

Abstract
Chromosome conformation capture-based methods such as Hi-C have become mainstream techniques for the study of the 3D organization of genomes. These methods convert chromatin interactions reflecting topological chromatin structures into digital information (counts of pair-wise interactions). Here, we describe an updated protocol for Hi-C (Hi-C 2.0) that integrates recent improvements into a single protocol for efficient and high-resolution capture of chromatin interactions. This protocol combines chromatin digestion and frequently cutting enzymes to obtain kilobase (Kb) resolution. It also includes steps to reduce random ligation and the generation of uninformative molecules, such as unligated ends, to improve the amount of valid intra-chromosomal read pairs. This protocol allows for obtaining information on conformational structures such as compartment and topologically associating domains, as well as high-resolution conformational features such as DNA loops.
Keywords: Hi-C, chromosome conformation capture, paired-end sequencing
1. INTRODUCTION
The spatial organization of chromatin has been a topic of study for many years since chromatin conformation, and long-range associations between genes and distal elements are thought to play important roles in gene expression regulation and other genomic activities. The concept that dense matrices of chromatin interactions could be used to determine the spatial organization of chromatin domains, chromosomes and ultimately entire genomes, was first introduced in the original publication that described the chromosome conformation capture (3C) method [1]. This concept was then tested by development of the 3C technology, its application to yeast chromosomes, and analysis of interaction data using polymer models. This led to the first 3D model of a chromosome.
In 3C, chromatin is first fixed with formaldehyde to covalently link spatially proximal loci. This is essential for efficient detection of chromatin interactions, as leaving out cross-linking leads to dramatic loss of detected contacts and, in our hands, inability to detect chromatin conformation beyond a few Kb. Chromatin is then fragmented with a nuclease and ends are religated. This leads to unique ligation products between spatially proximal loci that can then be detected by PCR, ligation mediated amplification, or direct sequencing.
The concept of using matrices of contact frequencies to infer chromatin folding, and its proof-of-principle in yeast [1] has led to many new studies and the development of a range of 3C-based assays with increased throughput including 4C [2,3], 5C [4,5] and ChIA-PET [6]. Hi-C was introduced in 2009 [7] as a genome-wide version of 3C [1]. The incorporation of biotinylated nucleotides at the digested DNA ends prior to ligation allowed for the specific capture of digested and subsequently ligated chimeric molecules using streptavidin-coated beads. These chimeric molecules are then directly sequenced, e.g. on an Illumina platform. Since its introduction, the technique has gone through several stages of optimization. We have previously presented a base protocol that used incorporation of biotinylated dCTP in an overhang generated by HindIII digestion [8].
Here we describe Hi-C 2.0, a further optimized Hi-C protocol that integrates several recent technical improvements in one single protocol. One adaptation to the base protocol is the removal of a SDS solubilization step after digestion to better preserve nuclear structure. This prevents random ligation between released chromatin fragments by ligation in situ, i.e. in intact nuclei [9]. This adaptation was first introduced for 4C [10] and has since been used for single cell Hi-C [11,12] and more recently for Hi-C [13]. A second adaptation in recently developed protocols increases the resolution of Hi-C through the use of restriction enzymes that digest more frequently, such as MboI and DpnII, or nucleases such as DNaseI and Micrococcal nuclease [13,15–17]. Thirdly, experimental steps can be included to reduce the number of uninformative sequences such as unligated (“dangling”) ends. This is important because even though many topological structures, including compartments and topologically associating domains (TADs) can effectively be resolved by binning 100 million valid pair reads at 100 kb and 40 kb resolution respectively [18–21], detection of point-to-point looping interactions, e.g. between promoters and enhancers or between pairs of CTCF sites typically require >1 billion valid pairs [13]. Therefore, steps to increase the fraction of informative intra-chromosomal reads will help reduce cost by increasing the relative quantity of valid pairs. Removing dangling ends is also important because a sub-population of dangling ends can appear as valid interactions between adjacent restriction fragments. As we describe below, this subgroup can be as large as ~10% of reads, and can represent a large fraction of interactions detected over short distances. This in turn can influence subsequent data analysis (section 3.1).
Alternative approaches to determine chromatin interactions for specific regions of interest in a more cost-effective way include targeted approaches such as 4C [2,3], 5C [4,5], capture Hi-C [22] and Capture C [23,24].
Here we describe Hi-C 2.0 which uses the DpnII restriction enzyme, in situ ligation, and efficient unligated ends removal. A detailed step-by-step protocol is provided in the supplemental materials (“Hi-C 2.0 Protocol”).
2. CAPTURING CHROMOSOME CONFORMATION
2.1. CELL CULTURE & CROSSLINKING CELLS USING FORMALDEHYDE
The objective to increase the resolution of Hi-C requires a robust and efficient capturing of spatial DNA interactions, such as between enhancers and promoters. Digesting the genome into more and smaller pieces of DNA increases both the resolution and the complexity of a Hi-C library. To fully capture individual interactions within this complex library of pair-wise interactions, it is helpful to start with a large amount of cells. As such, even very infrequent interactions can still be captured, but in a statistically significant manner. In our Hi-C protocol, we start with 5 Million cells to ensure the generation of complex libraries. This improvement from 25 Million cells in our previous protocol, has successfully been implemented after adapting to in situ Hi-C [13]. Using a further adaptation to the protocol described here, we have successfully generated libraries with as little as 500,000 cells of starting material. However, the reduced amount of genome copies made these libraries less complex.
We use a final 1% concentration of formaldehyde to crosslink DNA-DNA interactions that are bridged by proteins (Figure 1A). Serum can affect the cross-linking efficiency because it is very rich in proteins and it will compete for formaldehyde. Therefore we replace serum containing medium with serum free medium before fixation. Although formaldehyde-based cross-linking biases have been proposed [25], our current fixation protocol has been the standard for immunoprecipitation and has remained unaltered from previous Hi-C or 3C based protocols [1,8]. Further, any such biases can be removed using several normalization strategies (section 3.1.2).
Figure 1. Overview of the Hi-C method.
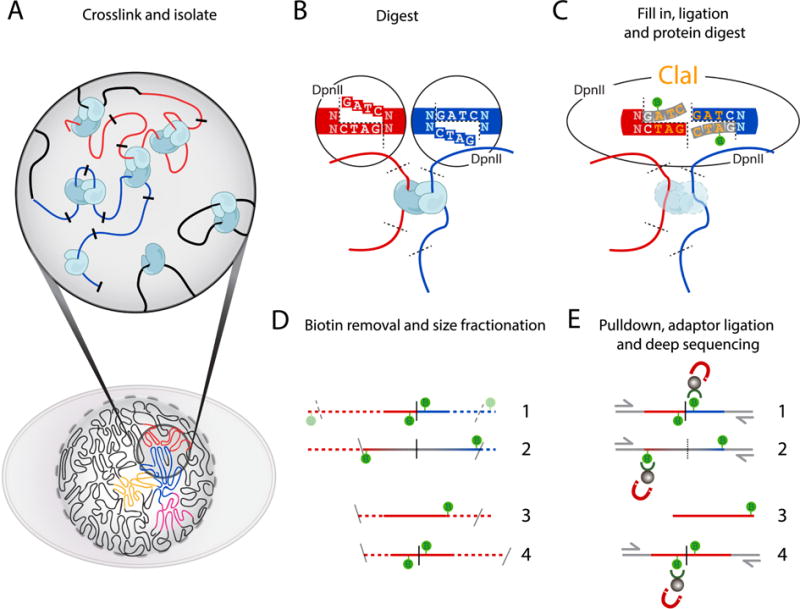
(A) Cells fixed with formaldehyde contain protein-mediated DNA-DNA interactions. (B) DNA digestion with DpnII, recognizing GATC, generates a 5′-GATC overhang. (C) Filling in of the 5′overhang with dNTPs and biotin-14-dATP blunts the overhang. Ligation of the blunted ends creates a new restriction site (ClaI), which can be used to assess fill-in efficiency. After ligation, crosslinks are reversed to remove proteins from DNA. (D) Removal of Biotin (green lollipops) from un-ligated ends. DNA is fragmented to 200–300bp DNA fragments to enable paired-end sequencing. Numbers 1–4 indicate the different ligation products observed: 1: valid interaction; 2: partial digest; 3: dangling end; 4: self-circle. Size fractionation results in fragment size reduction, indicated by dotted lines (E) Enrichment of ligation junctions by using the high affinity of streptavidin-coated beads for the incorporated biotin allows for ligation product enrichment prior to adapter ligation. Numbering of fragment types is as in (D).
We distinguish between adherent and suspension cells in order to fix them in their normal growth conditions. Adherent cells are washed once with the relevant serum-free medium before fixation. Fixation occurs by incubation in formaldehyde containing medium without detaching cells from their growth surface. For suspension cells we replace the wash medium with medium containing formaldehyde after centrifugation.
For both cell types, the formaldehyde is quenched with glycine to terminate the crosslinking. Cells are washed with PBS and pelleted cells can be snap-frozen with dry ice or liquid nitrogen. These cells can be stored at −80°C for up to a year before continuing Hi-C.
2.2. THE HI-C METHOD
2.2.1. Cell lysis and chromatin digestion
We perform Hi-C on lysed cross-linked cells. We use a douncer to lyse the cells in cold hypotonic buffer that is supplemented with protease inhibitors to maintain Protein-DNA complexes. After two rounds of douncing we pellet the material and wash twice with a cold buffer that we will use during digestion. At this point an aliquot of ~ 5% volume can be taken to check the integrity of DNA on an agarose gel.
Before digestion, we incubate the lysed cells in 0.1% of SDS to eliminate proteins that are not cross-linked to DNA, and open the chromatin for a better and more homogenous digestion. The reaction is terminated by addition of triton X-100 to a 1% final concentration. Now the DNA is accessible for digestion by an endonuclease of choice (Figure 1B). The restriction fragment size poses a hard limit to the maximum resolution for 3C-based methods. We previously described the use of HindIII, which has an average fragment length of ~4kb. Previous high resolution Hi-C libraries have used MboI or DpnII [13] to fragment DNA with restriction endonucleases to an average length of ~500 bp. Alternative ways of digestion include alternative enzymes, the use of micrococcal nuclease, which digests in between nucleosomes [16], and random breakage by sonication. Here an endonuclease is used that leaves a 5′overhang, which allows marking the sites of digestion with a biotinylated deoxyribonucleotide during overhang fill-in.
Both DpnII and MboI recognize and digest GATC, and leave a 5′-GATC overhang. We prefer the use of DpnII for eukaryotes, because unlike MboI it is insensitive to CpG methylation. The GATC sequence is frequently found genome-wide and should theoretically result in a median digestion into ~256 base pairs fragments for the 3×109 base pair (bp) human genome. To ensure maximal digestion, chromatin is incubated with DpnII overnight in a thermocycler with interval agitation. After digestion DNA forms a smear of 400–3000 bp on agarose gel (Figure 2A–2). Digestion is terminated by heat inactivation of the restriction enzyme at 65°C for 20 minutes.
Figure 2. Quality Control of Hi-C ligation products.
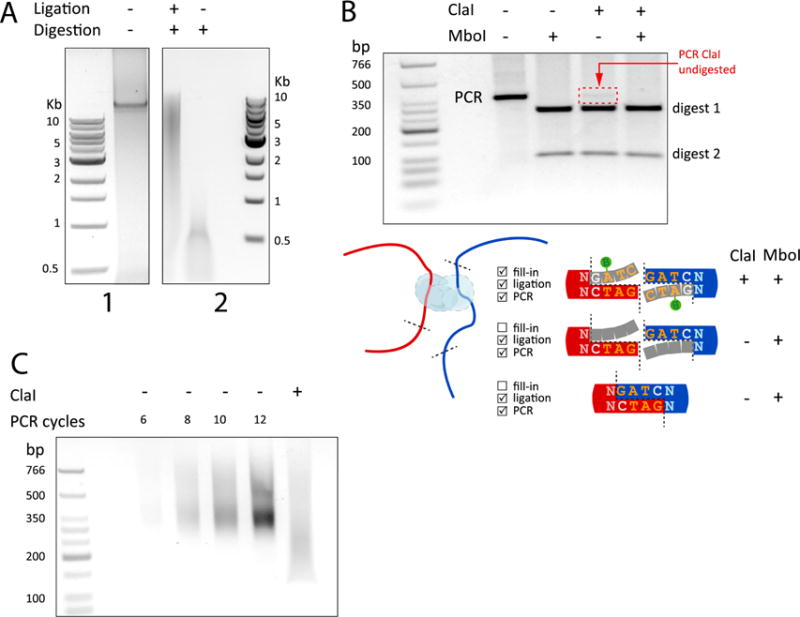
(A.1) Quality control of intact genomic DNA after cell lysis and before digestion. (A.2) Hi-C DNA after digestion and ligation (+,+) compared to unligated, digested control (−,+). Size is indicated by the 1Kb Molecular Weight Ladder from NEB (1 and 2). (B) PCR amplification of a specific ligation product to assess ligation efficiency. The PCR product (lane 1), PCR product digested with MboI (lane2), ClaI (lane3), or both ClaI and MobI (lane4). Only properly filled-in ligation products will be digested with ClaI (see cartoon). This allows for a qualitative comparison to MboI digestion, which cuts GATC sites that are present at the ligation junction of both properly filled-in and non-filled-in ligation products. Digestion of the PCR product using ClaI indicates efficient fill-in and the ClaI undigested fraction from the PCR can be used to estimate the fill-in efficiency (red arrow). The molecular weight ladder used is the Low Molecular Weight Ladder from NEB. (C) PCR titration of the final Hi-C library and quantification of the fill-in and ligation efficiency by ClaI digestion. PCR amplification is performed with primers that recognize the PE adaptors that were ligated to the Hi-C library before sequencing. With 6-cycles of PCR amplification enough DNA was produced for sequencing (lane #1). The last lane shows a downward shift of the amplified library after digestion with ClaI, indicative of efficient fill-in.
2.2.2. Marking of DNA ends with biotin
DNA digestion generates a 5′overhang that is then filled in with deoxyribonucleotides. By strategically replacing one of the deoxyribonucleotides with a biotin-conjugated variant, we can mark the site of digestion and enable enrichment for those sites in a later step. It is this specific fill-in that separates Hi-C from other chromosome conformation capture based methods. For DpnII, we incorporate biotin-14-dATP (Figure 1C). Although the incorporation of biotinylated dCTP is theoretically possible, we have found that this incorporation of a biotinylated nucleotide at the end the overhang leads to less efficient ligation (below).
Klenow fragment of DNA polymerase I is used to fill in the 5′ overhang for 4 hours at 23°C. This low temperature is crucial for efficient incorporation of the large biotinylated dATP and decreases 3′→5′exonuclease activity. Not all overhangs will be filled to completion; therefore, not all digested fragments can be properly ligated. In a later step, after DNA purification, unligated biotinylated ends are removed in a “dangling end removal” step to enrich for proper ligations.
2.2.3. In situ Ligation of proximal ends
Before starting ligation a 10 μl aliquot is taken that will be used to assess digestion efficiency on an agarose gel (Figure 2A, middle lane panel2). The size of the digested DNA is then compared to DNA that was kept aside after lysis before digestion and the DNA that is to be isolated from our ligated Hi-C library. While previous protocols used SDS to inactive the restriction enzyme prior to ligation, here we use an “in situ” ligation protocol [9–11,13], which leaves out this step and inactivates the restriction enzyme by heat. Leaving out this SDS step has previously been shown to better preserve nuclear structure and reduces random ligation [9]. Chromatin is then ligated for 4 hours at 16°C, which is efficient for most of Hi-C libraries. However, in some cases increasing the ligation time to improve the ligation efficiency may be needed. Note that prolonged ligation may increase random ligation. Ligation of the 2 blunted ends creates a new restriction site that can be used to assess the ligation efficacy (Figure 1C).
This blunt end ligation can lead to specific chimeric ligation products between ends that were in close spatial proximity. However, this process can also generate circularized ligation products of single restriction fragments. These are not informative and are not considered valid pairs (Figure 3B–3).
Figure 3. Possible products generated with Hi-C.
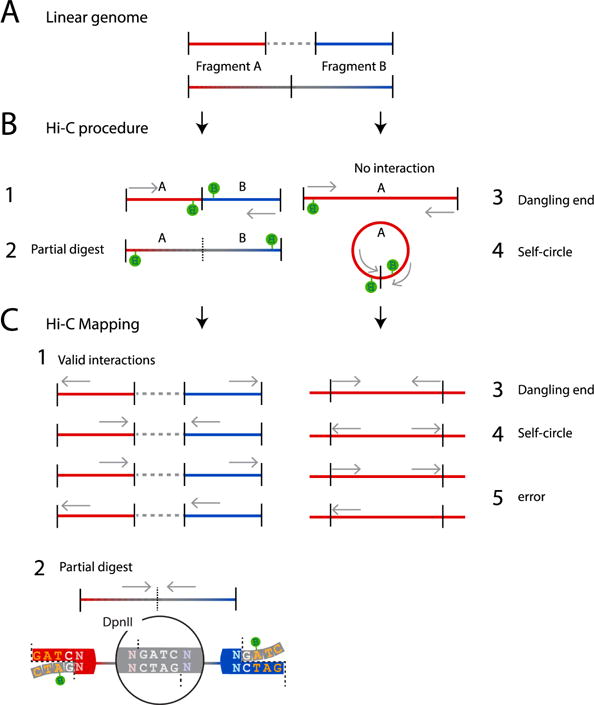
(A) Two fragments: A (red) and B (blue), are spatially separated in the linear genome (gray dotted line) or neighboring (red and blue to gray fading). (B) If fragment A and B are in close spatial proximity they can become cross-linked and ligated during the Hi-C procedure (1). Partial digests result from undigested neighboring fragments that were biotinylated (2). Other possible, non-valid products can be derived from non-ligated DNA (dangling-end; 3) or single fragments that have become circularized after ligation (self-circles; 4). The gray arrow indicate the orientation of the paired-end reads in the Hi-C library (C) Dangling ends can be removed from the Hi-C library prior to sequencing, as described in this protocol. Any remaining dangling-ends and self-circles can be filtered out from the sequenced library computationally after mapping and assessing the orientation of the DNA reads. After mapping, valid reads locate to different fragments in the reference genome and are either inward or outward oriented, or directed in the same direction (both pointing left or both pointing right) (1). Unligated partial digestion products cannot be distinguished from valid reads because the two reads will map to two (neighboring) restriction fragments. This category is characterized by an inward read orientation (2). Invalid reads have mapped to the same fragment in the reference genome and can be either inward (dangling ends; 3), outward (self-circles; 4) or same direction (error; 5). Gray arrows indicate the read orientation in the reference genome.
2.2.4. Reversal of crosslinking and DNA purification
Now that interacting loci are ligated into chimeric pieces of DNA, proteins that hold interacting fragments in close proximity can be removed. This is achieved by thermal reversion of cross-links and incubation with proteinase-K.
After proteinase K treatment DNA is isolated using 2 steps of phenol:chloroform (pH=7.9) and DNA is precipitated using a standard sodium acetate plus ethanol protocol. An Amicon column is used to wash pelleted DNA with low EDTA, tris-buffered water (TLE) to remove any excess of salt.
2.2.5. Quality Control of Hi-C ligation products
During the procedure described above, small aliquots were taken after three key steps in the protocol: lysis, digestion and ligation. DNA isolated from these aliquots can be run on an agarose gel to ascertain the intactness of the DNA prior to digestion, the extent of digestion and efficiency of subsequent ligation. The undigested genomic DNA typically runs as a tight band of over 20 Kb in size (Figure 2A-1). After digestion, the DNA runs as a smear with a size range specific for the applied restriction enzyme (Figure 2A-2). Both of these controls allow for a comparison with the actual library of DNA containing the chimeric ligated ends. These ligated chimeras should have a higher molecular weight than the digestion control and are most likely smaller in size than the undigested control. For DpnII digestion we usually obtain sizes ranging between 3Kb and 10kb (Figure 2A-2).
A second quality control involves quantification of the level of fill-in of overhangs prior to ligation. This is done by PCR amplification of a specific ligation product with primer pairs that were designed for 2 nearby digestion sites (e.g. adjacent restriction fragments) followed by digestion of the PCR product with a restriction enzyme that only cuts at the ligation junction when fill-in has occurred prior to ligation.
Specifically, PCR reactions are set up to detect head-to-head ligation products (Figure 2B–C). Primers are designed near neighboring restriction sites that have a high likelihood of being in close spatial proximity, which can only generate PCR products when properly ligated chimeras are present (Figure 2B). For some endonucleases, including HindIII and DpnII, ligation of the 2 blunt ends generates a new digestion site that can be used to quantify the ligation and fill-in efficiency (NheI for HindIII and ClaI in the case of DpnII; Figure 1C, 2B). After PCR amplification of a specific ligation product, the PCR product is digested with the enzyme that recognizes this newly generated ligation product. Typically the majority of the PCR product is cleaved indicating efficient fill-in (Figure 2B).
2.3. PREPARING CAPTURED CONFORMATIONS FOR DEEP SEQUENCING
2.3.1. Removal of Biotin from un-ligated ends
We have found that in most Hi-C experiments some digested sites will have remained unligated. For example, if the fill-in of some overhangs was incomplete, ligation to a proximal fragment will not occur and the overall ligation will not be 100% efficient. Such cases result in biotinylated but unligated ends. We prefer to remove these “dangling” ends from the Hi-C library, because they would make sequencing less efficient by generating uninformative reads (Figure 1D). Some of these uninformative reads can be readily recognized computationally as both reads will map to a single restriction fragment, and can be easily removed from the dataset (Figure 3C). However, given that digestion in Hi-C is not complete, sequencing of unligated but biotinylated partial digestion products can yield read pairs that map to different restriction fragments and appear as valid interactions. This can be a relatively large number of reads in a given library (below). These apparent valid interactions will be interactions between adjacent restriction fragments, and thus will contaminate very short-range interactions, and lead to over-estimation of the number of intra-chromosomal interactions in general. Experimentally removing these dangling ends of partial digestion products is therefore important.
Our biotin removal step uses T4 DNA polymerase and a low concentration of dNTPs to favor the 3′ to 5′ exonuclease activity over its 5′ to 3′ polymerase activity. By only providing dATP and dGTP, which are complementary to the inside of the 5′ overhang, the polymerase will not be able to complete re-filling the overhang after removing filled in bases. Dangling end removal reduces the level of unligated molecules (dangling ends at single, fully digested, fragments) in the Hi-C library to as low as 0.1–1.5% of read pairs. The in situ Hi-C protocol [13] also produces low amounts of such dangling ends (here 0.2–2.5%, Figure 4A), but we note that this frequency can be more variable between experiments (Figure 4A).
Figure 4. Dangling end removal to increase true valid pair reads.
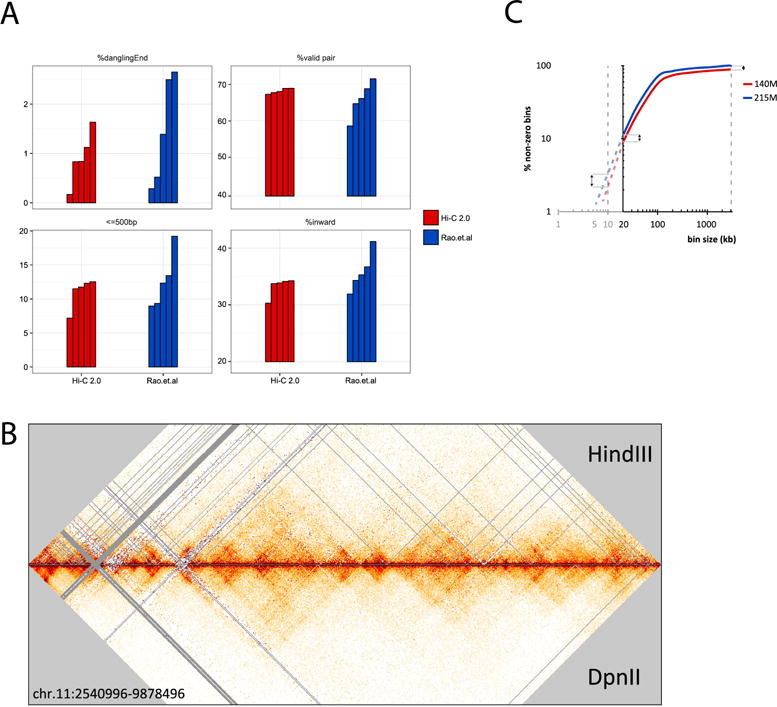
(A) Comparison of frequency of dangling ends, total valid pairs, valid pairs with inward read orientation for short range interactions between nearby fragments (separated by less than 500 bp), and frequency of total inward reads for datasets obtained with in situ Hi-C (SRR1658706, SRR1658593, SRR1658712, SRR1658671, SRR1658648) (Rao et al. [13]) and for datasets obtained with Hi-C 2.0. All datasets were analyzed by 100 bp paired end reads. Datasets from Rao et al. were selected solely based on their read depth that was comparable to datasets obtained with Hi-C 2.0 (100–200 million reads). All data were analyzed through our Hi-C mapping pipeline (available in Github: https://github.com/dekkerlab/cMapping). Hi-C 2.0 and removal of dangling ends results in a more consistent percentage of valid reads. Within the set of valid pair reads, we see a reduction in experiment-to-experiment variation of total amount of inward read pairs. The overrepresentation of inward reads appears due to a large extent to the fact that almost all read pairs between neighboring restriction fragments (separated by less than 500 bp) are inward and this category can be 10–20% of all valid pairs. Hi-C 2.0 reduces the experiment-to-experiment variation of interactions between fragments separated by less than 500 bp. This suggests that at least a subset of interactions between adjacent fragments (interactions separated by less than 500 bp) represent dangling ends. (B) A 10 kb resolution heatmap for chromosome 11 (hg19: 2,540,996–9,878,496 bp) derived from 2 libraries with 300M reads. Libraries were generated with HindII (top triangle) or DpnII (bottom triangle). Color scales are normalized and bins without reads are visualized as gray lines. More unfilled bins (gray) in HindIII are caused by larger fragment sizes (C) An increase in valid pair reads, scored as bins containing at least 1 read (i.e. non-zero), allows for analyses at a higher resolution. The plot compares 2 libraries generated with the same protocol, but with different numbers of valid pair reads (blue: 215 million; red: 140 million). Double arrows indicate that at a higher resolution (smaller bins), adding more valid pair reads (by deeper sequencing) becomes important. These libraries were binned with 20kb as the highest resolution from where dotted lines (red, blue) start extrapolating the data to higher resolutions.
2.3.2. Sonication
In order to sequence both ends of ligation products DNA is sonicated to reduce their size to 200–300 bp in preparation for paired-end sequencing. We prefer to use a Covaris sonicator, because its reproducibility in generating a tight range of DNA fragments. For sequenced reads to be mapped correctly, each end of a read pair should not pass the chimeric ligation junction, since this will result in a sequence that cannot be mapped to a reference genome. Fragments that are 200–300 bp are likely to contain enough mappable sequence at each end before reaching a ligation junction.
2.3.3. Size selection
Covaris sonication results in a relatively small size range of DNA fragments. Therefore, additional size selection could be omitted, but we prefer to use SPRI beads (AMpure) to create an even tighter distribution of fragments. Ampure is a mixture of magnetic beads and polyethylene glycol (PEG-8000). Adding AMpure to a DNA solution reduces the solubility of DNA, because PEG, a crowding agent, will effectively occupy the hydrogen bonds of aqueous solutions. As a result of this crowding, DNA will come out of the solution and bind to the coated magnetic beads. Since larger DNA molecules will come out of solution first, the final concentration of PEG can be used to generate a size cut-off. After sonication 2 consecutive size selections with Ampure are performed. The first AMpure selection will precipitate DNA larger than 300 bp. Using a magnet, bead-bound DNA is separated from the PEG supernatant, which contains fragments smaller than 300 bp. This supernatant undergoes an additional AMpure selection that precipitates DNA larger than 150 bp. Here after, the bead-bound DNA will be narrowly sized to 150–300 base pairs.
2.3.4. End repair
The shearing of DNA by sonication will inevitably damage DNA ends. To repair all the ends after sonication a mix of T4 and Klenow DNA polymerase is used together with T4 polynucleotide kinase (PNK). The first 2 enzymes will repair nicked DNA and single stranded ends, while T4 PNK phosphorylates 5′-ends allowing subsequent A-tailing and adaptor ligation.
2.3.5. Biotin pulldown
To enrich for Hi-C ligation junctions, we use streptavidin-coated beads with a high affinity for the incorporated biotin. This effectively eliminates any DNA without biotin, i.e. DNA that wasn’t properly digested, filled-in and ligated (Figure 1E).
As mentioned above, the step to remove biotin at DNA ends (see 2.3.1) reduces the pulldown of a large fraction of unwanted unligated fragments (Figure 1D–E). However, some unwanted fragments might still be captured. These include self-circled ligation products and other fragments that were insensitive to biotin removal (Figure 3B). For instance, during biotin incorporation, internally nicked DNA could be repaired with biotinylated nucleotides and when too far away from the DNA ends, these incorporated biotinylated nucleotides will not be removed by T4 Polymerase in our biotin removal step. Some of these read pairs (e.g. self-circles) can be removed during bioinformatic analysis of the data.
2.3.6. A-tailing and adaptor ligation
For DNA sequencing Illumina PE adaptors are ligated to both ends of the size selected ligation products. The PE adaptors were generated from DNA oligos, which after duplexing have a 5′-dTTP overhang. This overhang increases ligation efficiency when presented with a free 3′Adenyl. The 3′end of the ligation products are adenylated using dATP and a Klenow fragment lacking 3′ to 5′ exonuclease activity, and then adaptors are ligated using T4 DNA ligase. Depending on whether the preferred sequencing protocol includes multiplexing, one can either use indexed or non-indexed paired-end adapters. We have successfully used paired-end single index adaptors to sequence multiple libraries in a single lane. Strategically choosing the right combination of multiplex adaptors, as suggested by Illumina, at this step is essential when multiplexing is intended.
2.3.7. PCR titration and production
To obtain enough DNA for deep sequencing, the library of ligated fragments is amplified by PCR, using primers designed to anneal to the PE adaptors. Since over-amplification by PCR can result in reduced library complexity, a PCR titration is performed on an aliquot to find the optimal amount of PCR cycles. The smallest number of PCR cycles, producing enough DNA for sequencing will be chosen (Figure. 2C). After PCR, the PCR product is separated from the bead-bound DNA for a final AMpure cleanup. Generally, 6 to 10 PCR cycles will successfully produce enough DNA for sequencing.
As a final quality control an aliquot of the amplified library is digested with ClaI (Figure 2C). Ligation of blunted DpnII sites creates a new ClaI site at the ligation junction. When fill-in and ligation was successful and efficient one expects the majority of the PCR products to be cleaved, resulting in a shift in size compared to undigested PCR product which can be observed by running an aliquot of DNA on an agarose gel (Figure 2C).
2.4. Sequencing
We sequence Hi-C libraries using Illumina 50 bp or 100 paired-end sequencing. Using longer paired end reads (e.g. 100 bp instead of 50 bp) will increase the number of mappable reads. However, as a cautionary note we also found that longer reads will disproportionally increase the number of read pairs with inward read orientations leading to overestimation of very short-range interactions. As mentioned above, such partial digestion products can appear as valid interactions and cannot be filtered out computationally.
3. DATA ANALYSIS
3.1. MAPPING AND BINNING PIPELINE
The paired end sequencing information can be downloaded from the sequencing platform as standard fastq files. The reads are then mapped to a reference genome and valid interaction pairs are identified using read orientation (inward, outward, or same direction). Reads mapping to different fragments are used to assemble the Hi-C dataset. All 4 read strand combinations are possible and are expected to be observed in equal proportions (25% per combination). However, inward read pairs could be the result of undigested restriction sites (partial digests) [26]. Reads mapping to a single fragment are considered uninformative. There are several types of such uninformative single fragment read pairs (Figure 3C): self-ligations (self-circles), unligated fragments (dangling ends) and error pairs. These can be identified by the read orientation, as shown in Figure 3C. Inward pointing reads are considered unligated fragments (dangling ends). Outward pointing reads are classified as self-ligated fragments (“self-circles”). Same-strand reads are classified as “error pairs” as these products are a result of either a mismapping, random break, or an incorrect genome assembly products [26] (Figure 3). Improper ligations can be detected and filtered out by evaluating read orientation of the paired ends and whether the reads map to the same fragment [26]. However, one particular type of improper interaction that is derived from dangling ends cannot be accounted for by computational analysis alone because the two reads map to two (adjacent) restriction fragments. Dangling ends flanking an undigested restriction fragment (partial digest) will computationally be indistinguishable from a valid pair interaction with an inward orientation (Figure 3C). Such read pairs increase in frequency with decreasing restriction fragment size (i.e. when using more frequently cutting enzymes such as DpnII) (Figure 4A). We find that such “inward” read pairs are overrepresented in most Hi-C datasets (i.e. they occur more frequently than the expected 25%), and they represent almost all valid interactions between adjacent restriction fragments. A dangling end removal step can remove a subset of such problematic read pairs by removing unligated partial digest products.
3.1.1. Bias in inward read orientation
Unligated partial digestion products will always produce “inward” read pairs mapping to adjacent or very nearby restriction fragments (genomic distance <500 bp). We compared data we obtained with Hi-C 2.0 to the data published by Rao et al. [13] obtained with in situ Hi-C without dangling end removal (Figure 4A). We find that data obtained with both protocols display a bias in detection of interactions with inward read orientation (average 30–35% of all interactions). There can be quite a large experiment-to-experiment variation in the frequency of the inward read pairs. The number of inward reads between sites separated by less than 500 bp can represent up to 20% of all valid interactions in some datasets. Almost all such short-range interactions are in fact of the inward type, indicating that the general bias for inward read orientation is driven to a large extent by very short-range interactions. The experiment-to-experiment variation in this bias appears to be lower with Hi-C 2.0. This indicates that at least a subset of these reads may represent dangling ends of partial digestion products and also suggests that dangling end formation can be quite variable between experiments. In summary, Hi-C 2.0 and in situ Hi-C produce on average similar levels of such potentially problematic molecules (dangling ends, and inward read pair interactions), but Hi-C 2.0 appears to display less experiment-to-experiment variation at least for the set of experiments analyzed here.
3.1.2. Analysis of valid interaction pairs
Valid interaction pairs can be binned at a range of resolutions (e.g. 5–100 Kb bins) [26]. At some point the digestion frequency becomes the limiting factor for obtaining higher resolution. With an average digestion every 4kb for HindIII, a 10kb resolution heatmap will start showing unfilled bins; i.e. bins that are smaller than the restriction fragment covered. These smaller bins require more frequently cutting enzymes such as DpnII to fill them (Figure 4B).
Binned reads are stored as a symmetric matrix with each row and column representing a genomic location (bin). Interacting regions are represented by the number of reads for every bin within this matrix. We use the percentage of bins filled with at least one valid pair read (non-zero) to estimate the resolution that can be obtained (Figure 4C). Matrices are routinely displayed as heatmaps that display these interactions by coloring entries for the amount of reads they contains.
Binned interaction matrices are corrected for intrinsic biases in Hi-C such as read mappability, restriction site density and GC-content. Several approaches have been developed for bias removal. Yaffe and Tanay developed a computational approach to estimate each of these biases and then remove them [27]. We use an iterative correction approach, developed by Imakaev and co-workers to balance the matrix of interactions such that the sum of all interactions genome-wide for each bin adds up to the same number [28]. This correction should remove both known and unknown biases, such as potential cross-linking differences. We note that such balancing methods often exclude very short-range interactions (i.e. the first diagonal of a Hi-C interaction map) from analysis because these are contaminated with a variety of problematic read pairs, as we have outlined above. This reduces the ability to detect short-range interactions.
There are several public pipelines for processing Hi-C data, e.g. HOMER [29], HiCPro [30], HiCUP [31] and Juicer [32]. For a more detailed description of mapping and binning of data using our pipeline, we refer to in Lajoie et al., Methods (2015) [26]. A high quality Hi-C library for mammalian genomes typically has 50–70% of interactions mapping to intra-chromosomal interactions, less than 2 % dangling ends, less than 1% self-ligated circles, and less than 5% PCR redundant interactions per 400 million reads. We note that with the Illumina HiSeq 4000 platform additional apparently redundant reads on the flow cell can be produced that are not due to PCR amplification but the result of sample loading. Additional washing of libraries (on AMpure beads) and optimization of loading has helped us reduce such artifacts. Finally, these numbers can depend on biological state and therefore are only general guidelines for assessment of library quality.
3.2. USING HI-C DATA TO OBTAIN INSIGHTS INTO GENOME FOLDING
Recent research has shown that the genome is composed of several layers of structure, ranging from compartments to topologically associating domains (TADs) and loops (Figure 5). We will briefly describe how those features can be measured. For more details we refer to Lajoie et al., Methods (2015) [26].
Figure 5. Topological structures obtained at increasing resolution.
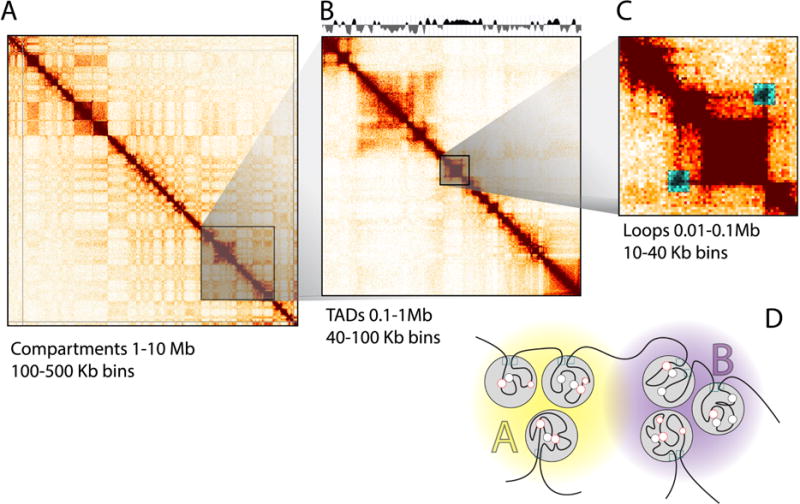
(A) Heatmaps generated from 100 kb binned Hi-C data for chromosome 14 show the alternating pattern of A and B compartments (yellow/purple) (B) On a sub-chromosomal level, heatmaps at 40 kb resolution show the location of TADs, as indicated by an insulation score on top (gray). (C) Within TADs, DNA loops can form that show up as “dots” of interactions in heatmaps of sufficient resolution (typically 10 Kb bins or less). (D) Interpretation of the topological hierarchy obtained from Hi-C. TADs (gray circles) within the same compartment (A or B) interact more frequently than those located in different compartments. TADs are bordered by insulating proteins (e.g. CTCF, cyan squares). DNA loops form between CTCF sites, enhancers and promoters (red/black circles).
3.2.1. Compartments
Compartments are defined as groups of domains, located along the same chromosome or on different chromosomes that display increased interactions with each other. In heatmaps generated from 100Kb bins, this is visible as a specific plaid pattern. These alternating blocks of high and low interaction frequencies represent A and B compartments [7]. Principal component analysis (PCA) readily identifies these compartments that tend to be captured by the first component. The active “A” compartments are gene-dense euchromatic regions, whereas the inactive “B”-compartments are gene-poor heterochromatic regions (Figure 5).
3.2.2. Topologically associated domains (TADs)
TADs are contiguous regions that display high levels of self-association and that are separated from adjacent regions by distinct boundaries [19,21]. The locations of TADs can be determined when interaction data is binned at 40 Kb or less. There are several computational approaches to identify the locations of TAD boundaries including the directionality index [19] or an insulation score algorithm [33] to determine the location of TADs (Figure 5).
3.2.3. Point to point interactions (loops)
Many point-to-point interactions or loops appear as off-diagonal “dots” in a heatmap. Typically, a 10Kb resolution or higher is required for identifying looping interactions. Mapping to smaller bins will allow for more specific interactions, but this comes at the cost of a decreased number of reads per bin. Specific interactions between for instance pairs of CTCF sites are expected to show up as increased signal compared to their surrounding area [13]. Rao et al describe a useful approach to detect such dots using a local background model (Figure 4) [13]. Other types of local interactions, e.g. lines in the heatmap can be detected using global background models [34,35]. Using HiCUP, we have observed loops at 10 kb resolution with a library containing 300M valid reads.
4. CONCLUSIONS
This Hi-C 2.0 protocol combines in situ ligation with dangling end removal to produce Hi-C libraries enriched in intra-chromosomal valid interaction pairs. This protocol can effectively be used to visualize chromosome conformation at Kb resolution genome-wide.
Supplementary Material
HIGHLIGHTS.
Recent innovations in Hi-C experimentation are combined in a single protocol
The rationale for each step in the procedure is provided
A detailed step-by-step protocol is described for performing Hi-C 2.0
Acknowledgments
We would like to thank Liyan Yang, Hakan Ozadam (Dekker lab) and Sarah Hainer (Fazzio lab) for their help in analysis and optimizing this protocol. Work in the Dekker lab is supported by the National Human Genome Research Institute (R01 HG003143, U54 HG007010, U01 HG007910), the National Cancer Institute (U54 CA193419), the NIH Common Fund (U54 DK107980, U01 DA 040588), the National Institute of General Medical Sciences (R01 GM 112720), and the National Institute of Allergy and Infectious Diseases (U01 R01 AI 117839). The authors declare that they have no competing interests. J.D. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science (80-) 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 2.Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 3.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 4.Ferraiuolo MA, Sanyal A, Naumova N, Dekker J, Dostie J. From cells to chromatin: Capturing snapshots of genome organization with 5C technology. Methods. 2012;58:255–267. doi: 10.1016/j.ymeth.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith EM, Lajoie BR, Jain G, Dekker J. Invariant TAD Boundaries Constrain Cell-Type-Specific Looping Interactions between Promoters and Distal Elements around the CFTR Locus. Am J Hum Genet. 2016;98:185–201. doi: 10.1016/j.ajhg.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fullwood MJ, Han Y, Wei CL, Ruan X, Ruan Y. Chromatin interaction analysis using paired-end tag sequencing. Curr Protoc Mol Biol Chapter. 2010;21:1–25. doi: 10.1002/0471142727.mb2115s89. Unit 21 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (80-) 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagano T, Várnai C, Schoenfelder S, Javierre B-M, Wingett SW, Fraser P. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biol. 2015;16:175. doi: 10.1186/s13059-015-0753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Splinter E, de Wit E, van de Werken HJ, Klous P, de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: From fixation to computation. Methods. 2012 doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagano T, Lubling Y, Yaffe E, Wingett SW, Dean W, Tanay A, Fraser P. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat Protoc. 2015;10:1986–2003. doi: 10.1038/nprot.2015.127. [DOI] [PubMed] [Google Scholar]
- 13.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, Geeting KP, Gnirke A, Melnikov A, McKenna D, Stamenova EK, Lander ES, Aiden EL. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma W, Ay F, Lee C, Gulsoy G, Deng X, Cook S, Hesson J, Cavanaugh C, Ware CB, Krumm A, Shendure J, Blau CA, Disteche CM, Noble WS, Duan Z. Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nat Methods. 2014;12:71–78. doi: 10.1038/nmeth.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell. 2015;162:108–119. doi: 10.1016/j.cell.2015.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh THS, Fudenberg G, Goloborodko A, Rando OJ. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods. 2016 doi: 10.1038/nmeth.4025. [DOI] [PubMed] [Google Scholar]
- 18.Valton AL, Dekker J. TAD disruption as oncogenic driver. Curr Opin Genet Dev. 2016;36:34–40. doi: 10.1016/j.gde.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernardi G. Chromosome Architecture and Genome Organization. PLoS One. 2015;10:e0143739. doi: 10.1371/journal.pone.0143739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Bluthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, Herman B, Happe S, Higgs A, LeProust E, Follows GA, Fraser P, Luscombe NM, Osborne CS. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. 2015;47:598–606. doi: 10.1038/ng.3286. [DOI] [PubMed] [Google Scholar]
- 23.Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, Higgs DR. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genet. 2014;46:205–212. doi: 10.1038/ng.2871. [DOI] [PubMed] [Google Scholar]
- 24.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, Chew EG, Huang PY, Welboren WJ, Han Y, Ooi HS, Ariyaratne PN, Vega VB, Luo Y, Tan PY, Choy PY, Wansa KD, Zhao B, Lim KS, Leow SC, Yow JS, Joseph R, Li H, Desai KV, Thomsen JS, Lee YK, Karuturi RK, Herve T, Bourque G, Stunnenberg HG, Ruan X, Cacheux-Rataboul V, Sung WK, Liu ET, Wei CL, Cheung E, Ruan Y. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavrilov A, Razin SV, Cavalli G. In vivo formaldehyde cross-linking: it is time for black box analysis. Brief Funct Genomics. 2015;14:163–5. doi: 10.1093/bfgp/elu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lajoie BR, Dekker J, Kaplan N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 2015;72:65–75. doi: 10.1016/j.ymeth.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat Genet. 2011;43:1059–1065. doi: 10.1038/ng.947. [DOI] [PubMed] [Google Scholar]
- 28.Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wingett S, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P, Andrews S. HiCUP: pipeline for mapping and processing Hi-C data. F1000Research. 2015;4:1310. doi: 10.12688/f1000research.7334.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, Aiden EL. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016;3:95–8. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, Meyer BJ. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015;523:240–244. doi: 10.1038/nature14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013 doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.