

Summary
Many intracellular pathogens co-opt actin in host cells, but little is known about these interactions in vivo. We study the in vivo trafficking and exit of the microsporidian Nematocida parisii, which is an intracellular pathogen that infects intestinal cells of the nematode Caenorhabditis elegans. We recently demonstrated that N. parisii uses directional exocytosis to escape out of intestinal cells into the intestinal tract. Here, we show that an intestinal-specific isoform of C. elegans actin called ACT-5 forms coats around membrane compartments that contain single exocytosing spores, and that these coats appear to form after fusion with the apical membrane. We performed a genetic screen for host factors required for actin coat formation and identified small GTPases important for this process. Through analysis of animals defective in these factors, we found that actin coats are not required for pathogen exit although they may boost exocytic output. Later during infection, we find that ACT-5 also forms coats around membrane-bound vesicles that contain multiple spores. These vesicles are likely formed by clathrin-dependent compensatory endocytosis to retrieve membrane material that has been trafficked to the apical membrane as part of the exocytosis process. These findings provide insight into microsporidia interaction with host cells, and provide novel in vivo examples of the manner in which intracellular pathogens co-opt host actin during their life cycle.
Introduction
Actin is a dynamic, filament-forming protein that contributes to many important cellular functions. To regulate the activity of actin, an army of over 100 accessory proteins control actin polymerization and depolymerization, including the Rho small GTPases (smGTPases) Cdc42 and Rac1 (Heasman and Ridley, 2008; Pollard and Cooper, 2009). Cell motility, cell division, organelle movement and vesicle trafficking are just a few examples of complex cellular processes that are facilitated by actin. For example, studies of clathrin-mediated endocytosis have shown that actin can contribute to plasma membrane invagination, vesicle neck constriction and scission of vesicle budnecks (Kaksonen et al., 2006). The role of actin in exocytosis is less well understood, and actin can be either a positive or a negative regulator of exocytosis depending on the experimental system. An emerging theme is that actin promotes the last step of exocytosis by forming ‘coats’ around unusually large or insoluble cargo (Porat-Shliom et al., 2013). For example, actin promotes exocytosis of large, endothelial-specific organelles called Weibel–Palade bodies (WPBs), which contain the von Willebrand factor (VWF), an enormous multimeric protein that regulates blood clotting (Nightingale et al., 2011). During exocytosis of large cargo such as VWF, vesicles first fuse with the target membrane, then actin forms a coat to stabilize the fused vesicle and promote expulsion of the cargo (Nightingale et al., 2012). It is not well understood which proteins regulate actin coat formation in exocytosis (Porat-Shliom et al., 2013).
Intracellular pathogens exploit host actin machinery to facilitate multiple stages in their life cycles (Haglund and Welch, 2011). For example, the bacterial pathogen Listeria monocytogenes stimulates actin-dependent restructuring and clathrin-mediated endocytosis to trigger its invasion into host cells (Pizarro-Cerda et al., 2012). After invasion, several pathogens, including Listeria, exploit actin polymerization for intracellular motility, as well as cell-to-cell spread. The last stage within the life cycle of an intracellular pathogen is to egress from the host cell in order to propagate infection to new hosts. Here, too, there are examples of actin facilitating exit. Although exocytosis has been implicated in pathogen exit, the role of actin in this process has not been clear, particularly in vivo (Chrisman et al., 2010; Estes et al., 2011; Friedrich et al., 2012; Szumowski et al., 2014).
We have been studying the in vivo life cycle and host cell exit strategy of an intracellular parasite called Nematocida parisii, which naturally infects the intestine of the nematode host Caenorhabditis elegans (Troemel et al., 2008; Felix et al., 2013). N. parisii belongs to the microsporidia phylum, which includes more than 1400 species of intracellular parasites that are related to fungi (Williams, 2009; Texier et al., 2010). Microsporidia can infect a wide range of hosts, including agriculturally relevant hosts such as fish and insects, as well as humans, where they can cause lethal diarrhoea in immunocompromised patients (Didier and Weiss, 2011; Troemel, 2011). Microsporidia are obligate intracellular pathogens and thus undergo their entire replicative cycle inside of host cells (Keeling and Fast, 2002; Vavra and Lukes, 2013). Microsporidia commonly infect intestinal cells, and here, the transparent C. elegans host system provides an excellent model to investigate the life cycle of an intestinal pathogen in vivo. The C. elegans intestine is composed of 20 epithelial cells that have striking structural and functional similarity to human intestinal epithelial cells (McGhee, 2007; Pukkila-Worley and Ausubel, 2012). These cells are polarized, with microvilli on the apical side that are anchored into an actin-rich cytoskeletal structure called the terminal web. N. parisii spores invade the apical side of C. elegans intestinal cells, then replicate inside of these cells in a form called a meront, which then differentiates back into spores that exit exclusively out of the apical side of intestinal cells into the intestinal lumen (Troemel et al., 2008; Estes et al., 2011). Lumenal spores are then defecated out of the animal to infect new hosts as part of a fecal–oral life cycle.
Strikingly, a single live C. elegans animal can shed tens of thousands of spores in a few hours without lysis of intestinal cells, suggesting that the spore production and exit processes are carefully orchestrated inside of these cells (Estes et al., 2011). Indeed, we previously described extensive cytoskeletal restructuring of C. elegans intestinal cells that likely facilitates the N. parisii life cycle. For example, during meront development, gaps form in the terminal web, which may remove a barrier to exit for spores. Interestingly, we also found that at early life stages, meronts replicate in direct contact with the cytosol, but once they differentiate into spores, they are found in separate membrane-bound, spore-containing compartments (SCCs) (Szumowski et al., 2014). These SCCs then fuse with the apical plasma membrane for exit of spores into the intestinal lumen. The host smGTPase RAB-11 is a key player in this directional exocytosis: C. elegans RAB-11 localizes to SCCs and is required for fusion of these compartments with the apical membrane. Accordingly, RAB-11 is also required for spore exit into the lumen, and for host contagiousness (Szumowski et al., 2014). In summary, RAB-11-mediated directional exocytosis appears to be critical for N. parisii exit from C. elegans intestinal cells.
Here, we present data showing that an intestinal-specific isoform of C. elegans actin, ACT-5, forms actin coats around N. parisii SCCs that are exocytosing from intestinal cells into the lumen, similar to the formation of actin coats on large and insoluble kinds of endogenous cargo. We conducted an RNAi screen of C. elegans smGTPases to identify host factors that regulate ACT-5 coat formation on N. parisii SCCs. In particular, we found that formation of ACT-5 coats is dependent on the Rho GTPases cdc-42 and ced-10/Rac1, as well as the Rab GTPases rab-5 and rab-11 that we previously identified in a screen for genes required for spore exit (Szumowski et al., 2014). We found that actin coats form on exocytosing spores that are fused with the plasma membrane, and that ACT-5 coats may boost the exocytic output of spores, although these coats are not required for exit. We also show that later in infection there is clathrin-mediated endocytosis of spore-filled vesicles that are coated with ACT-5, likely as part of compensatory endocytosis to help maintain the balance of the intracellular and apical membrane pools. These findings demonstrate in vivo interactions of host actin machinery that may aid the pathogen in spreading to new hosts, and also help the host in tolerating the intracellular pathogen life cycle.
Results
Host actin forms coats around intracellular N. parisii spores at the apical membrane
Previously, we demonstrated a dose-dependent requirement for the actin isoform ACT-5 in N. parisii spore exit, although it was unclear how this host cytoskeletal structure promoted spore exit (Estes et al., 2011). Here we show that N. parisii spores near the apical membrane acquire an ACT-5 ‘coat’ and we investigated this phenomenon for a potential role in spore exit. Intracellular spores farther from The apical membrane or in the lumen do not exhibit ACT-5 coats (Fig. 1A and B). We observed ACT-5 coats with several transgenic ACT-5 markers, including YFP∷ACT-5 (Fig. 1A and B), as well as mCherry∷ACT-5 and GFP∷ACT-5 (see below). In order to confirm that the ACT-5 labelling was not due to an artefact from overexpressed transgenes, we also stained infected animals with antibodies specific for ACT-5 (MacQueen et al., 2005), and saw similar localization around spores near the apical side of intestinal cells (Fig. 1C). Additionally, we showed in earlier studies that YFP∷ACT-5 transgenic animals shed spores at the same frequency as wild-type animals, suggesting that labelled ACT-5 does not impair function (Estes et al., 2011). Using a strain that labels both ACT-5 and IFB-2, an intermediate filament component in the terminal web at the apical side of intestinal cells, we did not see IFB-2 colocalizing with ACT-5 around spores (Fig. S1A), indicating that actin, but not intermediate filaments, is recruited to these apical spores. To determine what fraction of animals contain actin-coated spores, we examined animals at the spore stage of infection and found that 85–90% of animals with spores had actin coating around some of the spores (Fig. S1B).
Fig. 1.
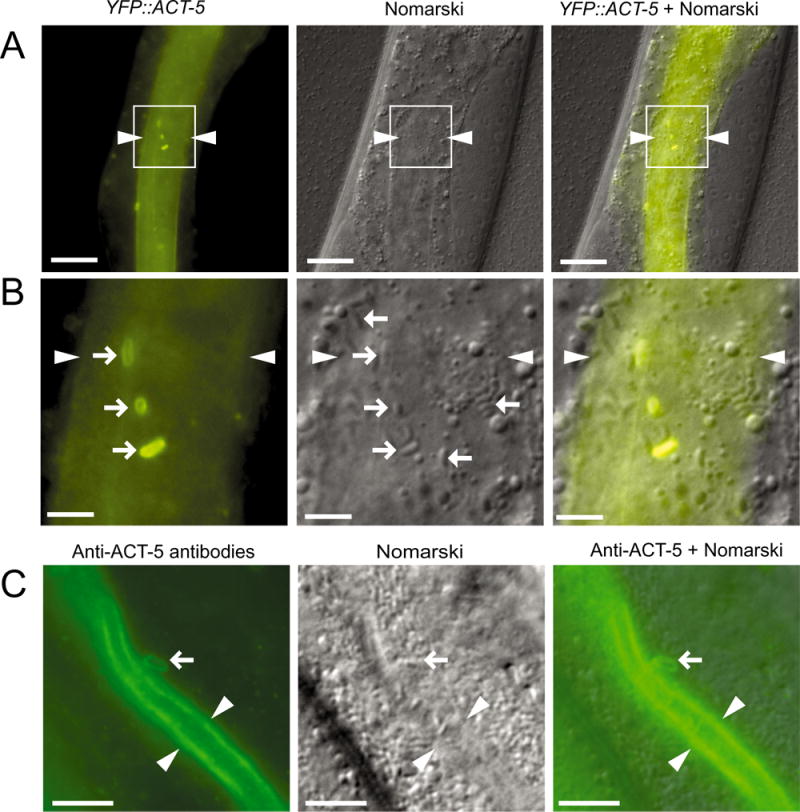
Intracellular N. parisii spores near the lumenal membrane of C. elegans intestinal cells acquire an actin ‘coat’.
A. YFP∷ACT-5 forms ‘coats’ around apically localized spores: region in white box is magnified in B. Arrowheads indicate lumen. Scale bars are 20 μm.
B. Right-pointing arrows indicate apical spores with a YFP∷ACT-5 coat. Left-pointing arrows indicate spores further from the membrane that lack a YFP∷ACT-5 coat. Arrowheads indicate lumen. Scale bars are 5 μm.
C. Anti-ACT-5 antibody staining of infected animals. Arrow indicates spore near lumen with ACT-5 staining similar to those observed with YFP∷ACT-5. Arrowheads indicate lumen. Scale bars are 10 μm.
A–C. Animals imaged at approximately 43 hpi.
ACT-5 forms coats on spores after they have fused with the apical membrane
Because we have been unsuccessful at visualizing pathogen trafficking using live imaging techniques, we assessed trafficking events using single time point analysis of spores with various markers. Our previous studies indicated that N. parisii SCCs fuse with the apical membrane of intestinal cells to ultimately exit out of these cells. Thus, we looked for colocalization of mCherry∷ACT-5-coated spores with the host apical membrane marker PGP-1∷GFP to determine whether these spores have fused with the host apical membrane. Indeed, we found that at 45 hpi, all spores with mCherry∷ACT-5 localization also showed localization of the apical membrane marker PGP-1∷GFP (Fig. 2A and B). In addition, some spores had PGP-1∷GFP localization but did not have mCherry∷ACT-5 coats. These observations suggest that SCCs first fuse with the apical membrane, and then recruit ACT-5 to form coats. Next, we stained with the cell-impermeable dye Calcofluor white (CW) to determine which SCCs had access to the lumen (Szumowski et al., 2014). In this assay, intracellular SCCs that have fused with the apical membrane and have access to the lumen via a fusion pore will be stained with CW as the dye diffuses into the SCC. Here, we found that some fused SCCs had access to the lumen without ACT-5 coats, whereas other fused SCCs had actin coats but no access to the lumen. However, most of the fused spores had both mCherry∷ACT-5 coats and CW staining (Fig. 2A and B). Notably, exocytosed spores that were free in the lumen never exhibited mCherry∷ACT-5 or PGP-1∷GFP, suggesting that these host factors remain inside the cell after exocytosis of the spore from the SCC. Altogether, these results suggest that SCCs likely fuse with the apical membrane before acquiring an ACT-5 coat or gaining access to the lumen. After SCC fusion, it appears that either ACT-5 coat formation or lumenal access can occur first, and that most fused spores are coated in ACT-5.
Fig. 2.

Actin-coated spores are labelled with host plasma membrane marker and have access to the lumen.
A. Two mCherry∷ACT-5-coated spores indicated with arrows are also labelled with the apical plasma membrane marker, PGP-1∷GFP, indicating the SCC has fused with the host apical membrane. Right-pointing arrow shows a spore also stained with CW (blue), indicating it has access to the lumen. Left-pointing arrow only shows the edge of spore staining with CW, perhaps due to partial access to the lumen. Asterisks mark structures that are likely non-specific aggregations of mCherry. Lumenal walls indicated with dotted lines and lumenal space indicated with L. Scale bars are 5 μm.
B. Quantification of marker localization to fused SCCs at 45 hpi. All 68 spores from six animals shown exhibit PGP-1 staining, and the colocalization of ACT-5 and CW staining to these spores is shown. Of note, all spores with actin coats also had PGP-1∷GFP membrane surrounding them.
smGTPases that regulate recycling endosomes and actin polymerization are required for actin coat formation
To investigate which host factors control ACT-5 coat formation, we conducted a feeding RNAi screen of 41 smGTPases, a class of genes involved in multiple intracellular trafficking events. After treatment with RNAi against these genes, the number of ACT-5-coated spores per animal was scored semi-quantitatively (Fig. 3A, and see Experimental procedures). Our screen identified the recycling endocytosis pathway components rab-5, rab-10 and rab-11, with RNAi against rab-5 and rab-11 significantly reducing the number of ACT-5 coats per animal (Fig. 3B). Previously, we found that rab-5, rab-10 and rab-11 were important for spore exit, with rab-11 required for fusion with the apical cell surface, and rab-5 and rab-10 not affecting fusion (Szumowski et al., 2014). Here we also found that the Rho GTPases cdc-42 and ced-10 (Rac1) were required for actin coat formation (Fig. 3B), although they were not previously identified in our screen for spore exit because they do not have a large effect on the level of spore shedding (see below). The Rho GTPases cdc-42 and ced-10/Rac1 are both known actin polymerization factors. We also examined ACT-5 coat formation at later time points (46 and 48 hpi) in animals treated with RNAi against cdc-42 and ced-10, and found similar phenotypes, suggesting animals defective in these smGTPases are not just delayed in forming ACT-5 coats around spores (data not shown). Because a defect in ACT-5 coat formation at the apical side of cells could be due to a disruption of cell polarity, we examined whether knocking down these smGTPases affected cell polarity, as assessed by apical localization of PGP-1∷GFP and mCherry∷ACT-5. Our previous analysis of spore exit indicated that rab-5, rab-10 and rab-11 RNAi do not affect cell polarity with this assay (Szumowski et al., 2014), and here we found that cdc-42 and ced-10 also do not affect cell polarity (Fig. S2). Altogether, these studies indicate that the smGTPases rab-5, rab-11, cdc-42 and ced-10/Rac1 are important for actin coat formation on SCCs.
Fig. 3.
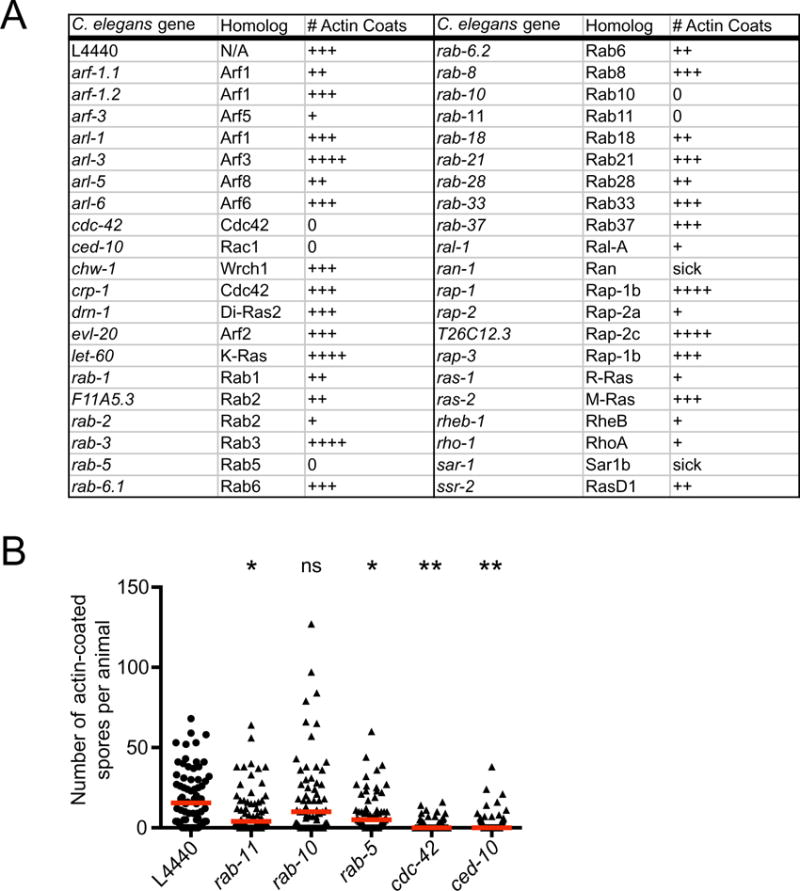
RNAi screen of smGTPases identifies factors required for ACT-5 coat formation on N. parisii spores.
A. Feeding RNAi screen of smGTPases. Infected animals were analysed at 43 hpi, when ACT-5 spore coats are first forming, and the number of ACT-5 spore coats per animal was determined using fluorescence microscopy. The number of + marks (0, +, ++, +++, ++++) indicates the fraction of animals examined (n > 60 per treatment) that had more than 20 actin coats per animal. The empty vector control L4440 received a score of +++, and screen hits with a score of 0 had a substantially reduced number of spore coat dense animals (see Experimental procedures for more information).
B. The number of actin-coated spores in screen hits was quantitatively analysed in a blinded, independent biological replicate experiment. The y-axis changes scale at y = 50 to enable visualization of data points close to 0. Each data point represents the number of actin-coated spores in a single animal (n ≥ 60 animals per sample). Red bar indicates the median value. P-value significance levels comparing each sample to L4440 using analysis of variance are: ns, not significant; *P < 0.05; **P < 0.001. rab-10 RNAi did not have a statistically significant effect, whereas rab-5, rab-11, cdc-42 and ced-10 RNAi did have statistically significant effects.
RAB-11 and CED-10/Rac1 localize to spores, but do not colocalize with ACT-5
To investigate whether the smGTPases identified in our screen had a direct role in the actin coat formation process, we next examined their localization to spores. In our earlier work we found that RAB-5 and RAB-10 did not localize to spores, but that RAB-11 did (Szumowski et al., 2014). Here, we compared RAB-11 localization to ACT-5 localization and did not observe any overlap between spores with RFP∷RAB-11 localization and spores with GFP∷ACT-5 or PGP-1∷GFP localization (Fig. 4A). This observation is consistent with the genetic findings that RAB-11 acts upstream of actin coat formation (Fig. 3B) and fusion with the apical membrane (Szumowski et al., 2014). Next, we used a GFP∷CED-10 transgene to examine CED-10/Rac1 localization around N. parisii spores (Lundquist et al., 2001). Here we found that GFP∷CED-10 formed coats around spores, and like RAB-11-coated spores, these CED-10/Rac1-coated spores were distinct from the ACT-5-coated spores (Fig. 4B). When comparing CED-10/Rac1 and RAB-11 localization in the same animals, we found that there were some spores with both CED-10/Rac1 and RAB-11 staining (Fig. 4C), although these were less common than spores with only CED-10/Rac1 or only RAB-11 staining (Fig. 4D). Specifically, of GFP∷CED-10-coated spores, only 29% (n = 15/52 spores, n = 5 animals) also had RFP∷RAB-11 localization, while of RFP∷RAB-11-coated spores, 5% (n = 15/277 spores, n = 5 animals) also had GFP∷CED-10 localization.
Fig. 4.
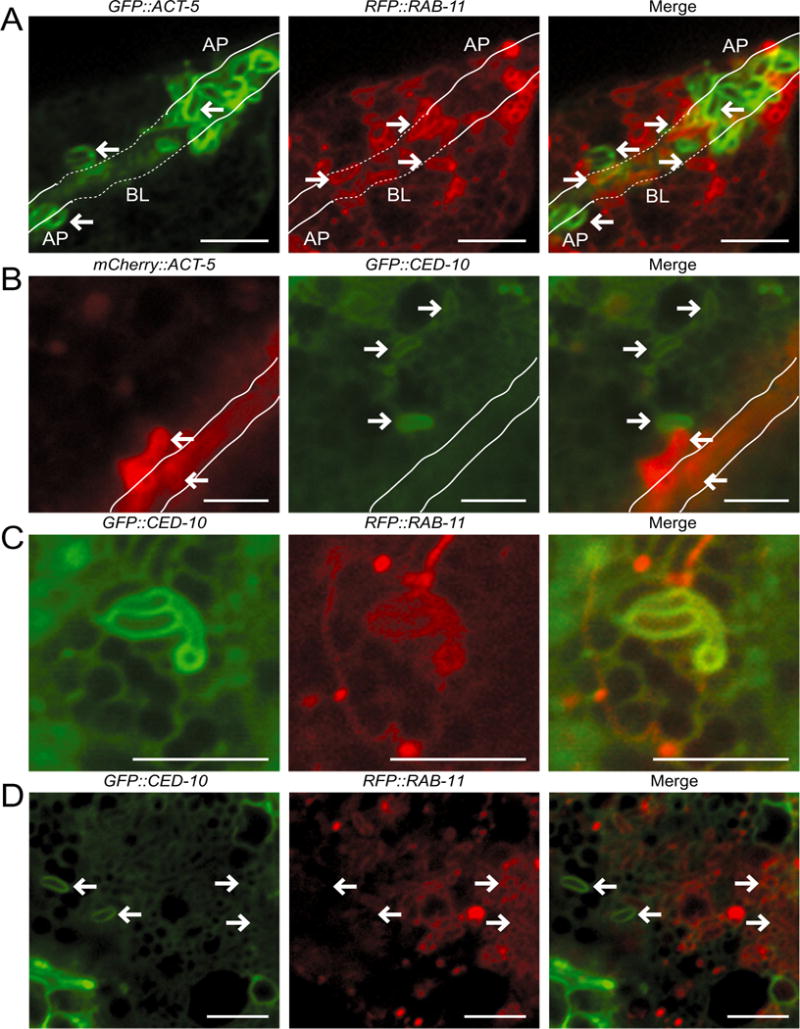
RAB-11 and CED-10 localize to N. parisii spores distinct from those where ACT-5 localizes.
A. In GFP∷ACT-5; RFP∷RAB-11 transgenic animals left-pointing arrows indicate examples of ACT-5-coated spores, which are distinct from the right-pointing arrows that mark RAB-11-positive spores. Because of the three-dimensional nature of the intestine, the lumen dips in and out of the Z-plane of focus. This tissue depth is indicated with the solid line marking apical regions (AP) and dotted line marking more basolateral (BL) regions of the intestine.
B. In mCherry∷ACT-5; GFP∷CED-10 transgenic animals left-pointing arrows indicate examples of ACT-5-coated spores, which are distinct from the right-pointing arrows that mark CED-10-positive spores. Solid white lines indicate the lumen.
C. In GFP∷CED-10; RFP∷RAB-11 transgenic animals spores often exhibit only a GFP∷CED-10 coat (left-facing arrows), or a RFP∷RAB-11 coat (right-facing arrows).
D. In GFP∷CED-10; RFP∷RAB-11 transgenic animals CED-10 and RAB-11 occasionally colocalize on spores.
A–D. All animals were infected with microsporidia and observed at 43–46 hpi. Scale bars are 5 μm.
Formation of ACT-5 coats correlates with spore exit but is not required for spore exit
Several lines of evidence suggested that ACT-5-coated spores could be exiting from host cells. First, as mentioned earlier, ACT-5 is required for spore exit (Estes et al., 2011). Second, ACT-5-coated spores are localized apically and have access to the lumen (Fig. 2). Third, we found that ACT-5 coats appear at about 43 hpi (Fig. S1), shortly after spores have differentiated and roughly when animals begin excreting spores (Estes et al., 2011). Fourth, many of the smGTPases that regulate ACT-5 coat formation also were previously shown to regulate spore exit (Szumowski et al., 2014). Therefore, we next examined contagiousness to assess spore exit from individual animals that either had ACT-5-coated spores or did not have ACT-5-coated spores. Although contagiousness is an indirect measure of spore exit, it addresses the role of ACT-5 coats in pathogen exit by enabling direct comparison of the presence of ACT-5 coats and the ability to transmit infection in the same animal.
To assess contagiousness, infected YFP∷ACT-5 transgenic donor animals were individually placed on a plate with 200 uninfected, recipient animals. After exposure to recipients, these infected donor animals were removed and examined for the presence of ACT-5 spore coats; after an incubation period, the recipients were observed to determine whether or not they became infected (see Experimental procedures for more information). If the recipients became infected, this result indicated that they had been exposed to a contagious donor that was excreting spores. We found that 74% of contagious donors had ACT-5-coated spores inside their intestinal cells, while 26% did not. Of non-contagious donors, none had ACT-5-coated spores (Fig. 5A). Together, these results indicate that ACT-5 coats are correlated with contagiousness, but are not required for contagiousness. Further supporting this correlation, we found that donors with actin spore coats were more contagious than donors without actin spore coats: 81% of donors with actin spore coats caused larval arrest, a phenotype of severe infection, in recipient animals, whereas only 44% of contagious animals lacking ACT-5-coated spores caused larval arrest. Therefore, donors with YFP∷ACT-5-coated spores are more contagious. The fact that 26% of contagious animals did not exhibit ACT-5-coated spores could be explained if ACT-5 coats are transient and the animal was examined after an ACT-5 coat had formed and disappeared. However, another interpretation of these results is that ACT-5 coats are not required for spore exit, and the presence of ACT-5 coats is simply correlated with a stage of infection when animals excrete more spores.
Fig. 5.
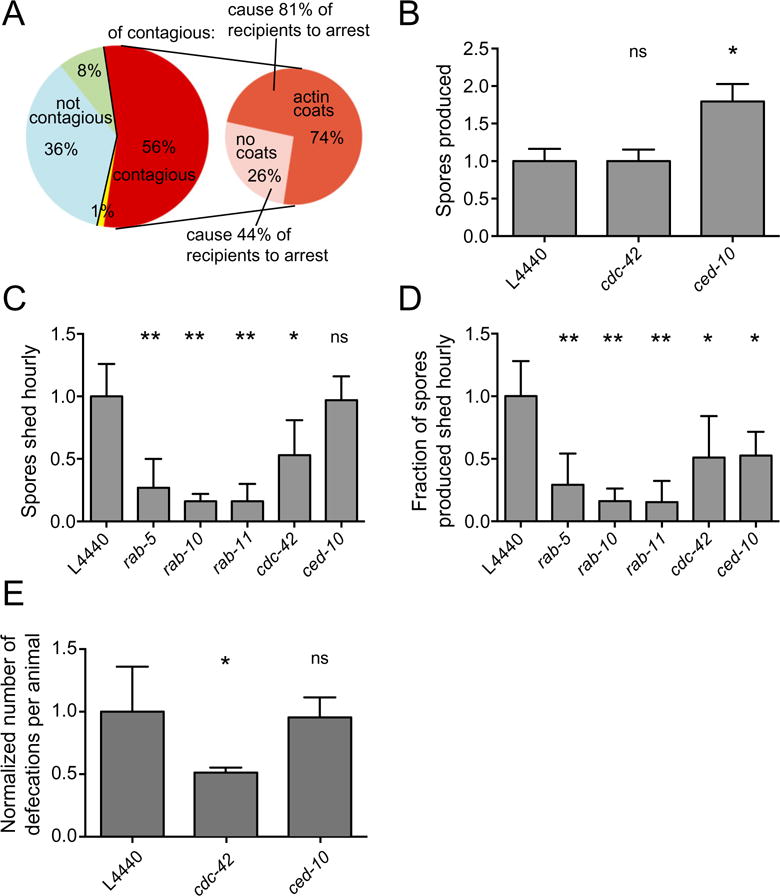
ACT-5 coats are correlated with spore exit and contagiousness, but are not required.
A. Contagiousness of 84 infected donor animals was compared with the presence of spores and ACT-5 coats after exposure to recipients. Non-contagious donors are shown in blue (no spores present, n = 30, 36%) and green (spores present, no ACT-5 coats, n = 7, 8%). One donor (yellow) was recorded to have no spores but was contagious, likely due to the presence of spores that were missed during visual inspection. Contagious donors (n = 46, 56%) are shown in red. These donors are subdivided into those that had ACT-5 spore coats (dark orange, n = 34, 74% of contagious animals) and those that did not have ACT-5 spore coats (pink, n = 12, 26% of contagious animals). Donors with ACT-5 coats caused recipients to become more heavily infected, leading to larval arrest 81% of the time, as opposed to 44% recipient larval arrest caused by donors without ACT-5 coats. Data combined from two biological replicate experiments.
B. Spore production in N2 RNAi-treated animals was quantified at 44 hpi. The total number of spores released from 50 lysed infected animals was quantified and normalized to the values from L4440 (vector alone) control RNAi-treated animals. Mean is from three independent experiments.
C. Quantification of spore shedding in N2 animals treated with RNAi against genes identified in Fig. 3 as regulators of ACT-5-coated spores. The normalized number of spores excreted per animal per hour is shown on the y-axis (mean is from three to four independent experiments for each sample), RNAi clone is shown on the x-axis.
D. Fraction of total spores produced that are excreted hourly. Normalized spore-shedding data for each sample (B) were divided by the normalized total number of spores produced for each sample (panel C and Szumowski et al., 2014 for Rab values) to give the fraction of the total spores produced that are excreted hourly. Propagation of error from combined experiments was calculated by taking the square root of the sum of standard deviations from all experiments squared, n = 6.
E. Defecation counts of RNAi-treated animals normalized to control L4440 values. The normalized number of defecation events over a 35 min period for groups of eight worms over three independent experiments is shown on the y-axis.
B–E show mean and standard deviation of independent experiments normalized to control L4440 values. Analysis of variance was used to calculate P-values indicated above graphs, comparing each sample to the L4440 control: ns, not significant; *P < 0.05; **P < 0.001.
To more directly examine whether ACT-5 coats are required for spore exit specifically, we measured spore production, spore shedding and defecation in animals treated with RNAi against genes important for formation of ACT-5 coats. First we measured spore production, because one possible explanation for a reduction of ACT-5-coated spores is that there are fewer spores made under these RNAi conditions. We previously had shown that knock-down of rab-5, rab-10 and rab-11 did not block spore production (Szumowski et al., 2014). Here we show that cdc-42 and ced-10/Rac1 knock-down also does not block spore production. In fact, ced-10/Rac1 RNAi caused a doubling in spore production (Fig. 5B). Thus, these RNAi clones do not cause a reduction in ACT-5-coated spores due to a reduction in spore production. Next, we examined spore shedding. Previously, we had shown that rab-5, rab-10 and rab-11 RNAi-treated animals had strong spore exit defects (Szumowski et al., 2014), a result that suggests ACT-5 coats play a role in spore exit, because rab-5, rab-10 and rab-11 are important for coat formation (Fig. 3). Surprisingly, however, here we found that RNAi against ced-10/Rac1 caused no significant decrease in spore shedding, whereas RNAi against cdc-42 caused only about a 50% decrease in spore shedding (Fig. 5C), despite the fact that cdc-42 and ced-10/Rac1 RNAi cause a nearly complete block in the number of ACT-5 coats (Fig. 3B). When spore exit is adjusted by the number of spores produced (which is greater in ced-10/Rac1-defective animals), the fraction of intracellular spores that are shed for both cdc-42 and ced-10/Rac1 RNAi-treated animals was about half that of the control animals (Fig. 5D). In order to determine if spore-shedding defects were due to defecation defects caused by RNAi treatment, we measured the number of defecation events occurring in RNAi-treated animals. Previously, we had examined rab-5-, rab-10- and rab-11-defective animals and demonstrated that their role in exit was not due to defecation defects (Szumowski et al., 2014). Although ced-10 RNAi-treated animals had normal defecation, cdc-42 RNAi-treated animals exhibited a defecation defect (Fig. 5E). Therefore, defecation defects may be the cause of the decrease in spore shedding observed in cdc-42 RNAi-treated animals. However, cdc-42 is required for a later phenotype as well, which confounds analyses (see below). In summary, these results indicate that although ACT-5 coat formation strongly correlates with spore exit, actin coats are not required for spore exit.
ACT-5 forms coats on spore-filled clathrin-coated vesicles, which appear later during infection
While analysing ACT-5 spore coats, we noticed that later during infection (∼ 47–48 hpi) ACT-5 surrounded large vesicles containing multiple spores. These spore-filled vesicles are distinct from the ACT-5-coated SCCs described earlier, which contain only a single spore. To characterize these vesicles and compare them to ACT-5-coated SCCs, we examined colocalization of the apical membrane marker PGP-1∷GFP and the cell-impermeable dye CW. Here we found that like ACT-5-coated SCCs, both markers were present on spore-filled vesicles (Fig. 6A). We next assayed whether the same genes that regulate ACT-5 coat formation on SCCs are responsible for ACT-5 on spore-filled vesicles. Interestingly, we found that although both cdc-42 and ced-10 RNAi treatment abolish ACT-5 localization around individual spores, only cdc-42 RNAi affected the ACT-5 localization around vesicles, while ced-10 RNAi did not (Fig. 6B and C). Even though ACT-5 was absent from the spore-filled vesicles in animals treated with cdc-42 RNAi, these vesicles still formed and were marked with PGP-1∷GFP indicating that ACT-5 localization is not required for the formation of these structures (Fig. 6D). The fact that ced-10 is required for ACT-5 coats on SCCs but not on vesicles suggests that SCCs and spore-filled vesicles are likely distinct structures, and do not simply represent two different stages in the development of the same structure.
Fig. 6.
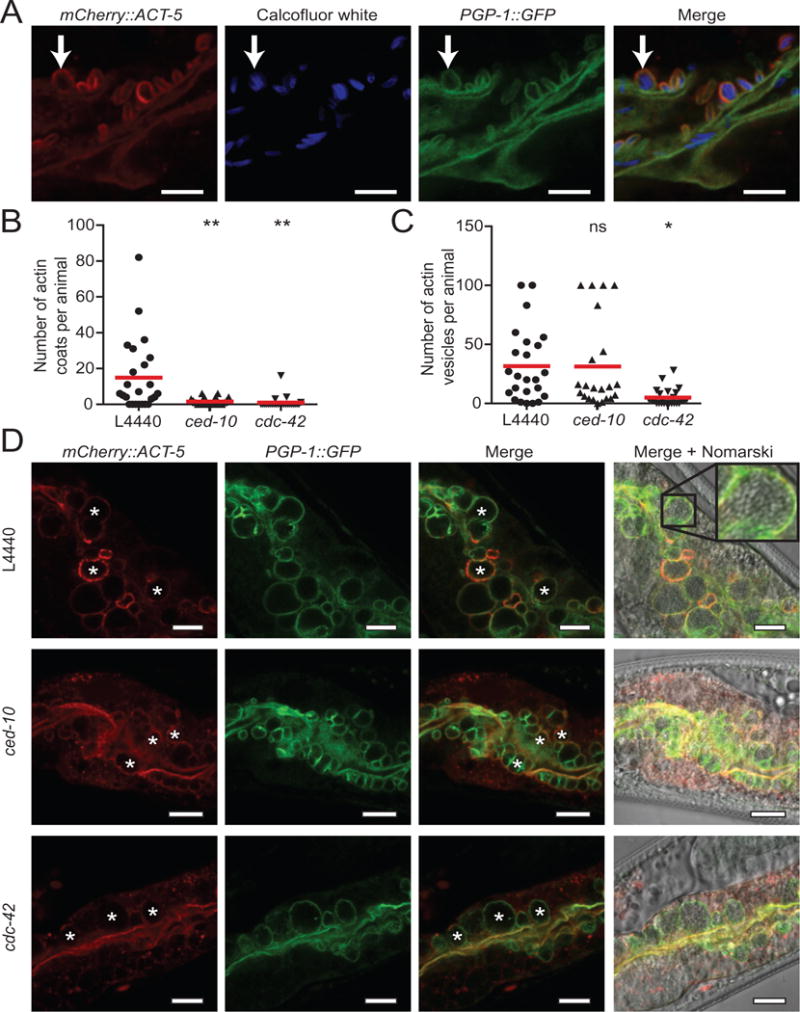
Spore-filled vesicles are coated in ACT-5, which requires cdc-42, but not ced-10.
A. Vesicles containing multiple spores (arrow) stain with mCherry∷ACT-5, PGP-1∷GFP and CW. Scale bar is 5 μm.
B. RNAi knock-down of Rho GTPases ced-10 or cdc-42 blocks actin coat formation.
C. RNAi knock-down of ced-10 does not affect ACT-5 localization to vesicles, but RNAi knock-down of cdc-42 blocks ACT-5 localization to vesicles. Quantification was capped at 100 vesicles per animal. For panels B and C, red bar is the mean number of ACT-5 coats or vesicles per animal across the population examined. n = 22–25 animals per sample. Analysis of variance was conducted on the data, comparing each sample to the L4440 control. P-values are indicated above each sample: ns, not significant; *P < 0.05; **P < 0.001. Data are representative of three independent assays.
D. Spore-filled vesicles (asterisks) marked with mCherry∷ACT-5 and PGP-1∷GFP are visible in animals treated with L4440 empty vector control or with ced-10 RNAi. L4440 panel shows magnification of an ACT-5 vesicle containing dozens of spores. Vesicles in animals treated with cdc-42 RNAi are marked with PGP-1∷GFP but lack ACT-5 staining. Scale bars are 10 μm.
Spore-filled vesicles are likely endocytic in origin, but actin-coated SCCs are not
To further study the nature of ACT-5-labelled spore-filled vesicles, and compare them to ACT-5-coated individual SCCs, we investigated the role of endocytosis in the formation of these structures. We found that actin-labelled vesicles colocalize with the endocytosis marker clathrin heavy chain, CHC-1, indicating that these actin-coated vesicles may be the result of endocytosis from the spore-dense lumen at later time points in infection (Fig. 7A). To further test if spore-filled vesicles are the result of endocytosis, we fed late stage-infected animals Tetramethylrhodamine isothiocyanate (TRITC)-conjugated Bovine serum albumin (BSA), which is known to enter cells via endocytosis (Grant and Audhya, 2005). We observed TRITC-BSA present inside of spore-filled vesicles, and verified that this staining was not an artefact of intestinal autofluorescence (Fig. 7B). To test whether spore-filled vesicles matured in the endocytic pathway, we examined actin-coated vesicles for the early endosome marker RAB-5, but did not observe RAB-5 to label the vesicles (Fig. S3). Together these results suggest that spore-filled vesicles are endocytic in origin, although they do not mature into early endosomes.
Fig. 7.
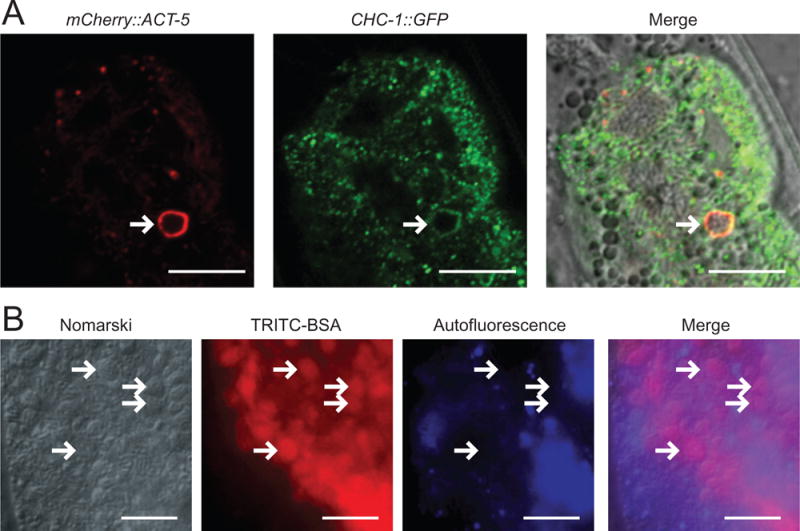
Actin-coated spore-filled vesicles are labelled with clathrin and take up the endocytosis marker TRITC-BSA.
A. Spore-filled vesicle (arrow) marked with mCherry∷ACT-5 colocalizes with clathrin, CHC-1∷GFP. Scale bar is 10 μm.
B. TRITC-BSA fed to animals accumulates in spore-filled vesicles. Arrows indicate examples of spore-filled vesicles that contain TRITC-BSA. Blue channel shows that intestinal cell autofluorescence does not overlap with red channel signal indicating that staining of vesicles is TRITC-BSA, not autofluorescence. Scale bar is 10 μm.
We next used the same endocytosis assays described earlier to examine ACT-5-coated SCCs earlier during infection. In contrast to the spore-filled vesicles, we observed that clathrin does not co-localize with ACT-5-coated SCCs, indicating these spores are not in the process of entering the host cell via clathrin-mediated endocytosis (Fig. S4A). Consistent with the lack of clathrin localization to individual ACT-5-coated spores, we could not detect any TRITC-BSA staining in actin-coated SCCs, nor did we observe RAB-5 localization to these spores (Fig. S4B). These data, together with the timing of ACT-5 coat formation on SCCs (Fig. S1), support the model that actin-coated spores are in the process of exiting from host cells rather than entering. On the other hand, spore-filled vesicles appear to be the result of late stage endocytosis of spores from the lumen.
Discussion
Exit from the host cell is a key stage in the spread of intracellular pathogens. Here, we describe investigations into the role of actin in the directional exocytosis and reuptake of N. parisii, a microsporidian pathogen that naturally infects the intestinal cells of C. elegans. In earlier studies we have shown that N. parisii spores are enclosed in membrane-bound compartments that are directed for exocytosis out of the apical side of intestinal cells into the lumen by the smGTPase RAB-11. We now show data supporting the model that after spores traffic to and fuse with the host apical membrane, most of these fused spores become coated with the intestinal-specific actin, ACT-5 (Fig. 2), in a process that is dependent on the host smGTPases rab-5, rab-11, cdc-42 and ced-10/Rac1 (Fig. 3). Based on actin coat quantification and assays of spore exit, we conclude that although actin coats often form on exiting spores, actin coating is not required for exit of N. parisii spores (Fig. 8, left panel). RNAi against actin polymerization factors cdc-42 and ced-10/Rac1 almost completely eliminated ACT-5 coats on spores, but only reduced spore exit in half, and this phenotype is confounded by additional pleiotropies in cdc-42-defective animals (Figs 5D, 5E and 7). Because RNAi against the recycling endosome factors rab-5, rab-10 and rab-11 caused a stronger block in spore exit, we propose they act upstream of both routes of exit, whereas cdc-42 and ced-10/Rac1 are only important for promoting actin coat formation on spores that make only a minor contribution to the exiting population. Based on the minor spore-shedding defect caused by RNAi against these actin polymerization factors, we propose that the ability to form ACT-5 coats is not required for the exocytosis of spores, a process that we described earlier as being dependent on ACT-5 (Estes et al., 2011). A distinct role for ACT-5 in the N. parisii life cycle is the clathrin-dependent compensatory endocytosis of bulk membrane material from the apical cell surface (Fig. 8, right panel). Exocytosis and endocytosis are highly interdependent: this endocytosis may serve to maintain membrane homeostasis and compensate for the massive efflux of intracellular membrane material to the apical cell surface during the extensive exocytosis of spores.
Fig. 8.

Model for actin regulation of N. parisii spore exocytosis and compensatory endocytosis in C. elegans intestinal cells. Left panel: At 43 hpi, meronts have differentiated into spores that are enclosed in membrane-bound SCCs labelled with RAB-11. These SCCs can traffic up to the apical cell surface through two pathways to fuse with host membrane and acquire a PGP-1 membrane marker. After fusion, spores in both pathways have access to the lumen and are able to exit from the host cell, while in one pathway they also acquire an ACT-5 coat. Formation of ACT-5 coats is dependent on the Rho GTPases ced-10 and cdc-42, as well as rab-5 and rab-11. Right panel: By 48 hpi, pathogen replication has overtaken the host intestinal cell and the lumen is packed with exited spores. Large membrane-bound, endocytic vesicles that contain multiple spores are coated in PGP-1 apical membrane, ACT-5 and clathrin (CHC-1). By taking in excess membrane from the apical surface that likely accumulates during bulk exocytosis of spores, host cells are able to maintain balance of their membrane pools.
Our findings about actin coats forming around N. parisii spores are intriguingly similar to descriptions of actin structures surrounding secretory vesicles containing large or insoluble endogenous cargo, where actin can form ‘coats’ or ‘rings’ around these vesicles after fusing with the plasma membrane (Nightingale et al., 2012). For example, vascular endothelial cells secrete an extremely large glycoprotein called VWF, which is contained in enormous cigar-shaped organelles called WPBs that are up to 1–5 μm long (Metcalf et al., 2008), similar in length to the 2 or 3 μm long N. parisii spores (Troemel et al., 2008). WPBs acquire actin rings upon fusion with the apical membrane and recent studies have shown that in this context actomyosin contractility squeezes VWF out into the extracellular environment (Nightingale et al., 2012). As with exocytosis of N. parisii spores, Rabs are recruited to WPBs and are required for VWF secretion, although the exact Rabs involved in actin coat formation have not been well defined (Zografou et al., 2012). Our studies provide insight into smGTPases that regulate actin coat formation, identifying Rabs involved in recycling endocytosis and two Rhos involved in actin polymerization. Actin has been shown to form coats around exocytosing vesicles in other systems as well, such as lung surfactant-containing vesicles (Miklavc et al., 2012), cortical granules in frog oocytes (Sokac et al., 2003; Sokac and Bement, 2006) and amylase-containing vesicles in pancreatic cells (Turvey and Thorn, 2004; Jang et al., 2012). In these systems, actin coats formed after vesicles fuse with the membrane are often stable for over 30 s and likely serve to squeeze the cargo outside of the cell (Sokac and Bement, 2006). Similarly, the actin coats appear to form around N. parisii spores after fusion with the membrane and may serve to expel these pathogens out of the cell. Although we are currently unable to visualize spore release in real time, calculations based on approximately 2000 spores exiting per animal per hour [Fig. 5C, L4440 (vector alone) control RNAi prior to normalization] and approximately 19 actin-coated spores per animal at any time (Fig. 3B, L4440) suggest an approximate actin coat duration time of 35 s, which is consistent with the kinetics of exocytosis of large endogenous cargo.
Defining the precise role of actin in secretory events has been challenging for endogenous cargo, with some studies showing that actin promotes secretion and other studies showing that actin inhibits secretion (Cingolani and Goda, 2008; Trifaro et al., 2008). Recent imaging studies with WPBs have clarified the opposing roles that actin can play in secretion of VWF, with actin inhibiting the initial fusion event, but promoting release once fusion has occurred (Nightingale et al., 2011). Similarly, we suggest that ACT-5 may play opposing roles in the N. parisii exit process, with inhibition early during the exit process, and possible promotion later in the process (Estes et al., 2011). Our previous studies showed that during the early stage of N. parisii development ACT-5 ectopically localizes to the basolateral side of cells and is then followed by formation of gaps in the terminal web. Because actin knock-down causes gap formation in the absence of infection, we hypothesized that actin relocalization triggers gap formation, perhaps to remove a barrier to exit. These findings suggested that early during infection actin can obstruct exit. However, we found that a reduction of ACT-5 expression impaired overall spore exit, which we now show is likely not due solely to the role of ACT-5 forming coats around spores to promote expulsion. This dual role of actin first inhibiting and then promoting secretion appears to be common for exocytosis of vesicles containing large cargo such as the VWF protein or N. parisii spores. Interestingly, actin has also been shown to play opposing roles in the secretion of dense core vesicles, although here the mechanism of promoting secretion is likely distinct from the actin coats that form on N. parisii spores (Wollman and Meyer, 2012).
N. parisii is only one of more than 1400 species of microsporidia, and very little is known about the exit process for any of these species. In particular, it would be interesting to examine the exit process of microsporidian species that infect human intestinal cells to determine whether this large cargo also acquires actin coats during the process of exiting from host cells. In this way it will be possible to determine whether the mechanistic insights we have gained from studying microsporidia transmission in C. elegans are conserved in medically relevant microsporidia infections of humans.
Experimental procedures
Strains
C. elegans were maintained on nematode growth media (NGM) plates seeded with Escherichia coli OP50-1 as described (Brenner, 1974). We used N2 wild-type animals (see Table 1 for a complete list of strains used).
Table 1.
Strains used in this study.
| Strain | Transgene | Source | Figures used |
|---|---|---|---|
| N2 | N/A | Caenorhabditis Genetics Center | 5B, 5C, 5D, 5E |
| ERT38 | kcIs6[ifb-2p∷IFB-2∷CFP]; caIs[ges-1p∷YFP∷ACT-5] | Estes et al., 2011 | 1A, 1B, 5A, S1A–B |
| ERT60 | jyIs13[act-5p∷GFP∷ACT-5] | UV psoralen integration of act-5p∷GFP∷ACT-5 (Kang et al., 2009) | Used for crosses |
| ERT89 | jyIs13[act-5p∷GFP∷ACT-5]; pwIs480[vha-6p∷RFP∷RAB-5] | ERT60 × RT1239 | S3, S4B |
| ERT99 | jyIs13[act-5p∷GFP∷ACT-5]; pwIs428[vha-6p∷RFP∷RAB-11.1] | ERT60 × RT1102 | 4A |
| ERT106 | jyIs17[vha-6p∷mCherry∷ACT-5]; dkIs166 [opt-2p∷PGP-1∷GFP] | Szumowski et al., 2014 | 2, 3, S2A–C |
| ERT137 | dkIs8[vha-6p∷CHC-1∷GFP]; jyIs17[vha-6p∷mCherry∷ACT-5]IV | ERT104 × GK35 | 7A, S4 |
| ERT197 | jyIs17 [vha-6p∷mCherry∷ACT-5]IV; nEx1039[ced-10p∷GFP∷CED-10]; unc-76(e911)V | ERT104 × MT10865 | 4B |
| ERT202 | pwIs428[vha-6p∷RFP∷RAB-11.1]; nEx1039[ced-10p∷GFP∷CED-10]; unc-76(e911)V | RT1102 × MT10865 | 4C, 4D |
N. parisii spore preps and infection assays
Spore preps were prepared by culturing N. parisii inside of C. elegans, and when animals were heavily infected, they were mechanically disrupted with silicon beads to release spores, which were filtered to remove intact larvae and eggs. Aliquots of spores were quantified and stored at −80°C prior to use (see Estes et al., 2011 for more information on spore preps). Infection assays were conducted with synchronized L1 larvae grown for 24 h on 10 cm NGM plates seeded with OP50 at 20°C until approximately L3 stage, when they were infected with 2 × 106 N. parisii spores and then incubated at 25°C. To stain spores in contact with the lumen (Fig. 2), CW was fed to infected animals 2 h prior to imaging by applying 300 μl of CW to each 6 cm NGM/OP50 plate.
Microscope image acquisition
Symptoms of infection were tracked by mounting approximately 50 animals on agarose pads and then viewing animals with Nomarski optics and/or fluorescence on a Zeiss AxioImager.M1 at 630× magnification (Zeiss EC Plan-NEOFLUAR Oil, DIC, 1.4 NA objective), using Axiovision 4.8 software and a Zeiss AxioCam MRm camera. These imaging conditions were used to assess the presence of spores inside infected donor animals in contagiousness assays (Fig. 5A), and the number of ACT-5-coated spores in RNAi screen (Fig. 3). All other images were acquired on a confocal Zeiss LSM700 Observer.Z1 at 630× magnification (Zeiss PLAN-APOCHOMAT Oil, 1.4 NA objective) using ZEN2010 software and a LSM T-PMT camera.
Anti-ACT-5 antibody staining
N. parisii-infected animals were fixed in a mixture of formaldehyde, n-heptane and methanol; freeze-thawed in liquid nitrogen; and then treated with collagenase. Samples were blocked with goat serum, incubated with normal goat serum and BSA, incubated with 1:10 anti-ACT-5 rabbit polyclonal antibodies, then incubated with goat anti-rabbit conjugated with Alexa488 secondary antibodies diluted 1:300 and mounted on agarose pads with Vectashield for viewing (see MacQueen et al., 2005 for more details on ACT-5 antibodies).
Endocytosis assays
Animals were picked into a 15 μl drop of 5 mg ml−1 TRITC-BSA and incubated for 4 h. After TRITC-BSA feeding, animals were washed three times in M9 to dilute extracellular TRITC and then were immediately mounted on agarose pads for imaging.
Spore-shedding assays
Synchronized L3 animals were infected with 2.0 × 105 spores on 6 cm plates and incubated at 25°C for 39 h to allow the infection to progress until the spore-shedding stage of infection. Animals were then washed three times in M9 to remove spores from cuticles and placed in microfuge tubes with 500 μl of 1:1 E. coli OP50 and M9. The spores excreted by 50 animals from 40 to 48 hpi were collected and quantified (see Estes et al., 2011 for more details on spore-shedding assays).
RNA interference assays
Feeding RNAi experiments were performed as described (Estes et al., 2011). Briefly, synchronized populations of L1 animals were seeded on plates of RNAi bacteria. All RNAi clones were used undiluted except for rab-5 and rab-11.1, which were diluted 1:10 with L4440 (vector alone) control RNAi bacteria to allow for normal development.
Defecation assays
RNAi-treated animals were prepared as described earlier. Spore stage-infected animals were fed 0.5 μm fluorescent beads (Polysciences, Inc. Cat# 19507) top plated onto OP50 for 2 h. Animals were then removed from the bead-feeding plate, briefly transferred to a clean OP50 plate to remove beads from the cuticle and then transferred to a fresh OP50 plate and allowed to defecate undisturbed for 35 min, at which point they were removed from the plates and the number of defecation events was quantified by counting the number of bead spots on the plates at 630× after removal of the animals. Each RNAi clone was tested on three independent plates, each containing eight animals during each of three independent biological experiments.
Contagiousness assays
A synchronized population of ERT38 kcIs6 [IFB-2∷CFP], caIs[YFP∷ACT-5] animals were infected at the early L3 stage with 2 million spores per 6 cm plate and allowed to reach 38 hpi. At this time point, animals were washed three times in M9 to remove any spores for their cuticles, and replated onto a clean OP50 plate. From this plate individual donor animals were cloned out onto plates containing 200 N2 L1s. The infected donor and the recipient N2s were allowed to cohabit the plate for 8 h at 25°C, at which point the infected donor animal was removed from the plate and mounted onto an agarose pad for microscopy. Each individual donor animal was assessed for the presence of any intracellular spores using Nomarski optics, and the presence of YFP∷ACT-5 coats using fluorescence. Results were recorded on a spreadsheet and later compared with the degree of infection of the recipients. After removal of the donor animal, the recipients were maintained at 25°C for 7 days and then a 1 × 1 cm region of agarose plate containing animals was transferred to a fresh OP50 plate and allowed to grow for 7 days at 25°C. At the end of this period, plates were assessed for larval arrest and lack of food clearing, traits associated with heavily infected animals. This information was paired with the actin phenotypes observed from the donor animal. As controls, animals that did not yet have fully developed spores were verified to be non-contagious.
RNAi screen for smGTPases that block formation of actin coats
A library of 41 C. elegans smGTPase RNAi clones was divided roughly into quarters and prepared as four separate batches, each with the empty vector control, L4440. All clones were sequence verified. A synchronized population of 200 L1 animals containing mCherry∷ACT-5 and PGP-1∷GFP transgene markers were plated on a lawn of RNAi bacteria and allowed to develop to the L3 stage before infecting with 2 million N. parisii spores per 6 cm plate. Infection proceeded at 25°C for 43 h to the ACT-5 spore coat stage. Animals were fixed in 4% PFA and stored at 4°C until analysed. To determine whether an RNAi clone affected the formation of ACT-5 spore coats, approximately 60–80 animals were mounted on an agarose pad and examined under the microscope for the number of actin spore coats per animal. Hits were defined as reducing the proportion of the population with greater than 20 ACT-5 coats per animal by at least 70% relative to L4440. RNAi treatments were grouped into bins relative to the control (0–30% of control value = 0, 31–50% = +, 51–70% = ++, 71–130% = +++, 131–200% = ++++). After testing each clone once, hits were validated by performing a biological replicate experiment along with clones that were not initially hits used as negative controls. Of note, there are two rab-11 isoforms, rab-11.1 and rab-11.2, that have high sequence similarity. An RNAi clone against rab-11.2 was used during the screen. However, for validation rab-11.1 RNAi (diluted 1:10 with L4440) was used instead of rab-11.2 RNAi because (i) we found direct localization of RAB-11.1 protein to N. parisii spores; (ii) the rab-11.2 RNAi clone knocks down rab-11.1; and (iii) using rab-11.1 directly was found to have a stronger phenotype (Szumowski et al., 2014). To determine whether the clone was affecting cell polarity, the localization of the apically localized plasma membrane marker, PGP-1∷GFP, was assessed. The spore production of RNAi-treated animals was measured at 44 hpi by dissolving host tissue of infected animals and counting the spores released. Infected animals were fixed with acetone and 50 animals were hydrolysed for 30 min with 200 mM NaOH, 0.1% SDS and 1:100 CW (see Szumowski et al., 2014 for more details).
Supplementary Material
Fig. S1. Kinetics of actin coat formation around N. parisii spores.
A. YFP∷ACT-5-coated spores (arrows) are not coated with IFB-2∷CFP. Transgenic animals grown on L4440 empty vector RNAi bacteria were infected with microsporidia and observed at the actin spore coat stage, 43–46 hpi. Scale bars are 5 μm. We chose these time points because previous studies had demonstrated that spores do not form until 40 hpi, and spore-filled vesicles form at 47–48 hpi (which can confound analysis of spore coats). Thus, we chose time points in between spore formation and spore-filled vesicle formation.
B. Spore stage-infected animals carrying a YFP∷ACT-5 transgene were imaged at one of three time points in 11 independent experiments. n = 34–76 spore-containing animals examined per time point.
Fig. S2. Apical localization of PGP-1 and ACT-5 is not affected by smGTPase RNAi treatment.
A. Empty vector RNAi treatment of infected animals shows that PGP-1 is normally localized along the lumen and colocalizes with ACT-5 at the apical cell surface.
B–C. Animals treated with smGTPase screen hit RNAi clones from late L2 adulthood (the treatment used for the screen) exhibit no change in PGP-1∷GFP or mCherry∷ACT-5 apical localization. Scale bar in all images is 5 μm.
Fig. S3. Spore-filled vesicles do not label with the early endosome marker RAB-5. GFP∷ACT-5; RFP-RAB-5 transgenic animals were infected with microsporidia and observed at the spore-filled vesicle stage, 47–48 hpi. Spore-filled vesicles marked with ACT-5 (arrows) do not stain with the early endosome marker RAB-5. Scale bars are 10 μm.
Fig. S4. Actin-coated individual spores do not label with clathrin CHC-1 or the early endosome marker RAB-5.
A. mCherry∷ACT-5; CHC-1∷GFP transgenic animals were infected with microsporidia and observed at the actin spore coat stage, 43–46 hpi. The clathrin-mediated endocytosis marker CHC-1∷GFP does not localize to mCherry∷ACT-5-coated individual SCCs (arrows). Scale bars are 5 μm.
B. GFP∷ACT-5; RFP-RAB-5 transgenic animals were infected with microsporidia and observed at 47–48 hpi. SCCs marked with ACT-5 (arrows) do not stain with the early endosome marker RAB-5. Scale bars are 5 μm.
Acknowledgments
We thank Keir Balla, Robert Luallen, Kirthi Reddy, Aaron Reinke and Roy Wollman for helpful comments on the manuscript. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by NIH predoctoral training grant T32 GM008666 and a NSF Predoctoral Fellowship to S.C.S.; NIH Training in Immunology Grant 5T32AI060536 to K.A.E.; and NIAID R01 AI087528, the Searle Scholars Program, Packard Foundation and Burroughs Wellcome Fund fellowships to E.R.T.
Footnotes
Conflict of interest
We claim no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
References
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisman CJ, Alvarez M, Casadevall A. Phagocytosis of Cryptococcus neoformans by, and nonlytic exocytosis from, Acanthamoeba castellanii. Appl Environ Microbiol. 2010;76:6056–6062. doi: 10.1128/AEM.00812-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Didier ES, Weiss LM. Microsporidiosis: not just in AIDS patients. Curr Opin Infect Dis. 2011;24:490–495. doi: 10.1097/QCO.0b013e32834aa152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes KA, Szumowski SC, Troemel ER. Non-lytic, actin-based exit of intracellular parasites from C. elegans intestinal cells. PLoS Pathog. 2011;7:e1002227. doi: 10.1371/journal.ppat.1002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix MA, Jovelin R, Ferrari C, Han S, Cho YR, Andersen EC, et al. Species richness, distribution and genetic diversity of Caenorhabditis nematodes in a remote tropical rainforest. BMC Evol Biol. 2013;13:10. doi: 10.1186/1471-2148-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich N, Hagedorn M, Soldati-Favre D, Soldati T. Prison break: pathogens’ strategies to egress from host cells. Microbiol Mol Biol Rev. 2012;76:707–720. doi: 10.1128/MMBR.00024-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, Audhya A. The ins and outs of endocytic transport. Nat Cell Biol. 2005;7:1151–1154. doi: 10.1038/ncb1205-1051. [DOI] [PubMed] [Google Scholar]
- Haglund CM, Welch MD. Pathogens and polymers: microbe-host interactions illuminate the cytoskeleton. J Cell Biol. 2011;195:7–17. doi: 10.1083/jcb.201103148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Jang Y, Soekmadji C, Mitchell JM, Thomas WG, Thorn P. Real-time measurement of F-actin remodelling during exocytosis using lifeact-EGFP transgenic animals. PLoS ONE. 2012;7:e39815. doi: 10.1371/journal.pone.0039815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M, Toret CP, Drubin DG. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2006;7:404–414. doi: 10.1038/nrm1940. [DOI] [PubMed] [Google Scholar]
- Kang J, Shin D, Yu JR, Lee J. Lats kinase is involved in the intestinal apical membrane integrity in the nematode Caenorhabditis elegans. Development. 2009;136:2705–2715. doi: 10.1242/dev.035485. [DOI] [PubMed] [Google Scholar]
- Keeling PJ, Fast NM. Microsporidia: biology and evolution of highly reduced intracellular parasites. Annu Rev Microbiol. 2002;56:93–116. doi: 10.1146/annurev.micro.56.012302.160854. [DOI] [PubMed] [Google Scholar]
- Lundquist EA, Reddien PW, Hartwieg E, Horvitz HR, Bargmann CI. Three C. elegans Rac proteins and several alternative Rac regulators control axon guidance, cell migration and apoptotic cell phagocytosis. Development. 2001;128:4475–4488. doi: 10.1242/dev.128.22.4475. [DOI] [PubMed] [Google Scholar]
- McGhee JD. The C. elegans Intestine. WormBook; 2007. Mar 27, pp. 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen AJ, Baggett JJ, Perumov N, Bauer RA, Januszewski T, Schriefer L, Waddle JA. ACT-5 is an essential Caenorhabditis elegans actin required for intestinal microvilli formation. Mol Biol Cell. 2005;16:3247–3259. doi: 10.1091/mbc.E04-12-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf DJ, Nightingale TD, Zenner HL, Lui-Roberts WW, Cutler DF. Formation and function of Weibel-Palade bodies. J Cell Sci. 2008;121:19–27. doi: 10.1242/jcs.03494. [DOI] [PubMed] [Google Scholar]
- Miklavc P, Hecht E, Hobi N, Wittekindt OH, Dietl P, Kranz C, Frick M. Actin coating and compression of fused secretory vesicles are essential for surfactant secretion – a role for Rho, formins and myosin II. J Cell Sci. 2012;125:2765–2774. doi: 10.1242/jcs.105262. [DOI] [PubMed] [Google Scholar]
- Nightingale TD, White IJ, Doyle EL, Turmaine M, Harrison-Lavoie KJ, Webb KF, et al. Actomyosin II contractility expels von Willebrand factor from Weibel-Palade bodies during exocytosis. J Cell Biol. 2011;194:613–629. doi: 10.1083/jcb.201011119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale TD, Cutler DF, Cramer LP. Actin coats and rings promote regulated exocytosis. Trends Cell Biol. 2012;22:329–337. doi: 10.1016/j.tcb.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Pizarro-Cerda J, Kuhbacher A, Cossart P. Entry of Listeria monocytogenes in mammalian epithelial cells: an updated view. Cold Spring Harb Perspect Med. 2012;2:a010009. doi: 10.1101/cshperspect.a010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat-Shliom N, Milberg O, Masedunskas A, Weigert R. Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell Mol Life Sci. 2013;70:2099–2121. doi: 10.1007/s00018-012-1156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukkila-Worley R, Ausubel FM. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr Opin Immunol. 2012;24:3–9. doi: 10.1016/j.coi.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokac AM, Bement WM. Kiss-and-coat and compartment mixing: coupling exocytosis to signal generation and local actin assembly. Mol Biol Cell. 2006;17:1495–1502. doi: 10.1091/mbc.E05-10-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokac AM, Co C, Taunton J, Bement W. Cdc42-dependent actin polymerization during compensatory endocytosis in Xenopus eggs. Nat Cell Biol. 2003;5:727–732. doi: 10.1038/ncb1025. [DOI] [PubMed] [Google Scholar]
- Szumowski SC, Botts MR, Popovich JJ, Smelkinson MG, Troemel ER. The small GTPase RAB-11 directs polarized exocytosis of the intracellular pathogen N. parisii for fecal-oral transmission from C. elegans. Proc Natl Acad Sci USA. 2014;111:8215–8220. doi: 10.1073/pnas.1400696111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Texier C, Vidau C, Vigues B, El Alaoui H, Delbac F. Microsporidia: a model for minimal parasite-host interactions. Curr Opin Microbiol. 2010;13:443–449. doi: 10.1016/j.mib.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Trifaro JM, Gasman S, Gutierrez LM. Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol (Oxf) 2008;192:165–172. doi: 10.1111/j.1748-1716.2007.01808.x. [DOI] [PubMed] [Google Scholar]
- Troemel ER. New models of microsporidiosis: infections in Zebrafish, C. elegans, and honey bee. PLoS Pathog. 2011;7:e1001243. doi: 10.1371/journal.ppat.1001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Felix MA, Whiteman NK, Barriere A, Ausubel FM. Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol. 2008;6:2736–2752. doi: 10.1371/journal.pbio.0060309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turvey MR, Thorn P. Lysine-fixable dye tracing of exocytosis shows F-actin coating is a step that follows granule fusion in pancreatic acinar cells. Pflugers Arch. 2004;448:552–555. doi: 10.1007/s00424-004-1288-z. [DOI] [PubMed] [Google Scholar]
- Vavra J, Lukes J. Microsporidia and ‘the art of living together’. Adv Parasitol. 2013;82:253–319. doi: 10.1016/B978-0-12-407706-5.00004-6. [DOI] [PubMed] [Google Scholar]
- Williams BA. Unique physiology of host-parasite interactions in microsporidia infections. Cell Microbiol. 2009;11:1551–1560. doi: 10.1111/j.1462-5822.2009.01362.x. [DOI] [PubMed] [Google Scholar]
- Wollman R, Meyer T. Coordinated oscillations in cortical actin and Ca2+ correlate with cycles of vesicle secretion. Nat Cell Biol. 2012;14:1261–1269. doi: 10.1038/ncb2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zografou S, Basagiannis D, Papafotika A, Shirakawa R, Horiuchi H, Auerbach D, et al. Rab-genome analysis reveals novel insights in Weibel-Palade body exocytosis. J Cell Sci. 2012;125(Part 20):4780–4790. doi: 10.1242/jcs.104174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Kinetics of actin coat formation around N. parisii spores.
A. YFP∷ACT-5-coated spores (arrows) are not coated with IFB-2∷CFP. Transgenic animals grown on L4440 empty vector RNAi bacteria were infected with microsporidia and observed at the actin spore coat stage, 43–46 hpi. Scale bars are 5 μm. We chose these time points because previous studies had demonstrated that spores do not form until 40 hpi, and spore-filled vesicles form at 47–48 hpi (which can confound analysis of spore coats). Thus, we chose time points in between spore formation and spore-filled vesicle formation.
B. Spore stage-infected animals carrying a YFP∷ACT-5 transgene were imaged at one of three time points in 11 independent experiments. n = 34–76 spore-containing animals examined per time point.
Fig. S2. Apical localization of PGP-1 and ACT-5 is not affected by smGTPase RNAi treatment.
A. Empty vector RNAi treatment of infected animals shows that PGP-1 is normally localized along the lumen and colocalizes with ACT-5 at the apical cell surface.
B–C. Animals treated with smGTPase screen hit RNAi clones from late L2 adulthood (the treatment used for the screen) exhibit no change in PGP-1∷GFP or mCherry∷ACT-5 apical localization. Scale bar in all images is 5 μm.
Fig. S3. Spore-filled vesicles do not label with the early endosome marker RAB-5. GFP∷ACT-5; RFP-RAB-5 transgenic animals were infected with microsporidia and observed at the spore-filled vesicle stage, 47–48 hpi. Spore-filled vesicles marked with ACT-5 (arrows) do not stain with the early endosome marker RAB-5. Scale bars are 10 μm.
Fig. S4. Actin-coated individual spores do not label with clathrin CHC-1 or the early endosome marker RAB-5.
A. mCherry∷ACT-5; CHC-1∷GFP transgenic animals were infected with microsporidia and observed at the actin spore coat stage, 43–46 hpi. The clathrin-mediated endocytosis marker CHC-1∷GFP does not localize to mCherry∷ACT-5-coated individual SCCs (arrows). Scale bars are 5 μm.
B. GFP∷ACT-5; RFP-RAB-5 transgenic animals were infected with microsporidia and observed at 47–48 hpi. SCCs marked with ACT-5 (arrows) do not stain with the early endosome marker RAB-5. Scale bars are 5 μm.