

Abstract
Intratracheal administration of corticosteroids using a natural pulmonary surfactant as a delivery vehicle has recently received significant attention in hopes of treating premature newborns with or at high risk for chronic lung disease. As a new practice, both the surfactant preparation used as the carrier and the corticosteroid delivered as the anti-inflammatory agent, and their mixing ratios, have not been standardized and optimized. Given the concern that corticosteroids delivered via a pulmonary surfactant may compromise its surface activity and thus worsen lung mechanics, the present study was carried out to characterize the biophysical interaction between a natural surfactant preparation, Infasurf, and two commonly used inhaled corticosteroids, budesonide and beclomethasone dipropionate (BDP). Based on surface activity measurements by the Langmuir balance and lateral film structure studied by atomic force microscopy, our findings suggest that when Infasurf is used as a carrier, a budesonide concentration less than 1 wt% of surfactant or a BDP concentration up to 10 wt % should not significantly affect the biophysical properties of Infasurf, thus being feasible for pulmonary delivery. Increasing corticosteroid concentration beyond this range leads to early collapse of the surfactant film due to increased film fluidization. Our study further suggests that different affinities to the surfactant films are responsible for the different behavior of budesonide and BDP. In addition to the translational value in treating chronic lung disease, this study may also have implications in inhaled steroid therapy to treat asthma.
1. Introduction
Being a noninvasive method, pulmonary drug delivery has received growing attention in recent years.1,2 The viability of pulmonary drug delivery is mainly due to the large surface area of the human lungs (∼70 to 140 m2 for adults) and the extremely thin diffusion barrier in the peripheral lung (less than 0.5 μm).3 These anatomic features make the lungs an ideal absorptive site for inhaled/instilled macromolecules, and hence a promising portal for drug delivery.4,5 In addition to ease of drug delivery, compared to intravenous injection, topical administration to the lungs avoids first-pass metabolism and eliminates potential side effects caused by high systemic dosage.1,2,4,5 These advantages make pulmonary drug delivery an ideal method for treating respiratory diseases, such as asthma, chronic pulmonary infection, emphysema, cystic fibrosis, pulmonary hypertension, and lung cancer. In addition to local delivery, the lungs are also suitable for systemic delivery of therapeutic agents, especially peptide and protein drugs.1,2,4,5
Pulmonary delivery of corticosteroids has been attempted in the treatment of premature newborns with or at high risk for chronic lung disease (CLD).6–14 Because of their anti-inflammatory actions, corticosteroids, such as dexamethasone, have long been used systemically to slow the progression of CLD in ventilated premature infants.15,16 However, the use of systemic corticosteroids all but ceased after concern was raised regarding its adverse effects on the developing brain.17,18
Pulmonary delivery of corticosteroids to infants with or at high risk for CLD should provide effective anti-inflammatory therapy locally, thereby having fewer adverse systemic effects.6–14 Two different pulmonary delivery methods have been clinically tested. They are inhalation delivery of steroid aerosols,6–9 and very recently, intratracheal instillation of steroids using an exogenous surfactant as a spreading agent.10–14 For example, Yeh et al. reported a randomized clinical trial for early postnatal intratracheal instillation of budesonide using a natural surfactant preparation (Survanta) as a vehicle of spreading.10 They showed that this method of delivery significantly improved the combined outcome of death and CLD in very premature infants without causing immediate adverse effects. The lack of long-term adverse effects was recently reported in a follow-up study by the same group.11 Similar results were also reported by Dani et al.12 These researchers found that intratracheal administration of beclomethasone with a natural surfactant preparation (Curosurf) effectively reduced oxidative lung stress and improved respiratory function in a preterm lamb model.12
The rationale of using a natural surfactant as a delivering vehicle for corticosteroid drugs is at least threefold. First, a natural surfactant has ideal biocompatibility and biodegradability as it can be cleared from the alveolar space by endocytosis back into type II cells or taken up by the alveolar macrophages. Second, a natural surfactant can increase the aqueous solubility of these hydrophobic drugs via drug solubilization.19 Third, a natural surfactant can spread automatically along the surface tension gradient at the inner surface of the respiratory tract, a phenomenon known as the Marangoni effect.20-23 This allows corticosteroids mixed with natural surfactant to be easily carried to the peripheral lung after intratracheal instillation. Both numerical simulation and biomechanical experiments have demonstrated the feasibility of using exogenous surfactant as a spreading agent in the lungs.20–23 However, in addition to the ability of self-propelled spreading, the design of a drug delivery system based on an exogenous surfactant also requires in-depth understanding of molecular interaction between the surfactant and the carried drug; it is vital that the delivered drug does not inhibit biophysical function of the pulmonary surfactant.
The pulmonary surfactant (PS) is synthesized by alveolar type II epithelial cells and forms a thin film at the air-water interface of alveoli.24 It consists of ∼80 wt% phospholipids, 5-10% neutral lipids (primarily cholesterol), and 5-10% proteins.25 The main biophysical function of this PS film is to reduce alveolar surface tension, thus maintaining a large surface area of the lungs for effective respiration.26 Although limited, available evidence suggests that a specific molecular interaction occurs between corticosteroids and PS, and such an interaction depends on the type of corticosteroid, the dose, and the surfactant preparation.27–32 To date, the specific molecular interaction between different corticosteroids and surfactant preparations and its potential biophysical and physiological impacts have not been studied.
In the present paper, we report an in vitro biophysical study of interfacial molecular interactions between two commonly used inhaled corticosteroids, budesonide and beclomethasone dipropionate, and a clinical surfactant preparation, Infasurf, using the combination of Langmuir balance and atomic force microscopy (AFM). Langmuir balance was used to study the effect of corticosteroids added at different concentrations on surface activity (i.e., surface tension-lowering ability) of the PS. AFM was used to observe the effect of corticosteroids on the interfacial molecular organization and lateral structure of the PS films. This study is expected to shed light on the feasibility of pulmonary delivery of corticosteroids using PS as a vehicle.
2. Experimental
2.1. Materials
Infasurf® (calfactant) is a gift from ONY, Inc. (Amherst, NY). It is a modified natural surfactant prepared from lung lavage of newborn calves by centrifugation and organic solvent extraction. Infasurf contains all hydrophobic components of the natural surfactant, including ∼90 wt% phospholipids, 5–8% neutral lipids (mainly cholesterol), and ∼2% hydrophobic surfactant proteins (SP-B and SP-C).33 Hydrophilic surfactant protein (SP-A), however, is removed during the extraction process. Infasurf has been used in the United States for treating premature newborns with respiratory distress syndrome (RDS). Infasurf was extracted with chloroform-methanol using a method modified from Bligh and Dyer.34 The chloroform-methanol extracts were dried under a nitrogen stream and re-dissolved in chloroform to a final concentration of 1 mg phospholipids per mL. All stock solutions were stored at −20 °C until use.
Budesonide and beclomethasone dipropionate (BDP) were purchased from Sigma-Aldrich (St Louis, MO) and used without further purification. Chemical structures of budesonide and BDP molecules are shown in Fig. 1. Budesonide is a second generation corticosteroid with low systemic absorption. It has the chemical name of 16,17-butylidenebis(oxy)-11,21-dihydroxypregna-1,4-diene-3,20-dione.35 Budesonide has been used as an anti-inflammatory agent in the treatment of asthma, rhinitis, and inflammatory bowel disease. BDP is a first generation anti-inflammatory corticosteroid having the chemical name of 9-chloro-11(3,17,21-trihydroxy-16β-methylpregna-1,4-diene-3,20-dione 17,21-dipropionate.35
Fig. 1.
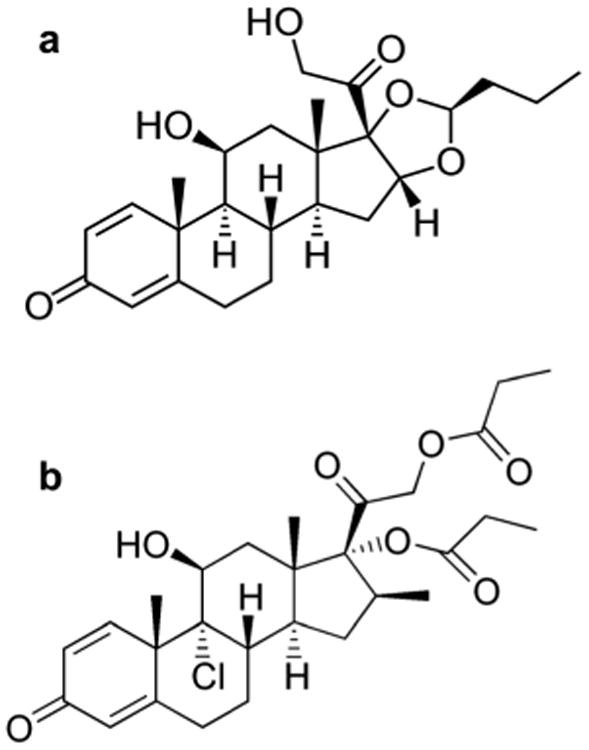
Chemical structures of two commonly used inhaled corticosteroids: (a) budesonide, and (b) beclomethasone dipropionate (BDP).
Each corticosteroid was studied at a wide range of concentration ratios with respect to phospholipids in Infasurf. For the stock solution of 1 mg mL−1 Infasurf, each corticosteroid was added at three concentrations of 0.001, 0.01, and 0.1 mg mL−1, corresponding to 0.1%, 1% and 10% of phospholipids in Infasurf, respectively. To date, intratracheally administrated corticosteroids in vitro,23,31 in animal models,12,14,23 and in clinical practice10 have commonly been in the range of 0.2–0.6% of pulmonary surfactant. Therefore, the present study covers all ranges of corticosteroids tested in the literature.
All solvents used were HPLC grade. The water used was Milli-Q ultrapure water (Millipore, Billerica, MA) which has a resistivity higher than 18 MΩ cm at room temperature.
2.2. Methods
Langmuir balance
Surface activity of Infasurf with and without corticosteroids was evaluated by measuring surface pressure-surface area isotherms using a Langmuir balance (KSV Nima, Coventry, UK) at room temperature (20 ± 1 °C). Surface pressure (π) and surface tension (γ) are linearly correlated by π = γ0 — γ, where, γ0 is the surface tension of a clean air-water interface, approximately equal to 72 mN m−1 at room temperature. Thus, the increase in π corresponds to the extent that a film decreases γ.
Detailed experimental procedures can be found elsewhere.36 Briefly, Infasurf films with/without corticosteroids were spread on pure water to increase π to 1–3 mN m−1, and were left undisturbed for 10 min to allow evaporation of solvent. The spread films were compressed at a rate of 20 cm2 min−1 (i.e., ∼0.1% area per second), with the π –area isotherms recorded. All compression isotherms were studied for at least three times to ensure reproducibility.
For AFM imaging, the surfactant film at the air-water interface was transferred to a solid substrate under controlled π using the Langmuir-Blodgett (LB) technique. Specifically, surfactant films, at characteristic π of 20, 30, 40, 50, and 60 mN m−1, were deposited onto freshly peeled mica surfaces at a dipping rate of 1 mm min−1. If 60 mN m−1 could not be reached, surfactant films at the highest possible π were studied.
Theoretically speaking, when all parameters are optimized, the LB technique should enable an accurate control of molecular organization and lateral structure of the surfactant film at the air–water interface.37 However, it has been long debated that the LB technique may introduce artifacts into the lateral structure and phase behavior of interfacial molecular films, thus complicating interpretation of AFM data.38,39 To the best of our knowledge, an unambiguous discussion of the potential artifacts associated with the LB technique is still not available.
Atomic force microscopy (AFM)
Topographical images of the LB samples were obtained using an Innova AFM (Bruker, Santa Barbara, CA). Samples were scanned in air at multiple locations with various scan areas to ensure detection of representative structures. Both contact mode and tapping mode were used. The different scan modes gave equivalent results. A silicon nitride cantilever with a spring constant of 0.12 N m−1 and a nominal tip radius of 2 nm was used in contact mode, and a silicon probe with a resonance frequency of 300 kHz and a spring constant of 40 N m−1 was used in tapping mode. Analyses of AFM images, including measurements of surface roughness, were carried out by the Nanoscope software (ver. 7.30). Quantitative data were expressed as mean ± standard deviation (n > 5).
3. Results
3.1. Surface activity evaluated by Langmuir balance
Fig. 2a and b compare the typical compression isotherms of pure Infasurf and Infasurf mixed with 0.1, 1, and 10% budesonide and BDP, respectively. For all isotherms, surface pressure (π) increased, corresponding to surface tension (γ) decrease, when the film was compressed (i.e., reducing surface area). All compression isotherms feature a plateau region at π 40- 50 mN m−1, corresponding to a monolayer-to-multilayer transition.36
Fig. 2.
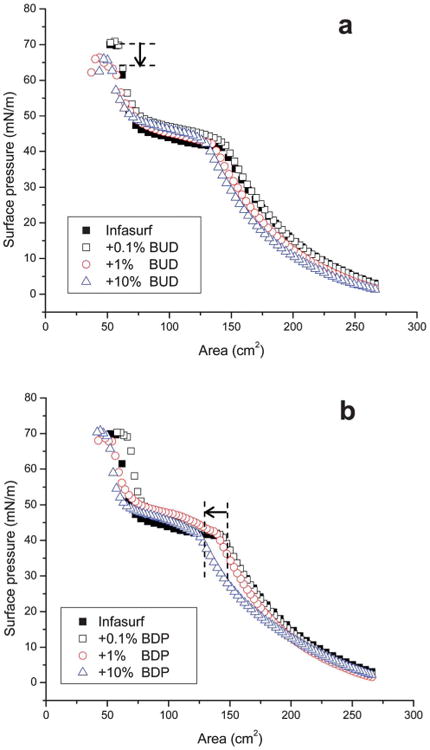
Effects of (a) budesonide (BUD) and (b) beclomethasone dipropionate (BDP) at various concentrations (0.1, 1, and 10 wt% of phospholipids in Infasurf) on the compression isotherms of Infasurf. Decrease of collapse pressure due to addition of 1% and 10% BUD, and shift of compression isotherm due to addition of 10% BDP are indicated in the figures with arrows.
As shown in Fig. 2a, addition of 0.1% budesonide does not significantly alter the compression isotherm of Infasurf. With increasing budesonide concentration to 1% and 10%, the compression isotherms slightly shift to the left, indicating moderate increase of film compressibility, i.e., more area reduction is needed to increase π. More appreciably, increasing budesonide concentration to 1% and 10% reduces the collapse pressure [i.e., the maximum surface pressure (πmax)] of Infasurf by approximately 5 mN m−1 (as indicated by the arrow shown in Fig. 2a). This result is in good agreement with the in vitro assessment by Palmer et al., who found that 1% budesonide increased the γmin of BLES, a natural surfactant preparation similar to Infasurf, as evaluated by a captive bubble surfactometer.31 Since the natural surfactant is expected to reduce alveolar surface tension to near-zero (i.e., π near 70 mN m−1), the decrease of πmax indicates surfactant inhibition by high concentration budesonide.
In contrast to budesonide, addition of BDP at all three concentrations (0.1, 1, and 10%) does not significantly affect the πmax of Infasurf. However, 10% BDP significantly shifts the compression isotherm of Infasurf monolayers to the low surface area region (i.e., to the left, as indicated by the arrow shown in Fig. 2b), which may indicate removal of surface material from the air-water interface during the compression process. It should be noted that the present study only considers the first compression after film formation. Our previous study has demonstrated that the shape of compression isotherms is relatively unchanged after repeated compression-expansion cycles, provided that the Langmuir balance in use is well prepared for minimizing film leakage.40,41
3.2. Interfacial molecular organization and lateral structure revealed by AFM
AFM images depicted in Fig. 3 compare the molecular organization and lateral structure of Infasurf with and without budesonide at increasing π. Different budesonide concentrations (0, 0.1%, 1% and 10% to Infasurf) are compared in columns. Various π (20, 30, 40, 50, and 60 mN m−1) of each preparation are compared in rows. These characteristic π were selected to cover the complete and detailed evolution of each film under compression. Infasurf with 10% budesonide failed to form a stable LB film at 60 mN m−1. All AFM images have the same scan area of 20 × 20 mm. The full z-range is 5 nm for all monolayers (i.e., π ≤ 40 mN m−1) and 20 nm for all multilayers (π > 40 mN m−1).
Fig. 3.
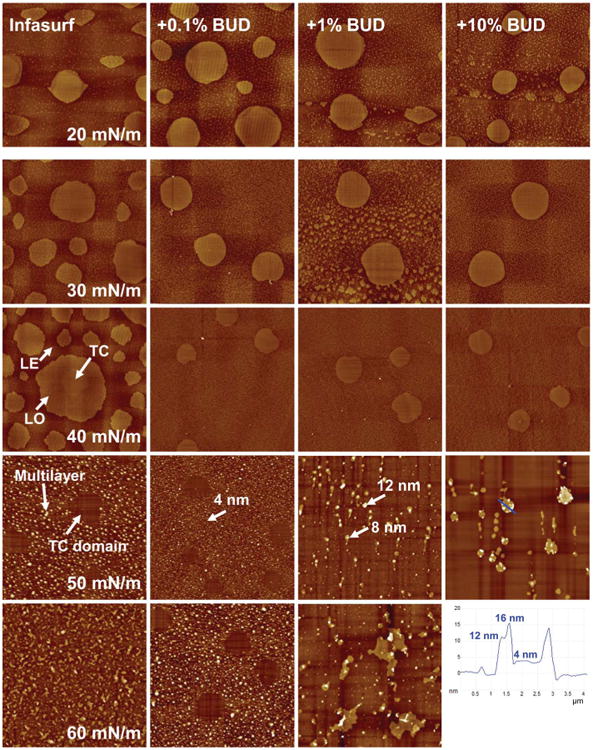
Effects of budesonide (BUD) at various concentrations on the molecular organization and lateral structure of Infasurf films. The first column shows AFM topographic images of pure Infasurf at five characteristic surface pressures (π) of 20, 30, 40, 50, and 60mN m−1. Columns 2–4 show AFM topographical images of Infasurf mixed with 0.1%, 1%, and 10% BUD, respectively. Infasurf + BUD films were compared to pure Infasurf at identical π in each row. Langmuir-Blodgett film transfer at 60 mN m−1 failed for Infasurf mixed with 10% BUD due to early film collapse. The AFM scan area was 20 × 20 μm for all images. The full z-range was set to be 5 nm for monolayers (i.e., π ≤ 40 mN m−1) and 20 nm for multilayers (i.e., π > 40 mN m−1). Three surface phases, i.e., tilted-condensed (TC), liquid-expanded (LE), and liquid-ordered (LO), are indicated in the Infasurf monolayer at 40 mN m−1. Characteristic relative heights of multilayers are indicated by arrows. The height profile along a line tracing for Infasurf + 10% BUD at 50 mN m−1 is shown in the bottom-right corner.
The first column of Fig. 3 shows the lateral structure of pure Infasurf at increasing π. A detailed description of these lateral structures and interpretation with respect to chemical composition and surface pressure can be found elsewhere.36 Briefly, at π ≤ 40 mN m−1, i.e., below the plateau region of the compression isotherm, Infasurf assumes a monolayer configuration. Three lipid phases, i.e., the liquid-expanded (LE), the tilted-condensed (TC), and the cholesterol-mediated liquid-ordered (LO) phases, are detected in the Infasurf monolayer, based on topography measurements using AFM. Note that when discussing phospholipid phase behavior we adopt the nomenclature proposed by Kaganer et al.,42 who suggest the use of the TC phase to replace the traditionally used liquid-condensed (LC) phase. As labeled in Fig. 3, coexistence of these surface phases in the Infasurf monolayer is indicated by formation of lipid domains of different relative heights, due to variation of lipid chain order in these phases. Lateral chemical analysis using time of flight-secondary ion mass spectroscopy (ToF-SIMS) has confirmed that the TC domains consist of disaturated phospholipids (mainly dipalmitoyl phosphatidylcholine), which extend approximately 1 nm higher than the surrounding LE phase that contains mainly unsaturated phospholipids and proteins.43,44 The lipid chain order of the LO domains, and hence their relative height, are intermediate between the TC and LE domains. Therefore, the three different lipid phases at the Infasurf monolayer can be distinguished by topography measurements with AFM.36
At π > 40 mN m−1, i.e., above the plateau region of the compression isotherms, the Infasurf monolayers are transformed into multilayers by selectively collapsing the LE and LO phases from the interfacial monolayers.45 Consequently, the solid-like TC domains at the interfacial monolayer appear as “holes” embedded within the collapsed fluid-like multilayers. Both the interfacial monolayer and the attached multilayer are closely packed at the highest π.
The second to fourth columns of Fig. 3 show the lateral structure of Infasurf with addition of 0.1%, 1% and 10% budesonide, respectively. Compared to pure Infasurf (the first column), three appreciable variations are detected.
First, the addition of budesonide at all three concentrations appears to inhibit the formation of the cholesterol-mediated LO phase at the surfactant monolayer (i.e., π ≤ 40). While the LO phase only appears with 0.1% and 1% budesonide at 20 mN m−1, it disappears at increasing π or increasing budesonide to 10%.
Second, the addition of budesonide at all three concentrations appears to have no significant effect on the TC phase. Both the size and area fraction of the TC domains at each π remain relatively unchanged with the addition of budesonide at different concentrations. The sizes of TC domains for all monolayers, with and without budesonide, are measured to be 4.8 ± 1.0 μm at 20 mN m−1, 5.2 ± 0.4 μm at 30 mN m−1, and 3.1 ± 0.2 μm at 40 mN m−1.
Third, and most important, although 0.1% budesonide does not significantly affect the multilayer structure, budesonide at 1% and 10% appreciably alters the multilayer structure at 50 and 60 mN m−1. At both concentrations, budesonide changes the multilayers from a uniform low matrix structure to isolated nonuniform high collapse patterns. At 50 mN m−1, with 0.1% budesonide, the multilayer is mainly a single phospholipid bilayer ∼4 nm in height, similar to pure Infasurf.36,45 With 1% budesonide, however, nonuniform multilayers of ∼8 nm and ∼12 nm appear, corresponding to 2 and 3 bilayer stacks. With 10% budesonide, isolated even higher collapse patterns appear. As shown by the line tracing in Fig. 3, these collapse patterns are bilayer stacks up to 4 bilayers in height. Variation of surfactant multilayers from a uniform matrix structure to isolated high collapse patterns is a strong indication of surfactant inhibition and reduction of whole-film stability, as the film preferably collapses from a few isolated nucleation sites.24,46 This structural variation due to 1% and 10% budesonide is consistent with the decrease of πmax found in Fig. 1.
Fig. 4 shows the effect of 0.1%, 1%, and 10% BDP on the lateral structure of Infasurf. For ease of comparison, lateral structures of pure Infasurf at controlled π are shown again in the first column. First, similar to the effect of budesonide, BDP inhibits formation of the LO phase but has no significant effect on the TC phase at the Infasurf monolayer (i.e., π ≤ 40). Second, at high π (50 and 60 mN m−1), different from budesonide, BDP does not significantly vary the matrix structure of the multilayer. Although 10% BDP appears to start inducing formation of nonuniform multilayers, a majority of multilayer patterns remain at similar height to those at low BDP concentrations, i.e., mainly 1 or 2 bilayers high. A very high local collapse phase, as in the case of 10% budesonide, does not appear. Third, a less obvious but notable difference between the Infasurf monolayer with budesonide and BDP is that BDP increases the surface roughness of the monolayer by ∼25%, compared to budesonide. In addition, during the AFM scan, the monolayer with BDP was noted to be appreciably stickier than the monolayer with budesonide. In combination with the shift of the compression isotherm to a lower surface area (Fig. 1b), this structural variation may indicate squeeze-out of BDP from the interfacial monolayer. Consequently, the influence of BDP on the multilayer structure at high π will be only limited.
Fig. 4.
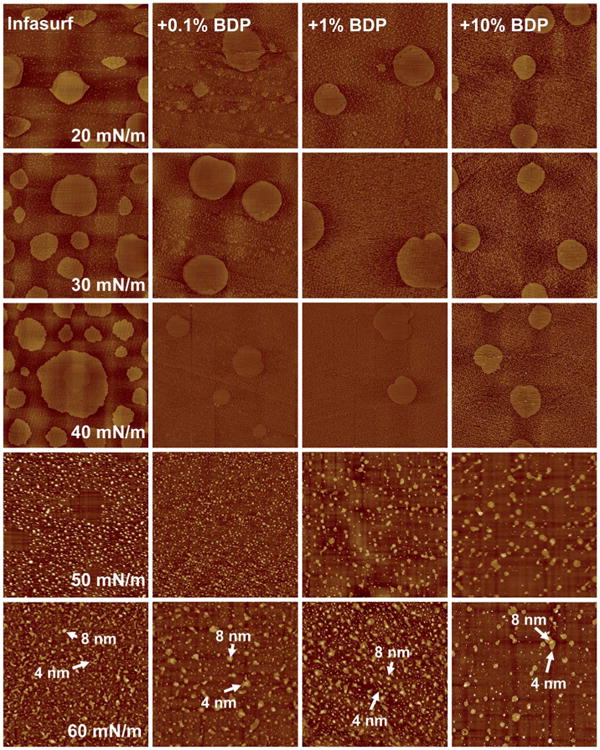
Effects of beclomethasone dipropionate (BDP) at various concentrations on the molecular organization and lateral structure of Infasurf films. For ease of comparison, the first column repeats AFM topographic images of pure Infasurf at five characteristic surface pressures (π) of 20, 30, 40, 50, and 60 mN m−1, as shown in the first column of the Fig. 3. Columns 2–4 show AFM topographical images of Infasurf mixed with 0.1%, 1%, and 10% BDP, respectively. Infasurf + BDP films were compared to pure Infasurf at identical π in each row. The AFM scan area was 20 × 20 μm for all images. The full z-range was set to be 5 nm for monolayers (i.e., π ≤ 40 mN m−1) and 20 nm for multilayers (i.e., π > 40 mN m−1). Characteristic relative heights of multilayers are indicated by arrows.
4. Discussion
The primary function of the lungs is gas exchange. In this process, PS plays the vital role of maintaining a large surface area by reducing alveolar surface tension. Therefore, a fundamental requirement of pulmonary drug delivery is that any macromol-ecules or drugs delivered via the lungs should not compromise respiratory mechanics in general, and surface activity of PS specifically. In this context, it has been reported that vitamin A, carried by an exogenous surfactant (BLES) for pulmonary delivery, significantly impaired the in vitro surface activity of this surfactant preparation.55 Corticosteroid molecules have an unsaturated ring moiety and hence are capable of interaction with PS phospholipids. It is therefore important to scrutinize the potential biophysical effect of corticosteroids on PS. This forms the rationale of the present study, in which we found both similarities and differences in the effect of two commonly used inhaled corticosteroids, budesonide and beclomethasone dipropionate (BDP), on surface activity and lateral structure of the modified natural surfactant Infasurf.
First, both corticosteroids inhibit formation of the LO phase at the interfacial monolayer. Formation and stability of the LO phase is due to cholesterol intercalation into disaturated phospholipid domains.47 Hence, cholesterol depletion would inhibit formation of this phase.48 Our present results may suggest that there is a direct interaction between the added corticosteroids and native cholesterol in Infasurf. Such an interaction must be strong enough to outweigh the molecular interaction between cholesterol and disaturated phospholipids, thus inhibiting formation of the LO phase. Davies et al. found that budesonide interacts with exogenous surfactant (Survanta) monolayers mainly through hydrophobic interactions.32 All corticosteroids are biochemically derived from cholesterol and, due to the structural similarity between corticosteroids and cholesterol, a stronger hydrophobic interaction between these two steroid molecules is not unexpected. However, it should be noted that despite the structural similarity, cholesterol differs from corti-costeroids in having a unique alkyl side chain at C-17 of the cholesterol molecule.49 This 8–10 carbon alkyl side chain is known to be related to the characteristic effects of cholesterol on regulating lipid phase behavior of model and biological membranes.50,51
Second, while 1% and 10% budesonide reduces the πmax of Infasurf, BDP at the same concentrations does not affect the πmax. Analysis of the lateral film structure confirms that budesonide at 1% and 10% significantly alters the multilayer structure of Infasurf at high π. In contrast, BDP only increases surface roughness of the Infasurf monolayer but does not significantly alter the multilayer structure. In addition, 10% BDP appreciably shifts the compression isotherms to the left at π < 40 mN m−1, while budesonide at the same concentration does not significantly affect the shape of the compression isotherm.
Our present findings consistently suggest a higher affinity of budesonide over BDP to Infasurf. While budesonide is associated with the surfactant film at even high π (>50 mN m−1), BDP is excluded from the interface at relatively low π (<40 mN m−1). Budesonide and BDP share close chemical and structural similarities. However, these two corticosteroids maintain very different in vitro physicochemical and pharmacokinetic characteristics; budesonide is more water soluble than BDP and it has a receptor affinity (measure of the strength with which the active molecule binds to the intracellular glucocorticoid receptor) 20-fold higher than that of BDP.35 If the significant difference in sensitivity of these glucocorticoids to the glucocorticoid receptor is related to their different affinities to Infasurf films, the mechanism of this interaction is presently unclear.52
Intratracheal administration of corticosteroids using an exogenous surfactant as a delivering vehicle is a relatively novel practice. Both the clinical surfactant used as the carrier and the corticosteroid delivered as the anti-inflammatory agent, and their mixing ratios, have yet to be standardized and optimized. Mixing ratios used in current clinical and animal trials ranges from 0.25 to 0.6 wt%.10,12,14,23 The present study, therefore, confirms the feasibility of using Infasurf as a carrier for either budesonide or BDP in this concentration range for pulmonary delivery. Moreover, we further suggest that when Infasurf is used as a carrier of BDP, a concentration beyond 1% might be feasible. However, if Infasurf is used to deliver budesonide at a concentration higher than 1%, budesonide may induce early collapse of surfactant films, thus ultimately disrupting optimal gas exchange within an already compromised system in most clinical practices. The mechanism of this early budesonide-induced collapse is likely due to increased film fluidization,49 which inhibits the formation of uniform and stable multilayer structures at high π.
It should be noted that the optimal mixing ratio may also be dependent on the exogenous surfactant preparation in use. In a separate study done by our group using the cholesterol-free surfactant preparation Curosurf, no significant variations in surface activity and lateral film structure were noted when used to carry budesonide at a 10% concentration (unpublished data). This suggests that optimal surfactant-corticosteroid pairs likely exist for pulmonary steroid delivery.
In addition to intratracheal administration using an exogenous surfactant as a carrier, our data may also have implications for the feasibility of inhaled corticosteroid therapy in general, which is widely used in treating chronic asthma53,54 and has been studied in treating CLD in premature newborns.6–9 Once inhaled, aerosolized steroids will reach pulmonary alveoli, where they will interact with the endogenous surfactant of the lungs. Given the low, and possibly unstable surfactant pool in preterm infants, caution should be taken when using inhaled steroids, as inhibition of endogenous surfactant may largely outweigh any benefits of the steroid anti-inflammatory effect.
5. Conclusions
The present in vitro biophysical study suggests an optimal concentration range of corticosteroids for pulmonary delivery when using natural surfactant as a delivering vehicle. Infasurf may carry less than 1% budesonide and up to 10% BDP without significantly compromising its surface activity. This difference in delivering capacity is due to different affinities of these cortico-steroids to surfactant films. Beyond this concentration range, beneficial anti-inflammatory effect of corticosteroids may be counteracted by their deleterious effect on surfactant layering and consequently result in worsening lung mechanics. These implications should be considered when using inhaled cortico-steroids, especially in the treatment of CLD in preterm infants.
Acknowledgments
The authors thank Dr Walter Klein at ONY Inc. for the generous donation of Infasurf samples. We thank Uyanga Tsedev for preliminary work. We also thank Dr Lloyd Hihara for sharing common facilities. This work was supported by the Leahi Fund to Treat & Prevent Pulmonary Disease (44936) from the Hawaii Community Foundation (Y.Y.Z.). H.Z. was supported by a faculty exchange program between Peking University and the University of Hawaii at Manoa, operated by the Center for Chinese Studies.
References
- 1.Patton JS, Byron PR. Nat Rev Drug Discovery. 2007;6:67–74. doi: 10.1038/nrd2153. [DOI] [PubMed] [Google Scholar]
- 2.Edwards DA, Ben-Jebria A, Langer R. J Appl Physiol. 1998;85:379–385. doi: 10.1152/jappl.1998.85.2.379. [DOI] [PubMed] [Google Scholar]
- 3.Weibel ER. Physiol Rev. 1973;53:419–495. doi: 10.1152/physrev.1973.53.2.419. [DOI] [PubMed] [Google Scholar]
- 4.Patton JS, Fishburn CS, Weers JG. Proc Am Thorac Soc. 2004;1:338–344. doi: 10.1513/pats.200409-049TA. [DOI] [PubMed] [Google Scholar]
- 5.Patton JS. Adv Drug Delivery Rev. 1996;19:3–36. [Google Scholar]
- 6.Cole CH, Colton T, Shah BL, Abbasi S, MacKinnon BL, Demissie S, Frantz ID., 3rd N Engl J Med. 1999;340:1005–1010. doi: 10.1056/NEJM199904013401304. [DOI] [PubMed] [Google Scholar]
- 7.Shah V, Ohlsson A, Halliday HL, Dunn MS. Cochrane database of systematic reviews (Online) 2007:CD001969. doi: 10.1002/14651858.CD001969.pub2. [DOI] [PubMed] [Google Scholar]
- 8.Halliday HL, Ehrenkranz RA, Doyle LW. Cochrane database of systematic reviews (Online) 2009:CD001146. doi: 10.1002/14651858.CD001146. [DOI] [PubMed] [Google Scholar]
- 9.Halliday HL. Paediatr Respir Rev. 2004;5(Suppl A):S245–S248. doi: 10.1016/s1526-0542(04)90046-2. [DOI] [PubMed] [Google Scholar]
- 10.Yeh TF, Lin HC, Chang CH, Wu TS, Su BH, Li TC, Pyati S, Tsai CH. Pediatrics. 2008;121:e1310–1318. doi: 10.1542/peds.2007-1973. [DOI] [PubMed] [Google Scholar]
- 11.Kuo HT, Lin HC, Tsai CH, Chouc IC, Yeh TF. J Pediatr. 2010;156:537–541. doi: 10.1016/j.jpeds.2009.10.049. [DOI] [PubMed] [Google Scholar]
- 12.Dani C, Corsini I, Burchielli S, Cangiamila V, Longini M, Paternostro F, Buonocore G, Rubaltelli FF. Pediatr Pulmonol. 2009;44:1159–1167. doi: 10.1002/ppul.21145. [DOI] [PubMed] [Google Scholar]
- 13.Bernhard W, Pynn CJ. Pediatr Pulmonol. 2009;44:1157–1158. doi: 10.1002/ppul.21123. [DOI] [PubMed] [Google Scholar]
- 14.Yang CF, Jeng MJ, Soong WJ, Lee YS, Tsao PC, Tang RB. Pediatr Neonatol. 2010;51:219–226. doi: 10.1016/S1875-9572(10)60042-3. [DOI] [PubMed] [Google Scholar]
- 15.Cummings JJ, D'Eugenio DB, Gross SJ. N Engl J Med. 1989;320:1505–1510. doi: 10.1056/NEJM198906083202301. [DOI] [PubMed] [Google Scholar]
- 16.Onland W, Offringa M, De Jaegere AP, van Kaam AH. Pediatrics. 2009;123:367–377. doi: 10.1542/peds.2008-0016. [DOI] [PubMed] [Google Scholar]
- 17.American Academy of Pediatrics, Committee on Fetus Newborn and Canadian Paediatric Society. Fetus and Newborn Committee. Pediatrics. 2002;109:330–338. [Google Scholar]
- 18.Jobe AH. Clin Perinatol. 2009;36:177–188. doi: 10.1016/j.clp.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiedmann TS, Bhatia R, Wattenberg LW. J Controlled Release. 2000;65:43–47. doi: 10.1016/s0168-3659(99)00230-8. [DOI] [PubMed] [Google Scholar]
- 20.Koch K, Dew B, Corcoran TE, Przybycien TM, Tilton RD, Garoff S. Mol Pharmacol. 2011;8:387–394. doi: 10.1021/mp1002448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grotberg JB, Gaver DP. J Colloid Interface Sci. 1996;178:377–378. [Google Scholar]
- 22.Zhang YL, Matar OK, Craster RV. Med Eng Phys. 2003;25:115–132. doi: 10.1016/s1350-4533(02)00190-x. [DOI] [PubMed] [Google Scholar]
- 23.Nimmo AJ, Carstairs JR, Patole SK, Whitehall J, Davidson K, Vink R. Clin Exp Pharmacol Physiol. 2002;29:661–665. doi: 10.1046/j.1440-1681.2002.03712.x. [DOI] [PubMed] [Google Scholar]
- 24.Zuo YY, Veldhuizen RA, Neumann AW, Petersen NO, Possmayer F. Biochim Biophys Acta. 2008;1778:1947–1977. doi: 10.1016/j.bbamem.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 25.Veldhuizen R, Nag K, Orgeig S, Possmayer F. Biochim Biophys Acta. 1998;1408:90–108. doi: 10.1016/s0925-4439(98)00061-1. [DOI] [PubMed] [Google Scholar]
- 26.Sirsi S, Pae C, Oh DKT, Blomback H, Koubaa A, Papahadjopoulos-Sternberg B, Borden M. Soft Matter. 2009;5:4835–4842. [Google Scholar]
- 27.Yu ZW, Zhang JH. Allergy Asthma Proc. 2008;29:486–492. doi: 10.2500/aap.2008.29.3155. [DOI] [PubMed] [Google Scholar]
- 28.Sims MW, Tal-Singer RM, Kierstein S, Musani AI, Beers MF, Panettieri RA, Haczku A. Respir Res. 2008;9:13. doi: 10.1186/1465-9921-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cogo PE, Simonato M, Mariatoffolo G, Stefanutti G, Chierici M, Cobelli C, Ori C, Carnielli VP. Pediatr Res. 2008;63:433–437. doi: 10.1203/PDR.0b013e3181659759. [DOI] [PubMed] [Google Scholar]
- 30.Gortner L, Wauer RR, Hammer H, Stock GJ, Heitmann F, Reiter HL, Kuhl PG, Moller JC, Friedrich HJ, Reiss I, Hentschel R, Jorch G, Hieronimi G, Kuhls E. Pediatrics. 1998;102:1153–1160. doi: 10.1542/peds.102.5.1153. [DOI] [PubMed] [Google Scholar]
- 31.Palmer D, Schurch S, Belik J. J Appl Physiol. 2000;89:884–890. doi: 10.1152/jappl.2000.89.3.884. [DOI] [PubMed] [Google Scholar]
- 32.Davies MJ, Brindley A, Chen X, Doughty SW, Marlow M, Roberts CJ. Colloids Surf B. 2009;73:97–102. doi: 10.1016/j.colsurfb.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 33.Notter RH, Wang Z, Egan EA, Holm BA. Chem Phys Lipids. 2002;114:21–34. doi: 10.1016/s0009-3084(01)00197-9. [DOI] [PubMed] [Google Scholar]
- 34.Bligh EG, Dyer WJ. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 35.Boobis AR. Respir Med. 1998;92(Suppl B):2–6. doi: 10.1016/s0954-6111(98)90434-6. [DOI] [PubMed] [Google Scholar]
- 36.Zhang H, Fan Q, Wang YE, Neal CR, Zuo YY. Biochim Biophys Acta. 2011;1808:1832–1842. doi: 10.1016/j.bbamem.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girard-Egrot AP, Blum LJ. In: Nanobiotechnology of Biomimetic Membranes. Martin DK, editor. Vol. 2. Springer; New York: 2007. pp. 23–74. [Google Scholar]
- 38.Leporatti S, Brezesinski G, Mohwald H. Colloids Surf A. 2000;161:159–171. [Google Scholar]
- 39.Hollars CW, Dunn RC. Biophys J. 1998;75:342–353. doi: 10.1016/S0006-3495(98)77518-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuo YY, Keating E, Zhao L, Tadayyon SM, Veldhuizen RA, Petersen NO, Possmayer F. Biophys J. 2008;94:3549–3564. doi: 10.1529/biophysj.107.122648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zuo YY, Tadayyon SM, Keating E, Zhao L, Veldhuizen RA, Petersen NO, Amrein MW, Possmayer F. Biophys J. 2008;95:2779–2791. doi: 10.1529/biophysj.108.130732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaganer VM, Mohwald H, Dutta PK. Rev Mod Phys. 1999;71:779–819. [Google Scholar]
- 43.Saleem M, Galla HJ. Biochim Biophys Acta. 2009;1798:730–740. doi: 10.1016/j.bbamem.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 44.Keating E, Waring AJ, Walther FJ, Possmayer F, Veldhuizen RA, Petersen NO. Biochim Biophys Acta. 2011;1808:614–621. doi: 10.1016/j.bbamem.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Wang YE, Fan Q, Zuo YY. Langmuir. 2011;27:8351–8358. doi: 10.1021/la201482n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan Q, Wang YE, Zhao X, Loo JS, Zuo YY. ACS Nano. 2011;5:6410–6416. doi: 10.1021/nn2015997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ipsen JH, Karlstrom G, Mouritsen OG, Wennerstrom H, Zuckermann MJ. Biochim Biophys Acta. 1987;905:162–172. doi: 10.1016/0005-2736(87)90020-4. [DOI] [PubMed] [Google Scholar]
- 48.Keating E, Rahman L, Francis J, Petersen A, Possmayer F, Veldhuizen R, Petersen NO. Biophys J. 2007;93:1391–1401. doi: 10.1529/biophysj.106.099762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghosh AK, Pore N, Basu R, De S, Nandy P. Colloids Surf B. 1996;7:65–68. [Google Scholar]
- 50.Vilcheze C, McMullen TP, McElhaney RN, Bittman R. Biochim Biophys Acta. 1996;1279:235–242. doi: 10.1016/0005-2736(95)00258-8. [DOI] [PubMed] [Google Scholar]
- 51.McMullen TP, Vilcheze C, McElhaney RN, Bittman R. Biophys J. 1995;69:169–176. doi: 10.1016/S0006-3495(95)79887-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marianecci C, Paolino D, Celia C, Fresta M, Carafa M, Alhaique F. J Controlled Release. 2010;147:127–135. doi: 10.1016/j.jconrel.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 53.Webb DR. Respir Med. 1998;92(Suppl B):7–14. doi: 10.1016/s0954-6111(98)90435-8. [DOI] [PubMed] [Google Scholar]
- 54.Adams N, Bestall JM, Jones PW. Cochrane database of systematic reviews (Online) 2002:CD003530. doi: 10.1002/14651858.CD003530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bronshtein V, Venkatesh V, Aulakh J, Chessex P. Drug Des Dev Ther. 2099;2:145–150. doi: 10.2147/dddt.s3255. [DOI] [PMC free article] [PubMed] [Google Scholar]