

Abstract
Basic helix-loop-helix (bHLH) transcription factor Twist is one of the key inducers of epithelial to mesenchymal transition (EMT) that is a transdifferentiation program associated with embryo development and tumor metastasis. High level of Twist expression is shown to be correlated with cancer malignancy. Although Twist has been reported to be degraded by F-box and leucine-rich repeat protein 14 (FBXL14), the molecular mechanisms by which Twist levels are regulated have not been fully elucidated. In the present study, we identified Twist to be a ubiquitin substrate of β-transducin repeat-containing protein (β-TRCP), the adaptor subunit of SCFβ-TRCP (Skp1-Cul1-F-box protein) E3 ligase complex. We observed that depletion of β-TRCP leads to an accumulation of Twist protein, which could enhance tumor cell motility and cancer metastasis. Moreover, phosphorylation of Twist by inhibitor of KappaB kinase β (IKKβ) at multiple sites triggers its cytoplasmic translocation and the destruction by SCFβ-TRCP. Thus, our results provide the potential molecular mechanism of how the mesenchymal marker Twist is degraded, thereby shedding lights into regulation of the EMT, and providing the rationale for development of new therapeutic intervention to achieve better treatment outcomes in human cancer.
Introduction
Epithelial to mesenchymal transition (EMT) is a phenomenon that happens during the embryogenesis (Kang and Massague, 2004). It is known that during the EMT process, epithelial cells become a fibroblast-like morphology and acquire the mesenchymal characteristics, leading to an enhanced invasive motility (Kang and Massague, 2004). In carcinogenesis, EMT allows tumor cells to migrate, invade, and metastasize to distant tissue sites (Brabletz, 2012). Therefore, EMT plays a pivotal role in promoting cancer metastasis. Given the adverse correlation between EMT and cancer survival, multiple molecular markers have been identified to distinguish the mesenchymal cells from the epithelial cells (Peinado et al., 2007). For example, E-cadherin is the best-characterized member of the epithelial markers. The mesenchymal markers include Twist, Snail, Slug, and ZEB1/2, all of which repress the expression of E-cadherin (Peinado et al., 2007).
Many growth factors and cytokines as well as cell signaling pathways have been demonstrated to regulate EMT (Kang and Massague, 2004). Recently, accumulating evidence has suggested that ubiquitin-mediated proteolysis plays a critical role in regulating the EMT process through destruction of key EMT proteins by the 26S proteasome complex (Voutsadakis, 2012c). Ubiquitination is a post-translational protein modification that is governed by the ubiquitin proteasome system (UPS) (Hoeller et al., 2006). UPS consists of the E1 ubiquitin-activating enzyme, the E2 ubiquitin-conjugating enzyme, and E3 ubiquitin-protein ligase (Frescas and Pagano, 2008). The specificity of target protein selection is largely determined by the individual E3 enzyme (Lipkowitz and Weissman, 2011). Among the E3 enzymes, the SCF (Skp1-Cul1-F-box protein) E3 ligase complex has been well studied. SCF consists of Skp1 (S-phase kinase-associated protein 1), Cul1, Rbx1/Roc1, and a variable F-box protein (Mujtaba and Dou, 2011).
The growing body of literature suggests that several E3 ligases govern EMT through degradation of proteins involved in regulating the EMT process (Figure 1). SCFβ-TRCP, one of the E3 ubiquitin ligases, regulates the Snail transcription factor (Zhou et al., 2004) and mesenchymal marker β-catenin (Latres et al., 1999; Winston et al., 1999). FBXW8/Cullin 7, on the other hand, plays an important role in EMT by degrading ZEB1 and Slug (Fu et al., 2010). MDM2 (mouse double minute 2) is an E3 ligase that promotes degradation of Slug (Wang et al., 2009). FBXL14 was also found to interact with Snail and promote its ubiquitylation and subsequent proteasomal degradation (Vinas-Castells et al., 2010). Taken together, these results indicated that E3 ubiquitin ligases play a critical role in regulating the EMT process and cancer metastasis.
Figure 1.
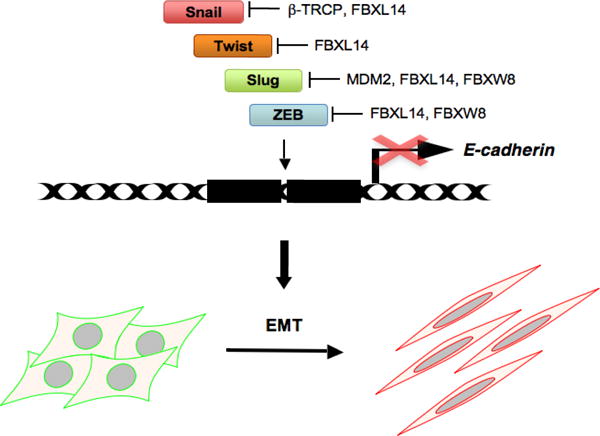
Epithelial to mesenchymal transition (EMT)-inducing transcription factors and the reported E3 ubiquitin ligases that govern their stability. The ubiquitin-proteasome pathway partly regulates EMT through proteolysis of EMT-inducing transcriptional factors. FBXL, F-box and leucine-rich repeat protein; FBXW, F-box and WD-40 domain protein; MDM2, mouse double minute 2.
In the present study, for the first time, we report that Twist is a potential substrate of β-transducin repeat-containing protein (β-TRCP). Our study showed that depletion of β-TRCP leads to an accumulation of Twist protein. Furthermore, inhibitor of KappaB kinase β (IKKβ)-mediated phosphorylation of Twist at multiple sites promotes its cytoplasmic translocation and destruction by SCFβ-TRCP. More importantly, IKKβ-dependent phosphorylation of Twist at T125 and S127 governs its nuclear localization. Hence, our current study supports the pivotal role of β-TRCP in IKKβ-mediated Twist degradation. Our results suggest that targeting β-TRCP could be a possible way to govern EMT process by accelerating Twist destruction.
Materials and Methods
Cell culture
HeLa cells were cultured in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 50 units/ml penicillin, and 50 mg/ml streptomycin. The cells were maintained in a 5% CO2-humidified atmosphere at 37°C.
Plasmids
Flag-β-TRCP1, shRNA-β-TRCP1+2, shTRCP1, shTRCP2, and shRNA-GFP constructs were described previously (Jin et al., 2003; Shirogane et al., 2005). Various Twist mutants were generated using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions. Hemagglutinin (HA)-tagged GSK3β construct was obtained from Dr. James DeCaprio (Dana-Farber Cancer Institute). The importin α1, importin α5, and importin α7 plasmids were obtained from the Dana-Farber/Harvard Cancer Center (DF/HCC) DNA Resource Core (Gao et al., 2009).
Antibodies and reagents
Anti-SP1 (SC-17824) and polyclonal anti-HA (SC-805) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Peroxidase-conjugated anti-mouse secondary antibody (A4416), peroxidase-conjugated anti-rabbit secondary antibody (A4914), polyclonal anti-FLAG (F2425), monoclonal anti-FLAG (F-3165), and anti-tubulin (T-5168) antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Monoclonal anti-HA antibody (MMS-101P) was purchased from Covance (Princeton, NJ). The anti-GFP (632380) antibody was purchased from Invitrogen. The anti-Twist (4119), anti-β-TRCP1 (4394), anti-IKKα (2682) antibodies, and IKKβ Kinase (7548) were purchased from Cell Signaling Technology (Danvers, MA). Proteasome inhibitor MG132 and phosphatase inhibitors (phosphatase inhibitor cocktail set I and II) were purchased from Calbiochem (Spring Valley, CA). Anti-GFP (632380) antibody, Oligofectamine, Lipofectamine and Plus reagents were purchased from Invitrogen.
Immunoblots and immunoprecipitation
HeLa cells were lysed in cell lysis buffer (50 mM Tris pH 8.0, 120 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors and phosphatase inhibitors. The protein concentrations of the lysates were measured using the Bio-Rad protein assay reagent on a Beckman Coulter DU-800 spectrophotometer. The lysates were then resolved by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and immunoblotted with indicated antibodies. For immunoprecipitation, 800 μg lysates were incubated with the appropriate antibody (1–2 μg) for 3–4 hours at 4°C followed by one hour-incubation with Protein A sepharose beads (GE Healthcare, Pittsburgh, PA). Immuno-complexes were washed five times with wash buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA and 0.5% NP-40) before being resolved by SDS-PAGE and immunoblotted with indicated antibodies.
In vitro kinase assay
The IKKβ in vitro kinase assay was performed as described previously (Inuzuka et al., 2010). Briefly, 5 μg of indicated glutathione-S-transferase-Twist fusion proteins (Soucy et al., 2009) were incubated with purified active IKKβ Kinase in the presence of 5 μCi [γ-32P] ATP and 200 μM cold, unlabeled ATP in the IKKβ reaction buffer for 30 minutes. The reaction was stopped by the addition of SDS-containing lysis buffer, resolved on SDS-PAGE, and detected by autoradiography.
Protein degradation analysis
Cells were plated into tissue-culture dishes 20 hours before transfection. When cells reached an appropriate confluence, they were transfected with various HA-Twist plasmids along with Flag-β-TRCP1, and GFP as a negative control, in the presence or absence of Flag-IKKβ. After 40 hours, cells were lysed and protein concentration was measured, and subsequently immunoblot analysis was performed.
Results
Depletion of β-TRCP increased Twist protein levels
It has been demonstrated that most β-TRCP substrates such as Snail typically contain the canonical DSGxxS phospho-degron (Figure 2A) (Zhou et al., 2004). Unexpectedly, Twist does not have a canonical DpSGxxpS degron that could be recognized by SCFβ-TRCP. However, Twist contains two derivative phospho-degron variants (Figure 2B). To identify Twist as a potential substrate of β-TRCP, we explored whether depletion of endogenous β-TRCP increases Twist protein abundance in HeLa cells. As expected, we found that depletion of either endogenous β-TRCP1 or β-TRCP1+2 caused an upregulation of Twist protein levels (Figure 2C). Consistent with the role of β-TRCP in governing Twist stability, we observed that Twist interacts with β-TRCP1 under ectopic overexpression conditions (Figure 2D). Taken together, these results indicate that β-TRCP might control the Twist stability, a critical regulator of the EMT process.
Figure 2.
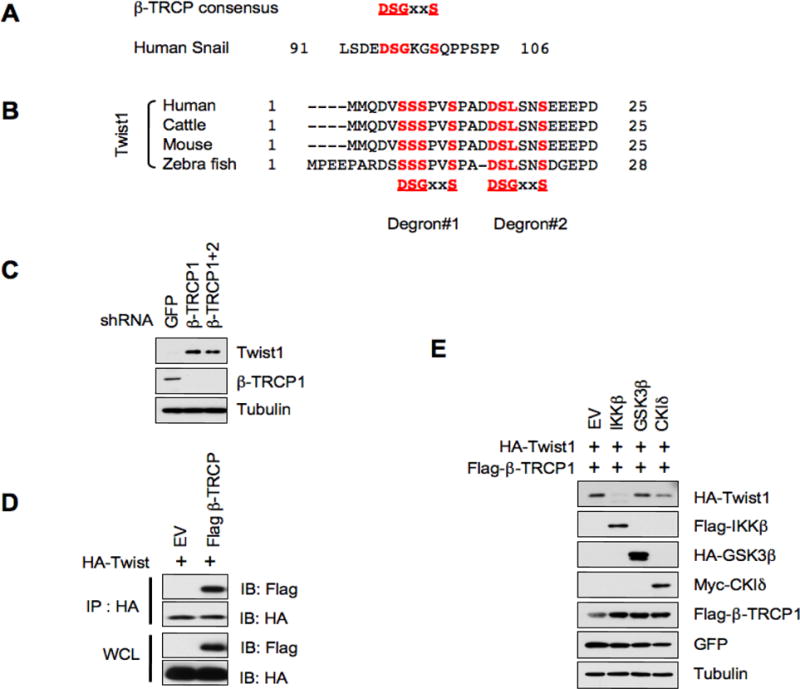
Identification of β-TRCP and IKKβ as upstream negative regulators of Twist. A. β-TRCP consensus phospho-degron motif and an amino acid sequence of human Snail β-TRCP degron. B. Alignment of evolutionarily conserved putative β-TRCP degron motifs at the N-terminal amino acid sequence of Twist1. C. HeLa cells were infected with the indicated lentiviral shRNA constructs for 24 hours. Uninfected cells were eliminated by selection with puromycin for 48 hours. Equal amounts of whole cell lysates were immunoblotted with the indicated antibodies. D. Immunoblot analysis of whole cell lysates and immunoprecipitates derived from HeLa cells transfected with hemagglutinin (HA)-Twist and the indicated Flag-β-TRCP1 constructs. After 30 hours posttransfection, cells were pretreated with 15 μM MG132 for 10 hours to block the proteasome pathway before harvest. E. Immunoblot analysis of whole cell lysates derived from HeLa cells transfected with the indicated constructs.
IKKβ controls Twist stability
It has been known that prior to the recognition and subsequent destruction by the SCFβ-TRCP E3 ligase complex, most characterized β-TRCP substrates need to be phosphorylated by one kinase or a combination of various kinases within their phospho-degron(s) (Frescas and Pagano, 2008). So far, it has several well-known kinases including GSK3β, CKI and IKKβ to phosphorylate degron sequence that could be recognized by the SCFβ-TRCP complex. To this end, we investigated whether these candidate kinases are involved in the degradation of Twist by SCFβ-TRCP. Indeed, we found that overexpression of IKKβ, but not GSK3, CKI, or the EV (empty vector) control, significantly reduced Twist protein level (Figure 2E). Interestingly, overexpression of CKId slightly decreased Twist expression (Figure 2E). These results demonstrated that IKKβ might play a critical role in destruction of Twist by SCFβ-TRCP.
IKKβ phosphorylates Twist at multiple sites
Our previous study has revealed that β-TRCP substrates often contain multiple suboptimal degron sequences if they have no canonical DSGxxS degron sequence (Inuzuka et al., 2010). To pinpoint the phospho sites by IKKβ within degrons, we generated multiple truncation mutants wherein the potential critical Ser/Thr sites for individual suboptimal degrons are located (Figure 3C). Using in vitro kinase assays, we observed that the region containing amino acids 112–140 possibly possesses the major IKK phosphorylation sites and the region in the extreme N-terminus contains weak IKK phosphorylation site(s) (Figure 3A and 3C). On the other hand, the other regions of Twist seem to contain no detectable IKK phosphorylation sites under this experimental condition. Next, we intended to pinpoint the exact IKK sites within these two regions. Notably, mutation of both Thr125 and Ser127 to Ala in Twist greatly reduced IKKβ-dependent Twist phosphorylation in vitro (Figure 3A). Similarly, mutation of all Ser7, Ser8, Ser11, Ser16, and Ser20 sites to Ala significantly decreased Twist phosphorylation by IKKβ (Figure 3B). Taken together, our results indicated that IKKβ could phosphorylate Twist at multiple sites to trigger Twist destruction.
Figure 3.
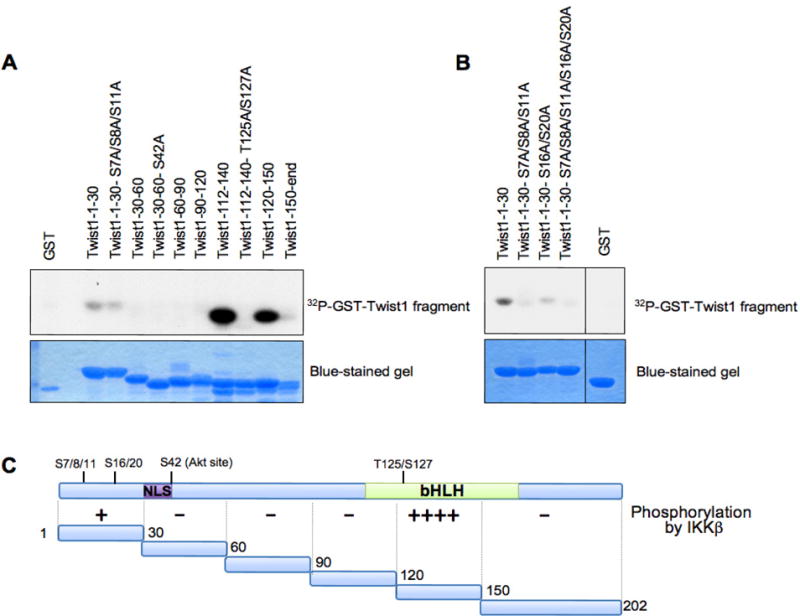
Mapping the putative IKKβ phosphorylation sites in Twist. A–B. Purified IKKβ kinase was incubated with 3 μg of the indicated GST-Twist proteins in the presence of γ-32P-ATP. The kinase reaction products were separated by SDS-PAGE, and phosphorylation was detected by autoradiography. GST was used as a negative control in this assay. C. Scheme of the GST-Twist1 constructs used in this assay. The extent of the phosphorylation by IKKβ is shown with +. NLS, nuclear localization signal; bHLH, basic helix-loop-helix.
Multiple phosphorylation sites are required for Twist degradation
To further confirm that these multiple phosphorylation sites are necessary for Twist destruction, we also performed the degradation assay. As illustrated in Figure 4, co-expression of IKKβ and β-TRCP significantly down-regulated the expression of wild-type HA-tagged Twist, a process that could be efficiently blocked by the treatment of the proteasome inhibitor, MG132. Notably, Ala substitutions of Ser and Thr in each degron motif partly protected the degradation by IKKβ and β-TRCP. More importantly, the IKKβ and β-TRCP-induced Twist degradation is completely blocked by the 7A (S7A/S8A/S11A/S16A/S20A/T125A/S127A) mutations that are defective for IKK-dependent phosphorylation as all the identified phosphorylation sites are mutated to Ala. This result strongly suggests that all seven identified IKK phosphorylation sites could be involved in β-TRCP-mediated degradation of Twist. However, we recognize that further studies are required to determine the contribution of each individual phosphorylation site to β-TRCP-mediated Twist destruction.
Figure 4.
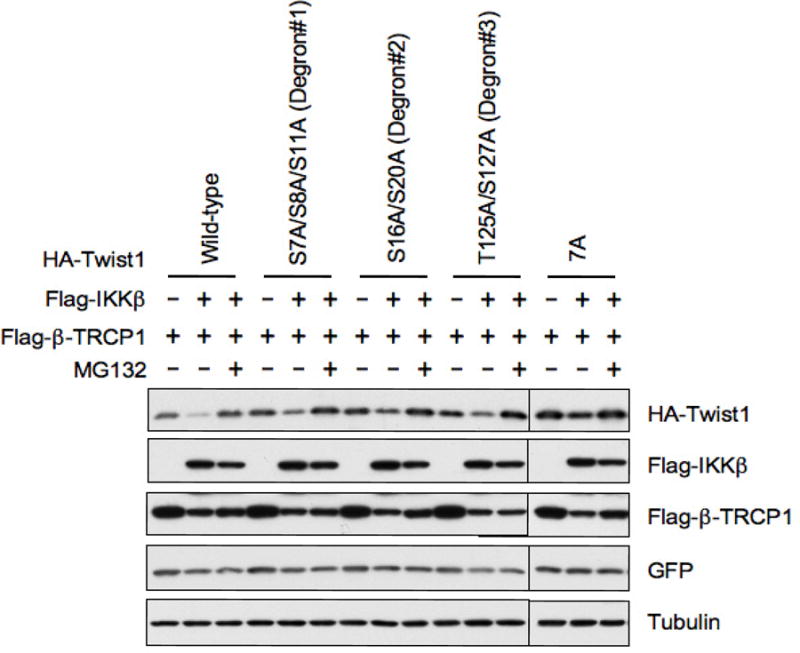
Identification of IKKβ phosphorylation sites that are critical for β-TRCP-mediated destruction of Twist. Immunoblot analysis of HeLa cells transfected with the indicated HA-Twist and Flag-β-TRCP plasmids in the presence or absence of Flag-IKKβ. A plasmid encoding GFP (green florescent protein) was used as an internal control for transfection efficiency. 7A, S7A/S8A/S11A/S16A/S20A/T125A/S127A.
IKKβ involved in Twist translocation into the nucleus
Since both Snail and Slug localizations are affected by glycogen synthase kinase 3 β (GSK3β)-mediated phosphorylation (Kim et al., 2012; Zhou et al., 2004), we also investigated whether the phosphorylation at Thr125 and Ser127 by IKKβ controls Twist localization. The phosphorylation sites at Thr125 and Ser127, which could affect dimerization and DNA binding ability (Firulli et al., 2005; Lu et al., 2011), are highly conserved in various species and other Twist family members (Figure 5A). To directly address this question, we evaluated the contribution of Thr125 and Ser127 residues for Twist nuclear localization. As illuminated in Figure 5B, like the wild-type Twist, phospho-deficient T125A/S127A mutation is largely retained in the nucleus, whereas phospho-mimic T125E/S127D mutation displayed a significant reduction in nuclear localization (Figure 5B). In further support of this notion, we found that Twist specifically binds to importin α5 and importin α7, but not importin α1 (Figure 5C). Moreover, the T125E/S127D and S42D mimetic mutations abolished the interaction with the importin complex (Figure 5C), which provides a possible mechanism for IKKβ-dependent Twist cytoplasmic translocation while further studies are warranted to fully understand this dynamic process.
Figure 5.
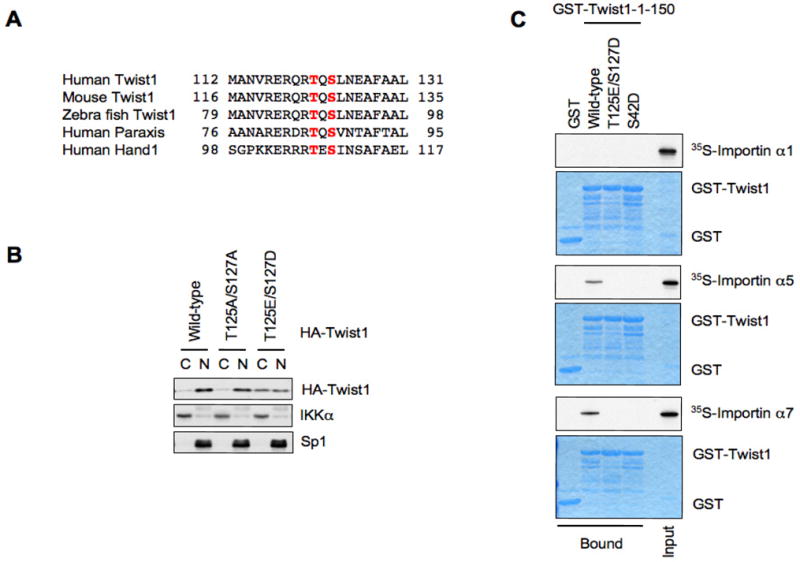
IKKβ-mediated phosphorylation of Twist promotes its cytoplasmic translocation. A. Identified phosphorylation sites at Thr125 and Ser127 are highly conserved across spices and other Twist family members. B. Immunoblot analysis of cellular fractionation of HeLa cells expressing wild-type, phospho-deficient (T125A/S127A) and phospho-mimic (T125E/S127D) HA-Twist. C, cytosolic fraction; N, nuclear fraction. C. Autoradiography of 35S-labelled importin α1, importin α5, or importin α7 bound to the indicated GST-Twist proteins.
Discussion
Twist, a basic helix-loop-helix (bHLH) transcriptional factor, is one of the EMT-inducing molecules that play a pivotal role in regulating the EMT process (Qin et al., 2012). It has been known that Twist contributes to metastasis by promoting an EMT through inhibition of the E-cadherin tumor suppressor, leading to activation of mesenchymal markers and subsequent induction of cell invasion (Yang et al., 2004). Moreover, overexpression of Twist has been found to correlate with tumor invasion and metastasis in human breast cancers (Yang et al., 2004). Consistent with this notion, Twist is required for tumor metastasis from the mammary gland to the lung nodules (Yang et al., 2006). To support the role of Twist in tumorigenesis, studies have shown that Twist enhanced the metastasis in several epithelial cancers including prostate and bladder cancers (Kang and Massague, 2004). More importantly, Twist has been considered as a prognostic biomarker in certain human cancers (Qin et al., 2012). Strikingly, Twist also plays a critical role in chemotherapeutic resistance of cancer stem cells (Qin et al., 2012). Therefore, Twist could be a novel potential drug target for achieving better treatment outcome for cancer patients.
Accumulating evidence suggests that different upstream signaling pathways govern Twist expression. For example, HIF-1 (hypoxia inducible factor-1) directly regulates Twist1 expression by binding directly to the hypoxic response element (HRE) in the Twist proximal promoter, leading to promotion of metastasis in response to intratumoral hypoxia (Yang et al., 2008). SRC-1 (steroid receptor coactivator-1) regulates Twist expression through PEA3 (polyomavirus enhancer activator 3), leading to breast cancer metastasis (Qin et al., 2009). STAT3 (signal transducer and activator of transcription 3) was found to induce Twist expression and exert its oncogenic function (Cheng et al., 2008; Lo et al., 2007). Recently, Notch1 was reported to promote the progression of human gastric cancer through activation of STAT3/Twist signaling axis (Hsu et al., 2012). Moreover, Bmi1 and Twist act cooperatively to inhibit expression of both E-cadherin and p16, resulting in induction of EMT and tumor invasion (Yang et al., 2010). Further studies showed that Bmi1 cooperates with Twist to repress let-7i expression, leading to upregulation of NEDD9 and DOCK3 as well as activation of RAC1 (Yang et al., 2012).
Recently, one study has suggested that Twist protein is regulated by the F-box protein FBXL14 (Lander et al., 2011). This group found that Twist is an unstable protein and is targeted for ubiquitination via the C-terminal WR domain. Moreover, FBXL14 is an endogenous regulator of Twist stability (Lander et al., 2011). Increased evidence indicated that most individual EMT proteins are regulated by multiple E3 ubiquitin ligases (Voutsadakis, 2012a; Voutsadakis, 2012b). For example, Snail is degraded by FBXL14, and β-TRCP, while ZEB1 is targeted by FBXW8 and FBXL14 (Voutsadakis, 2012a). Similarly, Slug stability is governed by MDM2, FBXW8, and FBXL14 (Voutsadakis, 2012c). However, little is known whether E3 ubiquitin ligases other than FBXL14 regulate Twist stability.
To this end, we found for the first time that depletion of either β-TRCP1 or β-TRCP1+2 markedly upregulates Twist expression (Figure 2C). Furthermore, we observed that Twist binds to β-TRCP1 under ectopic overexpression conditions. Moreover, overexpression of IKKβ significantly inhibited Twist expression. More importantly, IKKβ phosphorylates Twist at multiple sites to trigger Twist destruction, indicating that Twist protein, like β-TRCP1 substrate Mdm2, contains many suboptimal degron sequences that are phosphorylated by IKKβ. Interestingly, phosphorylation of Twist by IKKβ induces its translocation into the cytoplasm and β-TRCP-mediated destruction, which is of similar mechanism of Snail regulated by GSK3β (Zhou et al., 2004). However, we recognize that more biochemical studies including the in vitro ubiquitination and in vitro degradation assays might be required for us to draw a concrete conclusion regarding whether Twist, analogous to Snail and ZEB1, is controlled by SCFβ-TRCP. More importantly, we are also aware that most of our studies are using the in vitro cell culture system, thus much more studies, especially the mouse genetic models, are required to fully understand whether β-TRCP is the physiological E3 ligase to control Twist ubiquitination. Furthermore, immunohistochemistry studies of human clinical samples are preferred to examine whether there is an inverse correlation between the Twist expression and its negative regulators including β-TRCP or IKKβ, especially in those late stage samples with metastasis.
In summary, our results provide a possible molecular mechanism for the deregulation of Twist in HeLa cervical cancer cells (Figure 6). Therefore, activation of β-TRCP and/or IKKβ could be a novel strategy to treat various human cancers with high expression of Twist that is associated with EMT. Our work identifies β-TRCP and IKKβ as the possible upstream components to govern Twist stability, thus providing the rationale for development of β-TRCP and/or IKKβ inhibitors to achieve better treatment outcomes in human cancer patients.
Figure 6.
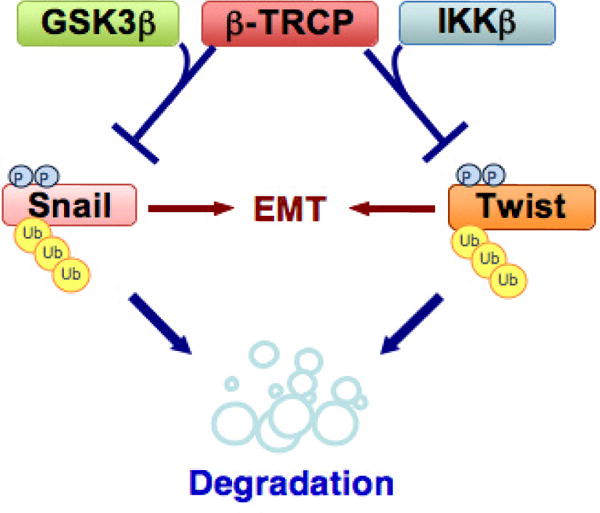
The putative model of the Snail and Twist stability controlled by β-TRCP. β-TRCP might negatively regulate EMT by degrading Snail and Twist in a phosphorylation dependent manner in HeLa cervical cells.
Acknowledgments
This work was supported by grants from the National Institute of Aging NIH K01 (AG041218) to H.I., and by NIH NRSA Fellowship to Z.W.
Footnotes
Disclosure
The authors report no conflicts of interest.
References
- Brabletz T. To differentiate or not–routes towards metastasis. Nat Rev Cancer. 2012;12(6):425–436. doi: 10.1038/nrc3265. [DOI] [PubMed] [Google Scholar]
- Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, Xu LM, Costanzo C, Cheng JQ, Wang LH. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem. 2008;283(21):14665–14673. doi: 10.1074/jbc.M707429200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli BA, Krawchuk D, Centonze VE, Vargesson N, Virshup DM, Conway SJ, Cserjesi P, Laufer E, Firulli AB. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat Genet. 2005;37(4):373–381. doi: 10.1038/ng1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8(6):438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Lv X, Lin H, Wu L, Wang R, Zhou Z, Zhang B, Wang YL, Tsang BK, Zhu C, Wang H. Ubiquitin ligase cullin 7 induces epithelial-mesenchymal transition in human choriocarcinoma cells. J Biol Chem. 2010;285(14):10870–10879. doi: 10.1074/jbc.M109.004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009;11(4):397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer. 2006;6(10):776–788. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- Hsu KW, Hsieh RH, Huang KH, Fen-Yau Li A, Chi CW, Wang TY, Tseng MJ, Wu KJ, Yeh TS. Activation of the Notch1/STAT3/Twist signaling axis promotes gastric cancer progression. Carcinogenesis. 2012;33(8):1459–1467. doi: 10.1093/carcin/bgs165. [DOI] [PubMed] [Google Scholar]
- Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, Wan L, Ang XL, Mock C, Yin H, Stommel JM, Gygi S, Lahav G, Asara J, Xiao ZX, Kaelin WG, Jr, Harper JW, Wei W. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell. 2010;18(2):147–159. doi: 10.1016/j.ccr.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17(24):3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118(3):277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim YM, Yang CH, Cho SK, Lee JW, Cho M. Functional regulation of Slug/Snail2 is dependent on GSK-3beta-mediated phosphorylation. FEBS J. 2012;279(16):2929–2939. doi: 10.1111/j.1742-4658.2012.08674.x. [DOI] [PubMed] [Google Scholar]
- Lander R, Nordin K, Labonne C. The F-box protein Ppa is a common regulator of core EMT factors Twist, Snail, Slug, and Sip1. J Cell Biol. 2011;194(1):17–25. doi: 10.1083/jcb.201012085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latres E, Chiaur DS, Pagano M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene. 1999;18(4):849–854. doi: 10.1038/sj.onc.1202653. [DOI] [PubMed] [Google Scholar]
- Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer. 2011;11(9):629–643. doi: 10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67(19):9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Nie J, Luan Q, Feng Q, Xiao Q, Chang Z, Shan C, Hess D, Hemmings BA, Yang Z. Phosphorylation of the Twist1-family basic helix-loop-helix transcription factors is involved in pathological cardiac remodeling. PLoS One. 2011;6(4):e19251. doi: 10.1371/journal.pone.0019251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba T, Dou QP. Advances in the understanding of mechanisms and therapeutic use of bortezomib. Discov Med. 2011;12(67):471–480. [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Z, Chen H, Xu J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 2009;69(9):3819–3827. doi: 10.1158/0008-5472.CAN-08-4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22(1):90–106. doi: 10.1038/cr.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirogane T, Jin J, Ang XL, Harper JW. SCFbeta-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J Biol Chem. 2005;280(29):26863–26872. doi: 10.1074/jbc.M502862200. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, Mcdonald A, Mizutani H, Narayanan U, Olhava EJ, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Vinas-Castells R, Beltran M, Valls G, Gomez I, Garcia JM, Montserrat-Sentis B, Baulida J, Bonilla F, De Herreros AG, Diaz VM. The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J Biol Chem. 2010;285(6):3794–3805. doi: 10.1074/jbc.M109.065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutsadakis IA. Epithelial to mesenchymal transition in the pathogenesis of uterine malignant mixed Mullerian tumours: the role of ubiquitin proteasome system and therapeutic opportunities. Clin Transl Oncol. 2012a;14(4):243–253. doi: 10.1007/s12094-012-0792-4. [DOI] [PubMed] [Google Scholar]
- Voutsadakis IA. The ubiquitin-proteasome system and signal transduction pathways regulating epithelial mesenchymal transition of cancer. J Biomed Sci. 2012b;19:67. doi: 10.1186/1423-0127-19-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutsadakis IA. Ubiquitination and the Ubiquitin-Proteasome System as regulators of transcription and transcription factors in epithelial mesenchymal transition of cancer. Tumour Biol. 2012c;33(4):897–910. doi: 10.1007/s13277-012-0355-x. [DOI] [PubMed] [Google Scholar]
- Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK, Li KC, Hong TM, Yang PC. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol. 2009;11(6):694–704. doi: 10.1038/ncb1875. [DOI] [PubMed] [Google Scholar]
- Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999;13(3):270–283. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA, Weinberg RA. Exploring a new twist on tumor metastasis. Cancer Res. 2006;66(9):4549–4552. doi: 10.1158/0008-5472.CAN-05-3850. [DOI] [PubMed] [Google Scholar]
- Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12(10):982–992. doi: 10.1038/ncb2099. [DOI] [PubMed] [Google Scholar]
- Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10(3):295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- Yang WH, Lan HY, Huang CH, Tai SK, Tzeng CH, Kao SY, Wu KJ, Hung MC, Yang MH. RAC1 activation mediates Twist1-induced cancer cell migration. Nat Cell Biol. 2012;14(4):366–374. doi: 10.1038/ncb2455. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]