

Abstract
Neurons affected in a wide variety of unrelated adult-onset neurodegenerative diseases (AONDs) typically exhibit a “dying back” pattern of degeneration, which is characterized by early deficits in synaptic function and neuritic pathology long before neuronal cell death. Consistent with this observation, multiple unrelated AONDs including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and several motor neuron diseases feature early alterations in kinase-based signaling pathways associated with deficits in axonal transport (AT), a complex cellular process involving multiple intracellular trafficking events powered by microtubule-based motor proteins. These pathogenic events have important therapeutic implications, suggesting that a focus on preservation of neuronal connections may be more effective to treat AONDs than addressing neuronal cell death. While the molecular mechanisms underlying AT abnormalities in AONDs are still being analyzed, evidence has accumulated linking those to a well-established pathological hallmark of multiple AONDs: altered patterns of neuronal protein phosphorylation. Here, we present a short overview on the biochemical heterogeneity of major motor proteins for AT, their regulation by protein kinases, and evidence revealing cell type-specific AT specializations. When considered together, these findings may help explain how independent pathogenic pathways can affect AT differentially in the context of each AOND.
1. INTRODUCTION
Adult-onset neurodegenerative diseases (AONDs) represent some of the most difficult human health challenges remaining. AONDs comprise a heterogeneous group of neurological disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD), as well as motor neuron diseases such as amyotrophic lateral sclerosis (ALS) and hereditary spastic paraplegias (HSPs). Despite their different etiology, all AONDs feature a sustained decline in the functionality of selected neuronal populations, with the identity of such populations playing a major role on the unique set of clinical symptoms that characterize each disease. As a major risk factor for most AONDs is aging, the longer lifespans in the modern era lead to an increased number of people affected by these diseases, making our need to understand pathogenic mechanisms imperative (Mattson and Magnus, 2006).
Novel insights on AONDs pathogenesis arrived with the identification of gene mutations associated with familial forms of AONDs (Jagmag et al., 2015; McGoldrick et al., 2013; Pouladi et al., 2013; Ribeiro et al., 2013; Wong et al., 1998). While clearly major breakthroughs, these genetic studies left investigators in the neurodegenerative disease research community with more questions than answers. More often than not, specific biological functions for most AOND-associated gene products remain unknown and for those gene products with an established function (i.e.; conversion of free superoxide radicals to peroxide by superoxide dismutase 1; SOD1), pathogenic mutations were found that did not affect such function (Pardo et al., 1995). Remarkably, many AOND-associated mutant gene products are widely expressed, yet only certain neuronal populations exhibit pathology while non-neuronal cells and many other neuronal cell types are spared. For example amyloid precursor protein (APP) and the microtubule-associated protein tau are abundant proteins found in all neurons and both are closely associated with pathology in AD (Rosenberg et al., 2016). However, AD-related mutations in APP preferentially affect neurons in the hippocampus and frontal cortex, while sparing neurons in other brain regions such as motor cortex and cerebellum (Gonzalez-Dominguez et al., 2014). On the other hand, mutations in tau protein are not implicated in AD, but are instead associated with a variety of different tauopathies and some forms of frontotemporal dementia; corticobasal degeneration; progressive supranuclear palsy; and Pick’s disease among others) that exhibit degeneration in different neuronal populations (Gonzalez-Dominguez et al., 2014). Alterations in the pattern of tau phosphorylation have also been reported in other unrelated AONDs, such as HD (Gratuze et al., 2016). However, the large number of phosphorylation sites in tau make it difficult to determine their specific contribution to pathogenesis in those diseases (Zerr and Bahr, 2016). Similarly, mutations in the highly abundant and ubiquitously expressed SOD1 cause familial forms of ALS, which primarily features dying back degeneration of upper and lower motor neurons (Ozdinler et al., 2011), leaving other neuronal populations largely spared (Fischer et al., 2004; Wong et al., 2002). Complicating matters further, idiopathic AONDs in patients with no family history cannot easily be distinguished from familial forms and mutations in multiple, structurally unrelated gene products may result in phenotypically indistinguishable forms of AONDs (Fink, 2013; Wong et al., 2002).
More revealing was the observation that most pathogenic gene mutations in familial AONDs are inherited in an autosomal dominant manner, and knocking out these genes failed to replicate AOND symptoms [see for example (Reaume et al., 1996; Zeitlin et al., 1995)]. Collectively, observations derived from genetic findings appear consistent with a scenario where multiple independent pathogenic pathways, typically involving a toxic gain of function, affect cellular processes critical for neuronal function. Identification of these processes and the cellular factors contributing to differential vulnerability of specific neuronal populations in each AOND will prove invaluable for the development of effective therapeutic strategies. As discussed below, an analysis of early pathogenic events common to AONDs provide hints on the identity of such processes.
Neurons affected in AONDs undergo a progressive loss of synaptic and neuritic connectivity
For decades, the significant neuronal loss observed in post-mortem brains of patients affected by AONDs at advanced disease stages focused primary research efforts on understanding of cell death-related mechanisms (Martin, 2001). To that end, identification of mutant genes associated with familial forms of AONDs allowed the development of animal models, which recapitulated major clinical hallmarks observed in human AONDs. These models provided researchers with an unprecedented opportunity to reveal early, presymptomatic pathogenic events in the context of specific AONDs. Remarkably, analysis of multiple unrelated AOND models revealed consistent alterations in neuronal connectivity that were concurrent or even preceded the manifestation of clinical symptoms (Adalbert and Coleman, 2013; Vickers et al., 2009). Phenotypically, such deficits manifested as behavioral and motor abnormalities in the absence of significant neuronal cell death, suggesting that clinical symptoms of AONDs result from neuronal dysfunction or disconnection, rather than loss of neurons (Brady and Morfini, 2010; Coleman, 2011). A significant body of pathological evidence provided a cellular basis for these functional abnormalities, documenting synaptic dysfunction (Henstridge et al., 2016; Wishart et al., 2006) and neuritic atrophy (Bellucci et al., 2016; Fischer and Glass, 2007; Gatto et al., 2015; Kanaan et al., 2013) in animal models of multiple unrelated AONDs. In some familial forms of AONDs, brain imaging-based studies highlighted the relevance of these findings, documenting microstructural alterations in white matter, axon-rich brain areas of living presymptomatic patients (Poudel et al., 2014; Rosas et al., 2010).
Collectively, the available data indicates that neurons affected in AONDs undergo a gradual loss of synaptic and neuritic connectivity, early pathogenic events that appear responsible for the disease-specific neurological symptoms. Accordingly, therapeutic strategies that successfully prevented neuronal cell death in various animal models of AONDs failed to prevent the progression of clinical symptoms (Djaldetti et al., 2003; Gould et al., 2006; Waldmeier et al., 2006) and targeting prevention of neuronal cell death in humans have been similarly ineffective (Waldmeier et al., 2006). Instead, the degeneration pattern of neurons affected in AONDs suggests that maintenance of neuronal connectivity may be a better target for therapeutic intervention than prevention of cell death (Cheng et al., 2010; Lingor et al., 2012). However, such strategies require knowledge of mechanisms underlying loss of connectivity in the context of each AOND (Conforti et al., 2007; Gerdts et al., 2016; Luo and O’Leary, 2005). Unfortunately, the study of mechanisms has been hampered in part due to the scarcity of experimental systems designed to study axon and synapse-specific molecular events in isolation (Grant et al., 2006; Leopold et al., 1994; Llinas et al., 1992).
Pathological hallmarks common to unrelated AONDs: commonalities amid diversity
As a group, AONDs share a number of common features (see Table 1). A major one includes the increased vulnerability of certain populations of projection neurons, which typically extend axons to anatomically distant targets. In contrast, interneurons that bear short axons usually confined to the boundaries of specific brain structures are commomly spared or affected very late in the course of AONDs. Along with other cellular factors, such morphological differences appear to contribute to the differential vulnerability of specific neuronal populations observed in each disease (Han et al., 2010; Mattson and Magnus, 2006; Parent and Parent, 2006; Prensa et al., 2009).
Table 1.
Common Features of Adult-onset Neurodegenerative Diseases.
| Etiological features |
| - Both hereditary and idiopathic forms may exist with the same pathology. |
| Clinical features |
| - Asymptomatic during development and maturation of the nervous system. |
| - Symptoms slowly progress for years after appearance of symptoms. |
| Anatomical features |
| - Projection neurons are affected primarily or exclusively affected. |
| - Select subpopulations of projection neurons are affected. |
| Cellular features |
| - Misfolded proteins are a common element of both familial and idiopathic forms. |
| - Altered synaptic function and neuritic pathology are early pathogenic events of the disease process. |
| Molecular features |
| - Affected neurons exhibit alterations in kinase activity and aberrant pattern of protein phosphorylation. |
| - Affected neurons exhibit deficits in axonal transport. |
At the cellular level, early pathological events common to neurons affected in all AONDs include synaptic dysfunction and neurite atrophy, characteristic features of neurons undergoing dying-back degeneration (Arendt, 2009; Fischer et al., 2004; Li and Conforti, 2013; Morfini et al., 2009a; Wishart et al., 2006). At the molecular level, another notable hallmark common to all AONDs is evidence of alterations in kinase-based signaling pathways, reflected by changes in the phosphorylation pattern of various neuronal protein substrates (Hu and Krieger, 2002; Krieger et al., 2003; Wagey and Krieger, 1998). Finally, data has accumulated indicating that many AONDs exhibit early alterations in axonal transport (AT), a cellular process critical for appropriate maintenance of synaptic function and neuritic connectivity (Morfini et al., 2009a; Roy et al., 2005). Significantly, many of the kinase pathways associated with AONDs also affect AT (Morfini et al., 2009a).
While the specifics of each AOND are distinctive (i.e. identity of affected neurons, which proteins are differentially phosphorylated, and kinase pathways involved), patterns have begun to emerge that connect these common hallmarks above, revealing disease-specific stories with a common mechanistic theme: aberrant activation of selected kinase pathways in each AOND promote a variety of AT alterations, which in turn promote synaptic dysfunction and neuritic pathology in affected neurons (Kanaan et al., 2013; Morfini et al., 2009a). Given the unique cellular topography of each AOND, such a mechanistic view would predict that cell type-specific features may render selected neuronal populations increasingly vulnerable to alterations in a given kinase pathway (Han et al., 2010; Perez-Navarro et al., 2006). Consistent with this notion, some drug treatments produce differential phosphorylation patterns in different neuronal cell types (Bateup et al., 2008; Bertran-Gonzalez et al., 2008). Also, many protein kinases and phosphatases display a unique tissue ditribution pattern (Boulanger et al., 1995) and even ubiquitously expressed protein kinases are expressed at different levels among different neuronal populations (Ackerley et al., 2004; Carboni et al., 1998).
Altered kinase-based signaling in AONDs
Under normal conditions, phosphorylation events are carefully regulated both spatially and temporally within neurons, but these functions are often disrupted in disease state(s) (Walaas and Greengard, 1991). Aberrant phosphorylation of neuronal proteins represents a consistent feature of AONDs that reflects abnormal activation of one or more kinase-based signaling pathways (Tenreiro et al., 2014). Classic examples of aberrantly phosphorylated proteins are tau and neurofilaments, major phosphoprotein components of the axonal cytoskeleton (Grant and Pant, 2000; Holmgren et al., 2012; Khan and Bloom, 2016; Stoothoff and Johnson, 2005).
In AD, for example, a variety of kinases have been associated with aberrant tau phosphorylation including MAP kinases, cyclin-dependent protein kinase 5 (cdk5), casein kinase 2 (CK2), casein kinase l (CK1) and MAP/microtubule affinity-regulating kinases (MARKs), but the kinase activity most consistently associated with tau phosphorylation in AD is glycogen synthase kinase 3 (GSK3) (Wang et al., 2007). Increased phosphorylation of tau is also seen in a variety of other neurodegenerative diseases including PD (Kawakami and Ichikawa, 2015), HD (Gratuze et al., 2015), ALS (Strong et al., 2006), FTDP17 and various tauopathies (Spillantini and Goedert, 2013; Stoothoff and Johnson, 2005). Significantly, recent studies indicate that GSK3p may be activated by pathogenic forms of tau and amyloid beta (Ap) (Hernandez et al., 2010; Kanaan et al., 2011; Pigino et al., 2009). Specifically, pathogenic forms of tau were shown to activate a pathway leading to GSK3 activation within both axons (Kanaan et al., 2011; Lapointe et al., 2009) and synapses (Moreno et al., 2016). In addition, oligomeric forms of amyloid beta (Ap) were found to activate CK2 within these compartments (Moreno et al., 2009; Pigino et al., 2009). Given the complexity and crosstalk among kinase pathways in both normal and pathological states, multiple kinases are likely to promote aberrant patterns of tau phosphorylation in AD and other AONDs (Wang et al., 2007), making the identification of pathologically relevant ones a major challenge for investigators.
Another set of neuronal proteins commonly exhibiting altered phosphorylation in AONDs are the abundant neurofilament subunits NFH and NFM (Grant and Pant, 2000; Grant et al., 2006; Julien and Mushynski, 1998). Accumulation of highly phosphorylated neurofilaments in the initial segment of motor neuron axons is a major hallmark of ALS (Ackerley et al., 2004; Xiao et al., 2006), and many other unrelated AONDs display changes in neurofilament phosphorylation (Holmgren et al., 2012). Findings of abnormal tau and neurofilament phosphorylation in multiple unrelated AONDs likely reflects the large number and variety of consensus phosphorylation sites in these proteins (Veeranna et al., 2008), which may or may not affect their functionality. Regardless, their abundance and degree of phosphorylation in neurons, combined with the availability of antibodies against many of their phospho-specific epitopes makes these proteins a uniquely sensitive reporter for alterations in neuronal kinase activity.
While many studies document aberrant phosphorylation of tau or neurofilaments in AONDs, less consideration has been given to detailing other consequences of altered kinase signaling. Indeed, most kinases have multiple cellular substrates, and a wide variety of cellular processes undergo kinase-based regulation including gene transcription and translation [see for examples (Gao and Roux, 2015; Thapar and Denmon, 2013; Whitmarsh, 2007)], channel functionality (Chahine and O’Leary, 2014), cytoskeleton organization (Rudrabhatla, 2014), and intracellular trafficking of proteins and cellular organelles (Gibbs et al., 2015; Morfini et al., 2009a). As discussed below, a cellular process affected by deregulated kinases with particular relevance for AOND pathogenesis may be AT. This is because the size and complex morphology of neurons make them uniquely vulnerable to changes in intracellular trafficking and, as discussed below, even minor disruptions in AT result in dying-back degeneration of neurons.
Axonal transport (AT): A complex set of intracellular trafficking events critical for the maintenance of neuritic connectivity
Major cellular processes involved in the survival and functionality of most cell types depend upon microtubule-based intracellular trafficking mechanisms. However, appropriate functionality of these mechanisms is particularly important in neurons, because their neuritic processes (i.e.; dendrites and axons), together comprise the overwhelming bulk of the total cell volume, yet the cellular machinery needed for the synthesis and packaging of proteins and lipids is mainly restricted to the somatodendritic compartment (Morfini et al., 2012; Morfini et al., 2001; Pigino et al., 2012). In addition, the remarkable biochemical and functional heterogeneity observed in specific neuritic subcompartments (i.e, pre- and post-synapses, nodes of ranvier, axon hillock, etc) makes the trafficking of molecular components within neuritic processes a major requirement for neuronal function and survival (Britt et al., 2016).
The term “axonal transport” (AT) refers to a wide variety of intracellular trafficking events, all required for appropriate maintenance of synaptic function and neuritic connectivity [reviewed in (Black, 2016; Britt et al., 2016; Maeder et al., 2014; Morfini et al., 2012)]. Such events include the regulated delivery of membrane- bounded organelles (MBOs) synthesized and packaged in the neuronal soma to pre- and post-synaptic compartments, as well as the removal and degradation of old materials. AT also mediates the delivery and turnover of non-synaptic membrane components, metabolic and cytoskeletal elements needed to generate and maintain the structural and biochemical heterogeneity that characterizes axonal and dendritic subdomains. Finally, AT carries neurotrophic signals that provide neuronal somata with information from distant target cells, including the integrity of synaptic connections. Accordingly, AT can be regarded as a major cellular process underlying the generation and maintenance of neuronal architecture and connectivity throughout the long lifetime of neurons (van den Berg and Hoogenraad, 2012). A brief overview of the heterogeneity of the major AT motor proteins reponsible for the execution of AT provides a glimpse on the remarkable complexity of this process.
Conventional kinesin and cytoplasmic dynein are major microtubule-based motor proteins powering axonal transport of membrane-bounded organelles
Microtubule-based molecular motor proteins recognize the intrinsic polarity of microtubules, selectively moving towards their plus or their minus ends (Falnikar and Baas, 2009). Based on this property, the organization of microtubules within neurites provides a basis for the vectorial transport of MBO cargoes in the retrograde (from the cell periphery towards the cell soma) and the anterograde (away from the neuronal soma) directions (Black, 2016; Morfini et al., 2012). Retrograde AT involves the translocation of signaling complexes and MBOs carrying components targeted for degradation from synaptic terminals and distal axons to the neuronal cell body by the multisubunit motor protein complex cytoplasmic dynein (CDyn) (Delcroix et al., 2004; Harrington and Ginty, 2013; Ito and Enomoto, 2016). On the other hand, anterograde AT is powered by members of the kinesin superfamily of motor proteins (KIFs) (Brady, 1995; Brady and Sperry, 1995; Hirokawa et al., 2010).
Based on phylogenetic analysis and sequence homology, KIF superfamily members have been classified into 15 KIF subfamilies, termed kinesin-1 to kinesin-14B (Lawrence et al., 2004). From all these, conventional kinesin represents the most abundant class of KIF superfamily members in the mature nervous system (Brady, 1995; Wagner et al., 1989), being involved in anterograde AT of a wide variety of MBOs including synaptic vesicle precursors, axolemmal proteins, and mitochondria, among others (Elluru et al., 1995; Feiguin et al., 1994; Leopold et al., 1992; Tanaka et al., 1998). As a holoenzyme, conventional kinesin exists as a rod-shaped heterotetramer composed of two heavy chain (KIF5s, kinesin-1s) and two light chain (KLCs) subunits (Deboer et al., 2008) (Fig. 1).
Figure 1.
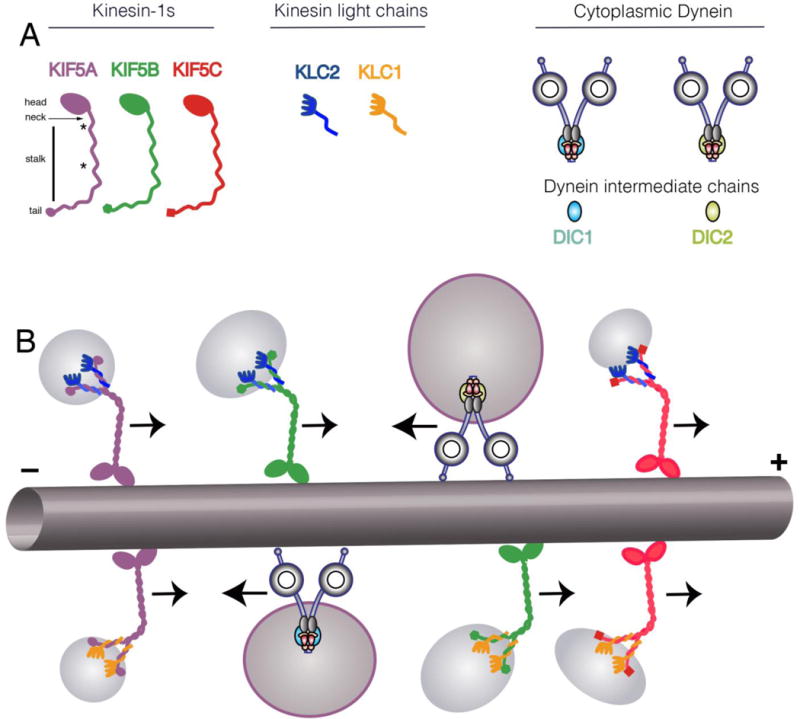
A) Schematic depicting the subunit organization of conventional kinesin holoenzymes. The head domain of kinesin heavy chains (kinesin-1, KIF5s) contains protein motifs for microtubule binding and ATP hydrolysis. Joined by a short neck linker region, the long stalk features coiled-coil and hinge domains mediating homodimerization of kinesin-1s. In addition, kinesin-1s contain a globular tail domain unique to each kinesin-1 subunit variant. On the other hand, KLC subunits associate with the tail domain of conventional kinesin through heteromeric coiled coil domains. Both the carboxy terminus of KLCs and the tail domain of kinesin-1s play a role on the targeting of conventional kinesin variants to MBOs of unique protein composition. Cytoplasmic dynein has a more complex composition with multiple subunits. Unlike kinesin, there is only one gene for the cytoplasmic dynein heavy chain, but other subunits are more diverse. For example, there are two dynein intermediate chain genes and they may exhibit alternative splicing as well as post-translation modification. Thus, molecular motor featuring unique subunit combinations may associate with specific cargoes and may be subject to different signaling pathways for regulation. B) Kinesin-1s and KLCs are organized as homodimers within the conventional kinesin holoenzyme (Deboer et al., 2008), potentially giving rise to six different conventional kinesin variants. In addition, KLC1 undergoes alternative splicing, potentially increasing the number and complexity of such variants. Similarly, cytoplasmic dynein may have either DIC1 or DIC2 as well as variability in other subunits and adapter proteins such as dynactin. The diversity in motors may allow interaction with different cargos (i.e. APP-containing vesicles preferentially interact with kinesin-1C (Szodorai et al., 2009) and signaling endosomes preferentially interact with cytoplasmic dynein containing DIC1 (Ha et al., 2008)) or may allow differential regulation (i.e. GSK3β preferentially modifies KLC2 (Morfini et al., 2002)).
Kinesin-1s are responsible for the mechanochemical properties of conventional kinesin, featuring both microtubule-binding and ATPase domains. In mammals, the kinesin-1 subfamily comprises three genes, which encode KIF5A KIF5B and KIF5C, (Hirokawa et al., 2010) (Fig. 1). KIF5A is thought to be primarily neuronal, while KIF5B and KIF5C are ubiquitously expressed [KIF5C is relatively enriched in many neurons (Kanai et al., 2000)]. Each kinesin-1 appears to fulfill a unique set of specialized functions, as suggested by their differential tissue distribution profiles and the diverse set of phenotypes associated with their deletion (Nakajima et al., 2012; Tanaka et al., 1998; Xia et al., 2003) (see also Fig. 2). However, all kinesin-1s feature a highly conserved domain organization [reviewed in (Jeppesen and Hoerber, 2012)]. A head motor region, located at the amino-terminus, comprises the microtubule binding and ATPase hydrolysis domains responsible for the mechanochemical properties of the conventional klnesin holoenzyme. Towards the carboxy terminus, a neck- linker follows, which plays an important role in processive movement and step coordination. The head and neck-linker domains are followed by a long stalk region containing a hinge and two coiled-coil domains with the first one responsible for dimerization of kinesin-1 subunits and the second involved in mediating interactions with KLC subunits. Finally, the extreme C-terminal tail of kinesin-1 comprises a small globular domain featuring the most divergent sequence stretches among kinesin-1 subfamily members (Jeppesen and Hoerber, 2012).
Figure 2.
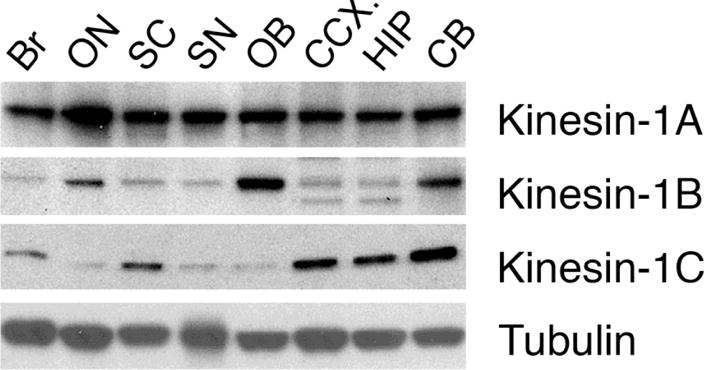
Three genes encoding kinesin-1 subunits are expressed in mammalian nervous system: kinesin-1A (KIF5A), kinesin-1B (KIF5B), and kinesin-1A (KIF5C). Antibodies that selectively recognize each one of these subunits (Deboer et al., 2008) reveal a unique distribution pattern for each subunit in different regions of the central nervous system, suggesting that different neurons display a unique complement of conventional kinesin heavy chain subunit variants: Br whole brain; ON optic nerve; SC spinal cord; SN sciatic nerve; OB olfactory bulb, CCX cerebral cortex; HIP hippocampus; and CB cerebellum. Note that kinesin-1A is widely expressed in nervous tissue, but that there are quantitative differences in different regions. Kinesin-1B and kinesin-1C show more variability among brain regions. This and other observations imply cell type-specific specializations of axonal transport rarely recognized in the literature, which may contribute to the selective vulnerability of specific neuronal populations in different AONDs.
More closely associated to the transported MBO cargo, KLC subunits localize to the end of the stalk domain of conventional kinesin (Fig.1). Two KLC genes are expressed in mammalian nervous tissue (KLC1 and KLC2) and KLC1 undergoes alternative splicing in its extreme carboxy-terminal sequence (Cyr et al., 1991; Rahman et al., 1998). At their amino terminus, KLC subunits feature a coiled-coil region that mediates their heterodimerization with kinesin-1 subunits. In addition, the central region of KLCs features highly conserved DnaJ-like domains (Tsai et al., 2000) [also termed as Tandem Repeat domains (TRs) (Gindhart and Goldstein, 1996)]. In combination with kinesin-1 tail domains, TRs appear to mediate the tight interaction of conventional kinesin with MBO cargoes, and can recruit and activate the chaperone hsc70 for its detachment from MBO surfaces (Tsai et al., 2000).
Co-immunoprecipitation studies demonstrated that KIF5s and KLCs are exclusively organized as homodimers within the conventional kinesin holoenzyme (Deboer et al., 2008). Thus, at least six different variants of conventional kinesin can be defined by their subunit composition (Fig.1B), although many more variants are predicted based on alternative splicing of KLC1, so the ultimate number of unique conventional kinesin motors is uncertain (Morfini et al., 2016) (Fig. 1). The functional significance of these conventional kinesin variants has not been fully established, but cumulative experimental evidence suggests a role in both the binding to selected MBO cargoes (Dahlström et al., 1991; Deboer et al., 2008; Elluru et al., 1995; Szodorai et al., 2009) and their delivery to discrete neuritic subcompartments (Morfini et al., 2016). The diversity of kinesin-1 motors, coupled with the wide variety of cargos and subcellular compartments in neurons, suggest that multiple regulatory pathways must exist to control the delivery of specific cargoes to selected subcellular domains (Brady, 1995; Brady and Sperry, 1995; Morfini et al., 2016; Morfini et al., 2001).
Biochemical hegeterogeneity also exists for the multisubunit motor protein CDyn [reviewed in (Brill and Pfister, 2000; Vallee et al., 2004)] (Fig. 1). In mammals, the main core of this multisubunit motor protein complex features two dynein heavy chain (DHCs), and two intermediate chain (DIC) subunits (Brill and Pfister, 2000). DHCs are responsible for the ATPase and microtuble-binding activities of CDyn, whereas DICs play a role on the binding of CDyn to a wide variety of MBO cargoes (Brill and Pfister, 2000; Vallee et al., 2004). In addition, CDyn can include various other subunits, including light intermediate chains (LICs) and various light chain subnits including LC8, roadblock and T-complex testis-specific protein 1 (TCTEX1) (Vallee et al., 2004), although the specific stoichiometries are unclear. There are also a variety of adaptors and modulators of CDyn such as the dynactin complex, Bic, as well as various proteins including NdI and LIS (Hoogenraad and Akhmanova, 2016). Highlighting the biochemical heterogeneity of CDyn, the two DIC genes found in mammals (DIC1 and DIC2) undergo extensive tissue-specific alternative splicing (Susalka and Pfister, 2000). As observed for conventional kinesin, these findings imply the existence of a large number of CDyn subunit variants (Pfister, 2015). The biochemical heterogeneity of CDyn appears consistent with the wide diversity of cargoes transported by this motor protein, ranging from signalling endosomes (Ha et al., 2008) to microtubules (He et al., 2005; Hoogenraad and Akhmanova, 2016), and the diversity of kinase pathways that regulate the transport of neurotrophins and turnover of membrane components at synapses. Supporting this notion, a specific DIC1 splice variant (IC1B) was found to mediate retrograde AT of neurotrophin receptor-contaning endosomes in hippocampal neurons (Ha et al., 2008).
One size does not fit all: cell type-specific specializations of axonal transport
The presence of isoforms and biochemically diverse subunit variants of molecular motors represent important parameters rarely addressed. The oversimplifications seen in simplified diagrams often obscure key aspects of fundamental mechanisms and lead to erroneous generalizations. In the context of AT, discussions and experiments addressing conventional kinesin or CDyn rarely consider subunit isotypes. Moreover, findings on the functionality and regulation of these molecular motors are often presented as relevant to all neuronal types. However, considerable diversity exists in the complement of microtubule-based motor subunits expressed in neurons, reflecting cell type-specific AT specializations (Fig. 2).
Initial hints of AT specializations stemmed from pulse-chase studies comparing the relative composition of proteins undergoing AT in optic and spinal axons (Brady, 1988; Oblinger et al., 1987), and others addressing transport rates of specific conventional kinesin variants along the rat optic nerve (Elluru et al., 1995). Studies on the composition of specific membrane-bounded cargo vesicles indicate selectivity as well. Thus, analysis of APP containing transport vesicles demonstrate an association with kinesin-1C-containing conventional kinesin motors (Szodorai et al., 2009), while studies on mitochondrial tranpsort have implicated kinesin-1B-based (Tanaka et al., 1998) and kinesin-1A is implicated in transport of GABA receptors (Nakajima et al., 2012). More recently, human pathologies associated with mutations in motor protein subunits extended these initial findings. For example, autosomal dominant loss-of-function mutations in KIF5A lead to a form of hereditary spastic paraplegia (SPG10-HSP) (Blair et al., 2006; Crimella et al., 2011; Reid et al., 2002; Tessa et al., 2008; Wang and Brown, 2010). Intriguingly, SPG10-HSP exhibits selective progressive dysfunction and dying-back degeneration of upper motor neurons, even though most neurons express KIF5A (Fig. 2). While the specific MBO cargoes transported by kinesin-1A-containing variants of conventional kinesin have not been established, these observations suggest a unique dependence of upper motor neurons on the trafficking of cargoes powered by kinesin-1A-contaning variants of conventional kinesin. Alternatively, different kinesin-1 isoforms may be subject to cell type-specific regulatory mechanisms (Morfini et al., 2016). Finally, other kinesin family members may play a greater role in some neuronal populations than others (Hirokawa et al., 2010), but this possibility is less well documented.
As observed for conventional kinesin, phenotypes elicited by mutations in specific CDyn subunits are variable, suggesting cell type-specific requirements in retrograde AT. For example, some mutations in the CDyn-associated protein p150glued primarily cause motor neuron disease (Puls et al., 2003; Puls et al., 2005), but other mutations in this protein are associated with Perry’s syndrome, which is characterized by degeneration of cortical and extrapyramidal neurons and lack motor neuron pathology (Eschbach and Dupuis, 2011; Farrer et al., 2009). Similarly, mutations in DHC subunits differentially promote a wide variety of neurological phenotypes associated with degeneration of specific neuronal populations. Indeed, while some mutations are associated with a peripheral sensory neuropathy, others promote preferentially affect spinal motor neurons or striatal neuron pathology (Braunstein et al., 2010; Hafezparast et al., 2003) Since genetic deletion of DHCs precludes organism development, these observations suggest that either regulation or interaction of CDyn with specific cargos may be differentially compromised in a mutation-specific manner.
In sum, the discovery of neuropathologies associated with mutations in ubiquitously expressed motor protein subunits provides evidence for cell type-specific AT specializations, while also highlighting the unique reliance of selected populations of neurons to discrete alterations in AT. However, most AONDs have not been associated with mutations in motor protein subunits, suggesting that alternative molecular mechanisms underlie the deficits in AT documented in these diseases. Within this context, the identification of kinase-based pathways involved in the regulation of motor proteins provided novel insights on mechanisms linking various neuropathogenic proteins to deficits in AT (Morfini et al., 2009a).
Phosphorylation-dependent regulation of motor proteins
To serve their function, MBOs transported by conventional kinesin need to be precisely delivered to spatially and functionally distinct subdomains within axons and dendrites (van den Berg and Hoogenraad, 2012). For example, in many CNS neurons, neurotransmitter-bearing synaptic vesicles are selectively delivered to hundreds or even thousands of presynaptic terminals en passant, whereas other vesicular MBOs containing ion channels need to be inserted at the plasma membrane within nodes of Ranvier. Such delivery of selected components at specialized neuritic subdomains long implied the existence of targeting mechanisms (Brady, 1995; Elluru et al., 1995; Morfini et al., 2001). The realization that motor proteins are phosphorylated in vivo suggested that phosphorylation may represent a molecular basis for targeted delivery in neurons (Donelan et al., 2002; Hollenbeck, 1990; Hollenbeck, 1993; Morfini et al., 2004; Morfini et al., 2002), but the challenge was to identify relevant kinases involved in the regulation of AT (Morfini et al., 2016; Morfini et al., 2001).
One preparation that provided key insights into candidate regulatory kinases for AT was the isolated squid axoplasm, a unique experimental model for the study of axon-specific molecular events, which earlier proved instrumental for the identification of molecular motors powering AT (Brady et al., 1982; Brady et al., 1990; Kang et al., 2016; Song et al., 2016). Unlike other experimental systems where AT deficits may be an indirect effect of genes in gene expression, the effect of active protein kinases on AT could be directly evaluated in this ex vivo preparation, owing to the lack of plasma membrane. Consistent with the heterogeneity of axonal MBOs and the need for delivering those to specific axonal compartments, studies in isolated axoplasm identified multiple candidate regulatory kinases for AT [reviewed in (Morfini et al., 2001; Morfini et al., 2009b)] (see Table 2). Further, specific phosphorylation events were identified, which differentially regulated the activities of conventional kinesin and CDyn (Gibbs et al., 2015; Martin, 2001; Morfini et al., 2009a).
Table 2.
Effects of selected kinases on AT
| Kinase | Kinesin-1 | Cytoplasmic Dynein |
|---|---|---|
|
| ||
| GSK3β | Inhibits | No effect |
|
| ||
| MAP kinase | ||
| -JNK1 | No effect | No effect |
| -JNK2 | Inhibits | |
| -JNK3 | Inhibits | Inhibits |
|
| ||
| MAP kinase | ||
| -P38α | Inhibits | No effect |
| -P38β | Inhibits | Inhibits |
| -P38γ | Slight inhibition | Slight inhibition |
| -P38δ | No effect | No effect |
|
| ||
| MAP kinase kinase kinase | ||
| -ASK1 | Inhibits | No effect |
| -Tak1 | Inhibits | Inhibits |
|
| ||
| CK1 | Inhibits | Inhibits |
|
| ||
| CK2 | Inhibits | Inhibits |
|
| ||
| PKCδ | Inhibits | Activates |
Through direct or indirect phosphorylation at selected residues, specific kinases were found that modulated non-enzymatic activities of conventional kinesin. For example, GSK3 selectively inhibited conventional kinesin-based anterograde AT, but not CDyn-based retrograde AT in isolated axoplasm (Morfini et al., 2002). Biochemical experiments showed this inhibitory effect involved direct phosphorylation of KLC2 subunits, an event associated with hsc70-mediated release of KLC2-containing conventional kinesin variants from their transported cargoes (Tsai et al., 2000) (Fig. 3). Extending these findings, other kinases were found to regulate AT indirectly by modulating localized GSK3 activation (Morfini et al., 2004; Ratner et al., 1998). Unlike most protein kinases, GSK3 activation requires dephosposphorylation of an inhibitory phosphorylation site (Grabinski and Kanaan, 2016). Interestingly, it was found that inhibition of the major protein kinase CDK5 promotes activation of axonal protein phosphatase 1 (PP1), which in turn dephosphorylated and activated GSK3 (Morfini et al., 2004; Ratner et al., 1998). Additional protein phosphatases may, directly or indirectly, modulate AT. For example, reduced calcineurin (PP2) activity has been reported in animal models of HD (Gratuze et al., 2015), which lead to increased phosphorylation of tau. Interestingly, activation of calcineurin has been reported to enhance glucose-dependent insulin release from beta cells through a dephosphorylation of kinesin 1b (Donelan et al., 2002).
Figure 3.
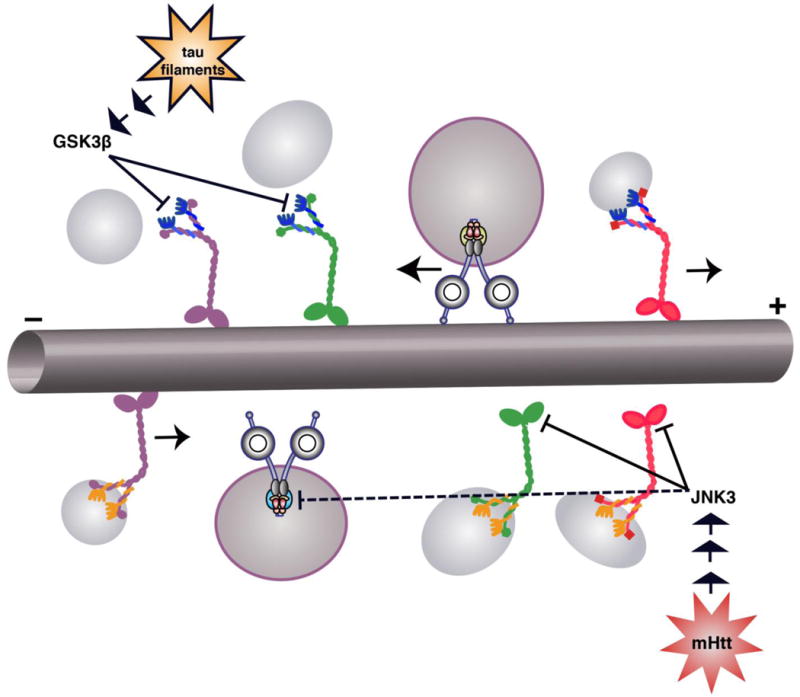
Axonal transport is disrupted by the presence of pathogenic forms of normal neuronal proteins, which lead to activation of specific kinase signaling pathways. For example, tau filaments activate a phosphatase PP1/GSK3P pathway (Kanaan et al., 2011), which preferentially phosphorylates conventional kinesin motors containing KLC2 (Morfini et al., 2002). This phosphorylation in turn leads to release of kinesin from the associated cargo vesicle. Similarly, pathogenic forms of huntingtin activate a MAP kinase pathway leading to JNK3, which preferentially phosphorylates motors containing kinesin-1B and -1C subunits and inhibits their interaction with microtubules (Morfini et al., 2009b). JNK3 also inhibits retrograde AT, but the specific cytoplasmic dynein subunit that is modified has not been identified.
Kinases were also identified that regulated anterograde AT by phosphorylating kinesin-1 subunits of conventional kinesin. One example is the neuron-specific JNK isoform JNK3 which directly phosphorylates a highly conserved serine residue within kinesin-1 head domains, inhibiting the processive movement of conventional kinesin among microtubules (DeBerg et al., 2013; Morfini et al., 2009b). In addition, JNK3 also inhibited retrograde AT, but a molecular basis for this inhibitory effect has not been evaluated. Notably, the ubiquitously expressed JNK isoform JNK1 did not modulate AT, despite sharing a wide variety of JNK3 substrates (Coffey, 2014). Similarly, the motor neuron-enriched p38a isoform (Ackerley et al., 2004; Carboni et al., 1998; Hu et al., 2003; Krieger et al., 2003) selectively inhibited anterograde AT by direct phosphorylation of selected kinesin-1 variants (Bosco et al., 2010; Morfini et al., 2013). In contrast the ubiquitously expressed p38 isoforms p38γ and p385 had little or no effect on AT, while p38β inhibited both anterograde and retrograde AT. Identification of kinase isoform-specific functions can prove difficult, but the isolated axoplasm preparation has proven remarkably useful in the context of AT. Other kinases, including PKC isoforms (Morfini et al., 2007), CK1 and CK2 (Ikeda et al., 2011; Karki et al., 1997; Morfini et al., 2001; Pigino et al., 2009; Pigino et al., 2003; Solowska et al., 2008), have simillarly been reported to affect anterograde and/or retrograde AT in normal and pathological conditions.
Neuropathogenic proteins associated with familial forms of AONDs trigger AT alterations through activation of selected kinase pathways
As a number of kinases that affect AT were identified, an interesting pattern emerged: Many of the kinases identified as regulator of AT were also associated with one or more AONDs (Table 3). Relevant to AD pathogenesis, multiple independent reports documented increased GSK3 activity in AD, as well as in various tauopathies (Cox et al., 2016; Crews and Masliah, 2010; Hernandez and Avila, 2008; Hernandez et al., 2010). Several different pathways may contribute to increased GSK3 activity in AD. For example, GSK3p is activated by pathogenic forms of tau was shown to involve increased exposure of the PAD domain located in the extreme amino terminus of of tau, which in turn promoted PP1-dependent activation of GSK3 via PP1 (Kanaan et al., 2011; Lapointe et al., 2009), changes in AT would be expected in familial forms of AD and tauopathies. Further, increased GSK3 activation and concomitant deficits in AT were also found in mutant APP or presenilin 1-based cellular (Pigino et al., 2003) and mouse-based models (Lazarov et al., 2007). Highlighting the disease relevance of these findings, immunohistochemical analysis of post mortem AD and various tauopathy brain samples demonstrated increased PAD exposure as an early pathogenic event common to all these diseases (Cox et al., 2016).
Table 3.
Signaling Pathways aberrantly activated in selected AONDs.
| Disease | Kinases implicated |
|---|---|
|
| |
| Both Familial and Sporadic forms | |
|
| |
| Alzheimer’s disease | |
| - AD | Cdk5, JNK, MARK, P38 MAPK, CK1, Fyn, Dyrk1A, GSK3 |
| - Aβ oligomers | CK2 |
| - Tau filaments | PP1/GSK3P pathway |
|
| |
| Parkinson’s disease | |
| - Synucleinopathies | Abl, PINK1, LRRK2 |
| - MPTP | PKC5, JNK |
|
| |
| Amyotrophic Lateral Sclerosis | P38 MAPK, TBK, |
| - SOD1 | ASK1, |
|
| |
| Familial forms only | |
|
| |
| Huntington’s disease | JNK3, GSK3P, lKKB, ASK1, PKC5 |
|
| |
| Hereditary Spastic Paraplegia (Spastin, SPG4) | CK2 |
|
| |
| Frontotemporal Dementia with Parkinson’s 17 (FTDP17) | PP1/GSK3P pathway |
Multiple independent reports document changes in AT in animal models of familial ALS (Borchelt et al., 1998; Bosco et al., 2010; Breuer and Atkinson, 1988; Collard et al., 1995; De Vos et al., 2007; Gonzalez de Aguilar et al., 1999; Marinkovic et al., 2012; Morfini et al., 2013; Morotz et al., 2012; Sasaki and Iwata, 1996; Sasaki et al., 2005). In contrast to AD, pathogenesis in ALS has been linked to activation of P38 MAP kinases (Ackerley et al., 2004; Bendotti et al., 2004; Bendotti et al., 2005). Activation of P38 MAP kinases is well documented in motor neurons in spinal cords of both sporadic and familial ALS (Bendotti et al., 2004; Bendotti et al., 2005; Bosco et al., 2010; Morfini et al., 2013). Again, changes in AT would be expected with activation of P38 MAP kinases in ALS (Bosco et al., 2010; Morfini et al., 2013).
Similarly, changes in PKC5 activity have been documented in Parkinson’s disease (Fan et al., 2014; Gordon et al., 2012; Gordon et al., 2016; Hanrott et al., 2008; Kaul et al., 2003) and increased PKC5 alters neuronal homeostasis by increasing retrograde AT and reducing anterograde AT (Morfini et al., 2007; Serulle et al., 2007). Other examples include pathogenic mutant forms of spastin that cause hereditary spastic paraplegia and activate CK2, which inhibits both directions of AT (Solowska et al., 2008). Interestingly. Aβ oligomers associated with AD also activate CK2 and inhibit AT (Moreno et al., 2009; Pigino et al., 2009). Finally, pathogenic forms of huntingtin activate JNK3 which reduces both directions of AT (Morfini et al., 2009b). In each case, kinases that were identified as candidate regulators of AT were misregulated in one or more AOND. As a result, one can expect changes in AT to be a feature of all these diseases and the available evidence validates this prediction.
Concluding remarks
For many years, the focus of research in AONDs was on preventing the loss of neurons. While progress was made in reducing neuronal cell death, there was little progress in affecting the progression of these diseases. As evidence accumulates that preservation of neuronal connectivity and function is a key to addressing these devastating diseases, new therapeutic approaches will need to emerge. Each AOND has a unique pathogenic progression, which means that a better understanding of the cell and molecular basis for specific AONDs is essential. The disruption in normal signaling pathways that is a common feature for many AONDs and the unique role of AT in maintaining neuronal connectivity and function may prove to be critical elements in developing these new therapeutic approaches.
Acknowledgments
The authors would like to acknowledge funding from CHDI and NIH grants RO1 NS066942A, R21 NS096642 (to G.M) and R01-NS023868 and R01-NS041170 and a Zenith Award from the Alzheimer’s Association (to STB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackerley S, et al. p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol Cell Neurosci. 2004;26:354–64. doi: 10.1016/j.mcn.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Adalbert R, Coleman MP. Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol. 2013;39:90–108. doi: 10.1111/j.1365-2990.2012.01308.x. [DOI] [PubMed] [Google Scholar]
- Arendt T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:167–79. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- Bateup HS, et al. Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat Neurosci. 2008;11:932–9. doi: 10.1038/nn.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci A, et al. Review: Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol. 2016;42:77–94. doi: 10.1111/nan.12297. [DOI] [PubMed] [Google Scholar]
- Bendotti C, et al. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J Neuropathol Exp Neurol. 2004;63:113–9. doi: 10.1093/jnen/63.2.113. [DOI] [PubMed] [Google Scholar]
- Bendotti C, et al. Inter- and intracellular signaling in amyotrophic lateral sclerosis: role of p38 mitogen-activated protein kinase. Neurodegener Dis. 2005;2:128–34. doi: 10.1159/000089617. [DOI] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, et al. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–85. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MM. Axonal transport: The orderly motion of axonal structures. Methods Cell Biol. 2016;131:119. doi: 10.1016/bs.mcb.2015.06.001. [DOI] [PubMed] [Google Scholar]
- Blair MA, et al. Mutation in KIF5A can also cause adult-onset hereditary spastic paraplegia. Neurogenetics. 2006;7:47–50. doi: 10.1007/s10048-005-0027-8. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, et al. Axonal transport of mutant superoxide dismutase 1 and focal axonal abnormalities in the proximal axons of transgenic mice. Neurobiol Disease. 1998;5:27–35. doi: 10.1006/nbdi.1998.0178. [DOI] [PubMed] [Google Scholar]
- Bosco DA, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger LM, et al. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neurosci. 1995;15:1532–44. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady S, Morfini G. A perspective on neuronal cell death signaling and neurodegeneration. Mol Neurobiol. 2010;42:25–31. doi: 10.1007/s12035-010-8128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady ST. Cytotypic specializations of the neuronal cytoskeleton and cytomatrix: Implications for neuronal growth and regeneration. In: Haber B, et al., editors. Cellular and Molecular Aspects of Neural Development and Regeneration. Springer-Verlag; New York City: 1988. pp. 311–322. [Google Scholar]
- Brady ST. A kinesin medley: Biochemical and functional heterogeneity. Trends in Cell Biol. 1995;5:159–164. doi: 10.1016/s0962-8924(00)88980-1. [DOI] [PubMed] [Google Scholar]
- Brady ST, et al. Fast axonal transport in extruded axoplasm from squid giant axon. Science. 1982;218:1129–1131. doi: 10.1126/science.6183745. [DOI] [PubMed] [Google Scholar]
- Brady ST, et al. Fast Axonal Transport in Isolated Axoplasm. Cell Motil Cytosk. 1990;17:22. Video Supplement 2. [Google Scholar]
- Brady ST, Sperry AO. Biochemical and functional diversity of microtubule motors in the nervous system. Curr Op Neurobiol. 1995;5:551–558. doi: 10.1016/0959-4388(95)80058-1. [DOI] [PubMed] [Google Scholar]
- Braunstein KE, et al. A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons. Hum Mol Genet. 2010;19:4385–4398. doi: 10.1093/hmg/ddq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer AC, Atkinson MB. Fast axonal transport alterations in amyotrophic lateral sclerosis (ALS) and in parathyroid hormone (PTH)-treated axons. Cell Motil Cytoskeleton. 1988;10:321–30. doi: 10.1002/cm.970100136. [DOI] [PubMed] [Google Scholar]
- Brill LB, 2nd, Pfister KK. Biochemical and molecular analysis of the mammalian cytoplasmic dynein intermediate chain. Methods. 2000;22:307–16. doi: 10.1006/meth.2000.1083. [DOI] [PubMed] [Google Scholar]
- Britt DJ, et al. Mechanisms of Polarized Organelle Distribution in Neurons. Front Cell Neurosci. 2016;10:88. doi: 10.3389/fncel.2016.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni L, et al. Differential expression of SAPK isoforms in the rat brain. An in situ hybridisation study in the adult rat brain and during post-natal development. Brain Res Mol Brain Res. 1998;60:57–68. doi: 10.1016/s0169-328x(98)00166-1. [DOI] [PubMed] [Google Scholar]
- Chahine M, O’Leary ME. Regulation/modulation of sensory neuron sodium channels. Handb Exp Pharmacol. 2014;221:111–35. doi: 10.1007/978-3-642-41588-3_6. [DOI] [PubMed] [Google Scholar]
- Cheng HC, et al. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67:715–25. doi: 10.1002/ana.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey ET. Nuclear and cytosolic JNK signalling in neurons. Nat Rev Neurosci. 2014;15:285–99. doi: 10.1038/nrn3729. [DOI] [PubMed] [Google Scholar]
- Coleman M. Molecular signaling how do axons die? Adv Genet. 2011;73:185–217. doi: 10.1016/B978-0-12-380860-8.00005-7. [DOI] [PubMed] [Google Scholar]
- Collard JF, et al. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- Conforti L, et al. Neuronal death: where does the end begin? Trends Neurosci. 2007;30:159–66. doi: 10.1016/j.tins.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Cox K, et al. Analysis of isoform-specific tau aggregates suggests a common toxic mechanism involving similar pathological conformations and axonal transport inhibition. Neurobiol Aging. 2016;47:113–126. doi: 10.1016/j.neurobiolaging.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet. 2010;19:R12–20. doi: 10.1093/hmg/ddq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimella C, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clinical genetics. 2011;8:157–164. doi: 10.1111/j.1399-0004.2011.01717.x. [DOI] [PubMed] [Google Scholar]
- Cyr JL, et al. Molecular genetics of kinesin light chains: Generation of isoforms by alternative splicing. Proc Nat Acad Sci USA. 1991;88:10114–10118. doi: 10.1073/pnas.88.22.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlström A, et al. The axonal transport motor kinesin is bound to anterogradely transported organelles: Quantitative cytofluorimetric studies of fast axonal transport in the rat. Acta Physiol Scand. 1991;141:469–476. doi: 10.1111/j.1748-1716.1991.tb09107.x. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–8. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerg HA, et al. Motor domain phosphorylation modulates kinesin-1 transport. J Biol Chem. 2013;288:32612–21. doi: 10.1074/jbc.M113.515510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deboer SR, et al. Conventional Kinesin Holoenzymes Are Composed of Heavy and Light Chain Homodimers. Biochemistry. 2008;47:4535–4543. doi: 10.1021/bi702445j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix JD, et al. Trafficking the NGF signal: implications for normal and degenerating neurons. Prog Brain Res. 2004;146:3–23. doi: 10.1016/s0079-6123(03)46001-9. [DOI] [PubMed] [Google Scholar]
- Djaldetti R, et al. Neuroprotection in progressive brain disorders. Isr Med Assoc J. 2003;5:576–80. [PubMed] [Google Scholar]
- Donelan MJ, et al. Ca2+-dependent dephosphorylation of kinesin heavy chain on beta-granules in pancreatic beta-cells. Implications for regulated beta-granule transport and insulin exocytosis. J Biol Chem. 2002;277:24232–24242. doi: 10.1074/jbc.M203345200. [DOI] [PubMed] [Google Scholar]
- Elluru R, et al. Fast axonal transport of kinesin in the rat visual system: functionality of the kinesin heavy chain isoforms. Molec Biol Cell. 1995;6:21–40. doi: 10.1091/mbc.6.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschbach J, Dupuis L. Cytoplasmic dynein in neurodegeneration. Pharmacology & therapeutics. 2011;130:348–63. doi: 10.1016/j.pharmthera.2011.03.004. [DOI] [PubMed] [Google Scholar]
- Falnikar A, Baas PW. Critical roles for microtubules in axonal development and disease. Results Probl Cell Differ. 2009;48:47–64. doi: 10.1007/400_2009_2. [DOI] [PubMed] [Google Scholar]
- Fan Y, et al. Protein kinase C delta mediated cytotoxicity of 6-Hydroxydopamine via sustained extracellular signal-regulated kinase 1/2 activation in PC12 cells. Neurol Res. 2014;36:53–64. doi: 10.1179/1743132813Y.0000000267. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41:163–5. doi: 10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin F, et al. Kinesin-mediated organelle translocation revealed by specific cellular manipulations. J Cell Biol. 1994;127:1021–1039. doi: 10.1083/jcb.127.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126:307–28. doi: 10.1007/s00401-013-1115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–40. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Glass JD. Axonal degeneration in motor neuron disease. Neurodegener Dis. 2007;4:431–42. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- Gao B, Roux PP. Translational control by oncogenic signaling pathways. Biochim Biophys Acta. 2015;1849:753–65. doi: 10.1016/j.bbagrm.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Gatto RG, et al. Analysis of YFP(J16)-R6/2 reporter mice and postmortem brains reveals early pathology and increased vulnerability of callosal axons in Huntington’s disease. Hum Mol Genet. 2015;2:5285–98. doi: 10.1093/hmg/ddv248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, et al. Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron. 2016;89:449–60. doi: 10.1016/j.neuron.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs KL, et al. Regulation of Axonal Transport by Protein Kinases. Trends Biochem Sci. 2015;40:597–610. doi: 10.1016/j.tibs.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Gindhart JG, Jr, Goldstein LS. Tetratrico peptide repeats are present in the kinesin light chain. Trends Biochem Sci. 1996;21:52–53. [PubMed] [Google Scholar]
- Gonzalez de Aguilar JL, et al. A mouse model of familial amyotrophic lateral sclerosis expressing a mutant superoxide dismutase 1 shows evidence of disordered transport in the vasopressin hypothalamo-neurohypophysial axis. Eur J Neurosci. 1999;11:4179–87. doi: 10.1046/j.1460-9568.1999.00840.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Dominguez R, et al. Region-specific metabolic alterations in the brain of the APP/S1 transgenic mice of Alzheimer’s disease. Biochim Biophys Acta. 2014;1842:2395–402. doi: 10.1016/j.bbadis.2014.09.014. [DOI] [PubMed] [Google Scholar]
- Gordon R, et al. Proteolytic activation of proapoptotic kinase protein kinase Cdelta by tumor necrosis factor alpha death receptor signaling in dopaminergic neurons during neuroinflammation. J Neuroinflammation. 2012;9:82. doi: 10.1186/1742-2094-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R, et al. Protein kinase Cdelta upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson’s disease. Neurobiol Dis. 2016;93:96–114. doi: 10.1016/j.nbd.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, et al. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–86. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabinski T, Kanaan NM. Novel Non-phosphorylated Serine 9/21 GSK3beta/alpha Antibodies: Expanding the Tools for Studying GSK3 Regulation. Front Mol Neurosci. 2016;9:123. doi: 10.3389/fnmol.2016.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant P, Pant HC. Neurofilament protein synthesis and phosphorylation. J Neurocytol. 2000;29:843–72. doi: 10.1023/a:1010999509251. [DOI] [PubMed] [Google Scholar]
- Grant P, et al. Squid (Loligo pealei) giant fiber system: a model for studying neurodegeneration and dementia? Biol Bull. 2006;210:318–33. doi: 10.2307/4134568. [DOI] [PubMed] [Google Scholar]
- Gratuze M, et al. Is Huntington’s disease a tauopathy? Brain. 2016;139:1014–25. doi: 10.1093/brain/aww021. [DOI] [PubMed] [Google Scholar]
- Gratuze M, et al. Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington’s disease. Hum Mol Genet. 2015;24:86–99. doi: 10.1093/hmg/ddu456. [DOI] [PubMed] [Google Scholar]
- Ha J, et al. A neuron-specific cytoplasmic dynein isoform preferentially transports TrkB signaling endosomes. J Cell Biol. 2008;181:1027–39. doi: 10.1083/jcb.200803150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezparast M, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–12. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Han I, et al. Differential vulnerability of neurons in Huntington’s disease: the role of cell type- specific features. J Neurochem. 2010;113:1073–91. doi: 10.1111/j.1471-4159.2010.06672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrott K, et al. Differential activation of PKC delta in the substantia nigra of rats following striatal or nigral 6-hydroxydopamine lesions. Eur J Neurosci. 2008;27:1086–96. doi: 10.1111/j.1460-9568.2008.06097.x. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Ginty DD. Long-distance retrograde neurotrophic factor signalling in neurons. Nat Rev Neurosci. 2013;14:177–87. doi: 10.1038/nrn3253. [DOI] [PubMed] [Google Scholar]
- He Y, et al. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol. 2005;168:697–703. doi: 10.1083/jcb.200407191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge CM, et al. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res Rev. 2016;28:72–84. doi: 10.1016/j.arr.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Avila J. The role of glycogen synthase kinase 3 in the early stages of Alzheimers’ disease. FEBS Lett. 2008;582:3848–54. doi: 10.1016/j.febslet.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Hernandez F, et al. GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol. 2010;223:322–5. doi: 10.1016/j.expneurol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, et al. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–38. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. Kinesin heavy and light chains are phosphorylated in vivo in neurons. J Cell Biol. 1990;115:390a. abstract. [Google Scholar]
- Hollenbeck PJ. Phosphorylation of neuronal kinesin heavy and light chains in vivo. J Neurochem. 1993;60:2265–2275. doi: 10.1111/j.1471-4159.1993.tb03513.x. [DOI] [PubMed] [Google Scholar]
- Holmgren A, et al. Neurofilament phosphorylation and their proline-directed kinases in health and disease. J Peripher Nerv Syst. 2012;17:365–76. doi: 10.1111/j.1529-8027.2012.00434.x. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, Akhmanova A. Bicaudal D Family of Motor Adaptors: Linking Dynein Motility to Cargo Binding. Trends Cell Biol. 2016;26:327–40. doi: 10.1016/j.tcb.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Hu JH, et al. Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J Neurochem. 2003;85:422–31. doi: 10.1046/j.1471-4159.2003.01669.x. [DOI] [PubMed] [Google Scholar]
- Hu JH, Krieger C. Protein phosphorylation networks in motor neuron death. Prog Drug Res. 2002;59:71–109. doi: 10.1007/978-3-0348-8171-5_3. [DOI] [PubMed] [Google Scholar]
- Ikeda K, et al. CK1 activates minus-end-directed transport of membrane organelles along microtubules. Mol Biol Cell. 2011;22:1321–9. doi: 10.1091/mbc.E10-09-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Enomoto H. Retrograde transport of neurotrophic factor signaling: implications in neuronal development and pathogenesis. J Biochem. 2016;160:77–85. doi: 10.1093/jb/mvw037. [DOI] [PubMed] [Google Scholar]
- Jagmag SA, et al. Evaluation of Models of Parkinson’s Disease. Front Neurosci. 2015;9:503. doi: 10.3389/fnins.2015.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen GM, Hoerber JK. The mechanical properties of kinesin-1: a holistic approach. Biochem Soc Trans. 2012;40:438–43. doi: 10.1042/BST20110768. [DOI] [PubMed] [Google Scholar]
- Julien JP, Mushynski WE. Neurofilaments in health and disease. Prog Nucl Acid Res Molec Biol. 1998;61:1–23. doi: 10.1016/s0079-6603(08)60823-5. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, et al. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, et al. Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp Neurol. 2013;246:44–53. doi: 10.1016/j.expneurol.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, et al. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. 2000;20:6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, et al. Biochemical analysis of axon-specific phosphorylation events using isolated squid axoplasms. Methods Cell Biol. 2016;131:199–216. doi: 10.1016/bs.mcb.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S, et al. Casein kinase II binds to and phosphorylates cytoplasmic dynein. J Biol Chem. 1997;272:5887–91. doi: 10.1074/jbc.272.9.5887. [DOI] [PubMed] [Google Scholar]
- Kaul S, et al. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18:1387–401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- Kawakami F, Ichikawa T. The Role of alpha-Synuclein and LRRK2 in Tau Phosphorylation. Parkinsons Dis. 2015;2015:734–746. doi: 10.1155/2015/734746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SS, Bloom GS. Tau: The Center of a Signaling Nexus in Alzheimer’s Disease. Front Neurosci. 2016;10:31. doi: 10.3389/fnins.2016.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger C, et al. Aberrant protein kinases and phosphoproteins in amyotrophic lateral sclerosis. Trends Pharmacol Sci. 2003;24:535–41. doi: 10.1016/j.tips.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Lapointe NE, et al. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J Neurosci Res. 2009;87:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CJ, et al. A standardized kinesin nomenclature. J Cell Biol. 2004;167:19–22. doi: 10.1083/jcb.200408113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, et al. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer’s disease-linked mutant presenilin 1. J Neurosci. 2007;27:7011–20. doi: 10.1523/JNEUROSCI.4272-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold PL, et al. The nervous system of Loligo pealei provides multiple models for analysis of organelle motility. In: Abbott NJ, et al., editors. Cephalopod Neurobiology: Neuroscience studies in squid, octopus and cuttlefish. Oxford Univ. Press; Oxford: 1994. pp. 15–34. [Google Scholar]
- Leopold PL, et al. Association of kinesin with characterized membrane-bounded organelles. Cell Motil Cytoskeleton. 1992;23:19–33. doi: 10.1002/cm.970230104. [DOI] [PubMed] [Google Scholar]
- Li JY, Conforti L. Axonopathy in Huntington’s disease. Exp Neurol. 2013;246:62–71. doi: 10.1016/j.expneurol.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Lingor P, et al. Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res. 2012;349:289–311. doi: 10.1007/s00441-012-1362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, et al. Microdomains of high calcium concentration in a presynaptic terminal. Science. 1992;256:677–9. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- Luo L, O’Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–56. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Maeder CI, et al. Axon and dendritic trafficking. Curr Opin Neurobiol. 2014;27:165–70. doi: 10.1016/j.conb.2014.03.015. [DOI] [PubMed] [Google Scholar]
- Marinkovic P, et al. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2012;109:4296–301. doi: 10.1073/pnas.1200658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. Neuronal cell death in nervous system development, disease, and injury (Review) Int J Mol Med. 2001;7:455–78. [PubMed] [Google Scholar]
- Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006;7:278–94. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGoldrick P, et al. Rodent models of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2013;1832:1421–36. doi: 10.1016/j.bbadis.2013.03.012. [DOI] [PubMed] [Google Scholar]
- Moreno H, et al. Tau Pathology Mediated Presynaptic Dysfunction. Neuroscience. 2016;325:30–8. doi: 10.1016/j.neuroscience.2016.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno HH, et al. Synaptic transmission block by presynaptic injection of oligomeric amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5901–6. doi: 10.1073/pnas.0900944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, et al. Axonal Transport. In: Brady ST, et al., editors. Basic Neurochemistry: Principles of Molecular, Cellular and Medical Neurobiology. Elsevier; Boston: 2012. pp. 146–164. [Google Scholar]
- Morfini G, et al. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci U S A. 2007;104:2442–2447. doi: 10.1073/pnas.0611231104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, et al. Conventional kinesin: Biochemical heterogeneity and functional implications in health and disease. Brain Res Bull. 2016;126:347–353. doi: 10.1016/j.brainresbull.2016.06.009. [DOI] [PubMed] [Google Scholar]
- Morfini G, et al. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, et al. Glycogen Synthase Kinase 3 Phosphorylates Kinesin Light Chains and Negatively Regulates Kinesin-based Motility. EMBO Journal. 2002;23:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, et al. Regulation of kinesin: implications for neuronal development. Dev Neurosci. 2001;23:364–376. doi: 10.1159/000048720. [DOI] [PubMed] [Google Scholar]
- Morfini GA, et al. Inhibition of Fast Axonal Transport by Pathogenic SOD1 Involves Activation of p38 MAP Kinase. PLoS One. 2013;8:e65235. doi: 10.1371/journal.pone.0065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, et al. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009a;29:12776–86. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat Neurosci. 2009b;12:864–871. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morotz GM, et al. Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet. 2012;21:1979–88. doi: 10.1093/hmg/dds011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, et al. Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron. 2012;76:945–61. doi: 10.1016/j.neuron.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Oblinger MM, et al. Cytotypic differences in the protein composition of the axonally transported cytoskeleton in mammalian neurons. J Neurosci. 1987;7:453–462. doi: 10.1523/JNEUROSCI.07-02-00453.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdinler PH, et al. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G(3)A transgenic ALS mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:4166–77. doi: 10.1523/JNEUROSCI.4184-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo CA, et al. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci U S A. 1995;92:954–8. doi: 10.1073/pnas.92.4.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent M, Parent A. Relationship between axonal collateralization and neuronal degeneration in basal ganglia. J Neural Transm Suppl. 2006:85–8. doi: 10.1007/978-3-211-45295-0_14. [DOI] [PubMed] [Google Scholar]
- Perez-Navarro E, et al. Cellular and molecular mechanisms involved in the selective vulnerability of striatal projection neurons in Huntington’s disease. Histol Histopathol. 2006;21:1217–32. doi: 10.14670/HH-21.1217. [DOI] [PubMed] [Google Scholar]
- Pfister KK. Distinct functional roles of cytoplasmic dynein defined by the intermediate chain isoforms. Exp Cell Res. 2015;334:54–60. doi: 10.1016/j.yexcr.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, et al. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5907–12. doi: 10.1073/pnas.0901229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, et al. Intracellular Trafficking. In: Brady ST, et al., editors. Basic Neurochemistry: Principles of Molecular, Cellular and Medical Neurobiology. Elsevier; Boston: 2012. pp. 119–145. [Google Scholar]
- Pigino G, et al. Alzheimer’s Presenilin 1 Mutations Impair Kinesin-Based Axonal Transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poudel GR, et al. White matter connectivity reflects clinical and cognitive status in Huntington’s disease. Neurobiol Dis. 2014;65:180–7. doi: 10.1016/j.nbd.2014.01.013. [DOI] [PubMed] [Google Scholar]
- Pouladi MA, et al. Choosing an animal model for the study of Huntington’s disease. Nat Rev Neurosci. 2013;14:708–21. doi: 10.1038/nrn3570. [DOI] [PubMed] [Google Scholar]
- Prensa L, et al. The nigrostriatal pathway: axonal collateralization and compartmental specificity. J Neural Transm Suppl. 2009:49–58. doi: 10.1007/978-3-211-92660-4_4. [DOI] [PubMed] [Google Scholar]
- Puls I, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–6. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Puls I, et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–94. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, et al. Two kinesin light chain genes in mice. J Biol Chem. 1998;273:15395–15403. doi: 10.1074/jbc.273.25.15395. [DOI] [PubMed] [Google Scholar]
- Ratner N, et al. A Role for Cdk5 Kinase in Fast Anterograde Axonal Transport: Novel Effects of Olomoucine and the APC Tumor Suppressor Protein. J Neurosci. 1998;18:7717–7726. doi: 10.1523/JNEUROSCI.18-19-07717.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaume AG, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–7. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- Reid E, et al. A Kinesin Heavy Chain (KIF5A) Mutation in Hereditary Spastic Paraplegia (SPG10) Am J Hum Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro FM, et al. Animal models of neurodegenerative diseases. Rev Bras Psiquiatr. 2013;35(Suppl 2):S82–91. doi: 10.1590/1516-4446-2013-1157. [DOI] [PubMed] [Google Scholar]
- Rosas HD, et al. Altered white matter microstructure in the corpus callosum in Huntington’s disease: implications for cortical “disconnection”. NeuroImage. 2010;49:2995–3004. doi: 10.1016/j.neuroimage.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg RN, et al. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016;73:867–74. doi: 10.1001/jamaneurol.2016.0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, et al. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol (Berl) 2005;109:5–13. doi: 10.1007/s00401-004-0952-x. [DOI] [PubMed] [Google Scholar]
- Rudrabhatla P. Regulation of neuronal cytoskeletal protein phosphorylation in neurodegenerative diseases. J Alzheimers Dis. 2014;41:671–84. doi: 10.3233/JAD-130794. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology. 1996;47:535–40. doi: 10.1212/wnl.47.2.535. [DOI] [PubMed] [Google Scholar]
- Sasaki S, et al. Impairment of axonal transport in the axon hillock and the initial segment of anterior horn neurons in transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol (Berl) 2005;110:48–56. doi: 10.1007/s00401-005-1021-9. [DOI] [PubMed] [Google Scholar]
- Serulle Y, et al. 1-Methyl-4-phenylpyridinium induces synaptic dysfunction through a pathway involving caspase and PKC{delta} enzymatic activities. Proc Natl Acad Sci U S A. 2007;104:2437–2441. doi: 10.1073/pnas.0611227104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowska J, et al. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J Neurosci. 2008;28:2147–2157. doi: 10.1523/JNEUROSCI.3159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, et al. Fast axonal transport in isolated axoplasm from the squid giant axon. Methods Cell Biol. 2016;131:331–48. doi: 10.1016/bs.mcb.2015.07.004. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–97. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Strong MJ, et al. Tau protein hyperphosphorylation in sporadic ALS with cognitive impairment. Neurology. 2006;66:1770–1. doi: 10.1212/01.wnl.0000218161.15834.db. [DOI] [PubMed] [Google Scholar]
- Susalka SJ, Pfister KK. Cytoplasmic dynein subunit heterogeneity: implications for axonal transport. J Neurocytol. 2000;29:819–29. doi: 10.1023/a:1010995408343. [DOI] [PubMed] [Google Scholar]
- Szodorai A, et al. APP anterograde transport requires Rab3A GTPase activity for assembly of the transport vesicle. J Neurosci. 2009;29:14534–44. doi: 10.1523/JNEUROSCI.1546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, et al. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Tenreiro S, et al. Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci. 2014;7:42. doi: 10.3389/fnmol.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessa A, et al. A novel KIF5A/SPG10 mutation in spastic paraplegia associated with axonal neuropathy. Journal of neurology. 2008;255:1090–2. doi: 10.1007/s00415-008-0840-8. [DOI] [PubMed] [Google Scholar]
- Thapar R, Denmon AP. Signaling pathways that control mRNA turnover. Cell Signal. 2013;25:1699–710. doi: 10.1016/j.cellsig.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MY, et al. Release of kinesin from vesicles by hsc70 and regulation of fast axonal transport. Molec Biol Cell. 2000;11:2161–2173. doi: 10.1091/mbc.11.6.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB, et al. Dynein: An ancient motor protein involved in multiple modes of transport. J Neurobiol. 2004;58:189–200. doi: 10.1002/neu.10314. [DOI] [PubMed] [Google Scholar]
- van den Berg R, Hoogenraad CC. Molecular motors in cargo trafficking and synapse assembly. Adv Exp Med Biol. 2012;970:173–96. doi: 10.1007/978-3-7091-0932-8_8. [DOI] [PubMed] [Google Scholar]
- Veeranna A, et al. Neurofilament tail phosphorylation: identity of the RT-97 phosphoepitope and regulation in neurons by cross-talk among proline-directed kinases. J Neurochem. 2008;107:35–49. doi: 10.1111/j.1471-4159.2008.05547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers JC, et al. Axonopathy and cytoskeletal disruption in degenerative diseases of the central nervous system. Brain Res Bull. 2009;80:217–23. doi: 10.1016/j.brainresbull.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Wagey RT, Krieger C. Abnormalities of protein kinases in neurodegenerative diseases. Prog Drug Res. 1998;51:133–83. doi: 10.1007/978-3-0348-8845-5_4. [DOI] [PubMed] [Google Scholar]
- Wagner MC, et al. Copurification of kinesin polypeptides with microtubule-stimulated Mg- ATPase activity and kinetic analysis of enzymatic processes. Cell Motil Cytoskeleton. 1989;12:195–215. doi: 10.1002/cm.970120403. [DOI] [PubMed] [Google Scholar]
- Walaas SI, Greengard P. Protein phosphorylation and neuronal function. Pharmacol Rev. 1991;43:299–349. [PubMed] [Google Scholar]
- Waldmeier P, et al. Recent clinical failures in Parkinson’s disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases. Biochem Pharmacol. 2006;72:1197–206. doi: 10.1016/j.bcp.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Wang JZ, et al. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. The European journal of neuroscience. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brown A. A hereditary spastic paraplegia mutation in kinesin-1A/KIF5A disrupts neurofilament transport. Molecular neurodegeneration. 2010;5:52. doi: 10.1186/1750-1326-5-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim Biophys Acta. 2007;1773:1285–98. doi: 10.1016/j.bbamcr.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Wishart TM, et al. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733–9. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]
- Wong PC, et al. Familial amyotrophic lateral sclerosis and Alzheimer’s disease. Transgenic models. Adv Exptl Med Biol. 1998;446:145–159. [PubMed] [Google Scholar]
- Wong PC, et al. Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci. 2002;5:633–9. doi: 10.1038/nn0702-633. [DOI] [PubMed] [Google Scholar]
- Xia CH, et al. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003;161:55–66. doi: 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, et al. Neuronal intermediate filaments and ALS: a new look at an old question. Biochim Biophys Acta. 2006;1762:1001–12. doi: 10.1016/j.bbadis.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Zeitlin S, et al. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11:155–63. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zerr I, Bahr M. Is there a role of Tau in Huntington’s disease? J Neurochem. 2016;139:9–10. doi: 10.1111/jnc.13762. [DOI] [PubMed] [Google Scholar]