron-poor dienophiles, the striking simplicity, excellent diastereo- and enantioselectivity, and high yields obtained with commodity acid chlorides including acryloyl and fumaroyl chloride render this a promising process for de novo, rapid synthesis of polycyclic scaffolds including heterocyclic rings with multiple stereocenters useful for diversity-oriented synthesis.
Supplementary Material
Acknowledgments
Support from NSF (CHE-1112397 to D.R.; CHE-030089 with XSEDE to D.J.T.), the Robert A. Welch Foundation (AA-1280 to D.R.), ACS PRF (52801-ND4 to D.J.T.), and partial support from NIH (GM 052964 to D.R.) is gratefully acknowledged. Drs. Nattamai Bhuvanesh and Joe Reibenspies (Center for X-ray Analysis, TAMU) secured X-ray data, and Dr. Bill Russell (Laboratory for Biological Mass Spectrometry, TAMU) provided mass data.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and characterization details for all new compounds including 1H and 13C NMR spectra, computational data, crystallographic data, chiral phase-HPLC traces. Representative examples of unsuccessful DAL reactions employing furan dienes with pendant alcohols.
References
- 1.Tobia D, Harrison R, Phillips B, White TL, DiMare M, Rickborn B. J Org Chem. 1993;58:6701. [Google Scholar]; Foster RW, Benhamou L, Porter MJ, Bučar DK, Hailes HC, Tame CJ, Sheppard TD. Chemistry. 2015;21:6107. doi: 10.1002/chem.201406286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warrener RN, Margetic D, Sun G. Tetrahedron Lett. 2001;42:4263. [Google Scholar]; Zubkov FI, Zaytsev VP, Nikitina EV, Khrustalev VN, Gozun SV, Boltukhina EV, Varlamov AV. Tetrahedron. 2011;67:9148. [Google Scholar]; Froidevaux V, Borne M, Laborbe E, Auvergne R, Gandini A, Boutevin B. RSC Adv. 2015;5:37742. [Google Scholar]; Lacerda TM, Carvalhoc AJF, Gandini A. RSC Adv. 2016;6:45696. [Google Scholar]
- 3.Evans DA, Barnes DM. Tetrahedron Lett. 1997;38:57. [Google Scholar]
- 4.Evans DA, Barnes DM, Johnson JS, Lectka T, von Matt P, Miller SJ, Murry JA, Norcross RD, Shaughnessy EA, Campos KR. J Am Chem Soc. 1999;121:7582. [Google Scholar]
- 5.Ryu DH, Kim KH, Sim JY, Corey EJ. Tetrahedron Lett. 2007;48:5735. [Google Scholar]
- 6.Liu D, Canales E, Corey EJ. J Am Chem Soc. 2007;129:1498. doi: 10.1021/ja068637r. [DOI] [PubMed] [Google Scholar]
- 7.Mimoto A, Nakano K, Ichikawa Y, Kotsuki H. Heterocycles. 2010;80:799. [Google Scholar]
- 8.Abbasov ME, Hudson BM, Tantillo DJ, Romo D. J Am Chem Soc. 2014;136:4492. doi: 10.1021/ja501005g. [DOI] [PMC free article] [PubMed] [Google Scholar]; Abbasov ME, Hudson BM, Tantillo DJ, Romo D. Chem Sci. 2017;18:1511. doi: 10.1039/c6sc04273b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bappert E, Muller P, Fu GC. Chem Commun. 2006;2604 doi: 10.1039/b603172b.Pandiancherri S, Ryan SJ, Lupton DW. Org Biomol Chem. 2012;10:7903. doi: 10.1039/c2ob26047f.Robinson ERT, Fallan C, Simal C, Slawin AMZ, Smith AD. Chem Sci. 2013;4:2193.Liu G, Shirley ME, Van KN, McFarlin RL, Romo D. Nat Chem. 2013;5:1049. doi: 10.1038/nchem.1788.Vellalath S, Van KN, Romo D. Angew Chem, Int Ed. 2013;52:13688. doi: 10.1002/anie.201306050.Fukata Y, Okamura T, Asano K, Matsubara S. Org Lett. 2014;16:2184. doi: 10.1021/ol500637x.Fukata Y, Asano K, Matsubara S. J Am Chem Soc. 2015;137:5320. doi: 10.1021/jacs.5b02537.Matviitsuk A, Taylor JE, Cordes DB, Slawin AM, Smith AD. Chemistry-A European Journal. 2016;22:17748. doi: 10.1002/chem.201603318.Robinson ER, Frost AB, Elías-Rodríguez P, Smith AD. Synthesis. 2017;49:409.; For a recent review on unsaturated acylammonium salts, see: Vellalath S, Romo D. Angew Chem, Int Ed. 2016;55:13934. doi: 10.1002/anie.201602217.. For a potentially related organocascade involving acyl cyanides, see: Goudedranche S, Bugaut X, Constantieux T, Bonne D, Rodriguez J. Chem Eur J. 2014;20:410. doi: 10.1002/chem.201303613.
- 10.Jung ME, Gervay J. Tetrahedron Lett. 1988;29:2429. [Google Scholar]
- 11.Jung ME, Gervay J. J Am Chem Soc. 1989;111:5469. [Google Scholar]
- 12.Şanli S, Altun Y, Şanli N, Alsancak G, Beltran JL. J Chem Eng Data. 2009;54:3014. [Google Scholar]
- 13.Steinreiber J, Faber K, Griengl H. Chem Eur J. 2008;14:8060. doi: 10.1002/chem.200701643. [DOI] [PubMed] [Google Scholar]
- 14.Córdova A, Ibrahem I, Casas J, Sundén H, Engqvist M, Reyes E. Chem Eur J. 2005;11:4772. doi: 10.1002/chem.200500139. [DOI] [PubMed] [Google Scholar]
- 15.Steinreiber J, Schürmann M, Wolberg M, van Assema F, Reisinger C, Fesko K, Mink D, Griengl H. Angew Chem Int Ed. 2007;46:1624. doi: 10.1002/anie.200604142. [DOI] [PubMed] [Google Scholar]
- 16.Frisch MJ, et al. Gaussian 09, Revision D.01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 17.Zhao Y, Truhlar D. Theor Chem Acc. 2008;120:215. [Google Scholar]
- 18.Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 19.Petersson GA, Bennett A, Tensfeldt TG, Allaham MA, Shirley WA, Mantzaris J. J Chem Phys. 1988;89:2193. [Google Scholar]
- 20.Petersson GA, Allaham MA. J Chem Phys. 1991;94:6081. [Google Scholar]
- 21.Marenich AV, Cramer CJ, Truhlar DG. J Phys Chem B. 2009;113:6378. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 22.Fukui K. Acc Chem Res. 1981;14:363. [Google Scholar]
- 23.Gonzalez C, Schlegel HB. J Phys Chem. 1990;94:5523. [Google Scholar]
- 24.Maeda S, Harabuchi Y, Ono Y, Taketsugu T, Morokuma K. Int J Quant Chem. 2015;115:258. [Google Scholar]
- 25.Ball-and-stick images of computed structures were created with CYLview, 1.0b. Legault, C. Y: Université de Sherbrooke; 2009. ( http://www.cylview.org) [Google Scholar]
- 26.Alabugin IV, editor. Stereoelectronic Effects: A Bridge Between Structure and Reactivity. 1. John Wiley & Sons Ltd; 2016. [Google Scholar]
- 27.Black KA, Wilsey S, Houk KN. J Am Chem Soc. 2003;125:6715. doi: 10.1021/ja021330h. [DOI] [PubMed] [Google Scholar]
- 28.Liu JF, Jiang ZY, Wang RR, Zheng YT, Chen JJ, Zhang XM, Ma YB. Org Lett. 2007;9:4127. doi: 10.1021/ol701540y. [DOI] [PubMed] [Google Scholar]
- 29.Linnanen T, Rist O, Grimstrup M, Frimurer T, Hoegberg T, Nielsen FE, Gerlach LO. WO 2008/092681 2008
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.