

Abstract
Background and Purpose
The psychostimulant cocaine induces complex molecular, cellular and behavioural responses as a consequence of inhibiting presynaptic dopamine, noradrenaline and 5‐HT transporters. To elucidate 5‐HT transporter (SERT)‐specific contributions to cocaine action, we evaluated cocaine effects in the SERT Met172 knock‐in mouse, which expresses a SERT coding substitution that eliminates high‐affinity cocaine recognition.
Experimental Approach
We measured the effects of SERT Met172 on cocaine antagonism of 5‐HT re‐uptake using ex vivo synaptosome preparations and in vivo microdialysis. We assessed SERT dependence of cocaine actions behaviourally through acute and chronic locomotor activation, sensitization, conditioned place preference (CPP) and oral cocaine consumption. We used c‐Fos, quantitative RT‐PCR and RNA sequencing methods for insights into cellular and molecular networks supporting SERT‐dependent cocaine actions.
Key Results
SERT Met172 mice demonstrated functional insensitivity for cocaine at SERT. Although they displayed wild‐type levels of acute cocaine‐induced hyperactivity or chronic sensitization, the pattern of acute motor activation was different, with a bias toward thigmotaxis. CPP was increased, and a time‐dependent elevation in oral cocaine consumption was observed. SERT Met172 mice displayed relatively higher levels of neuronal activation in the hippocampus, piriform cortex and prelimbic cortex (PrL), accompanied by region‐dependent changes in immediate early gene expression. Distinct SERT‐dependent gene expression networks triggered by acute and chronic cocaine administration were identified, including PrL Akt and nucleus accumbens ERK1/2 signalling.
Conclusion and Implications
Our studies reveal distinct SERT contributions to cocaine action, reinforcing the possibility of targeting specific aspects of cocaine addiction by modulation of 5‐HT signalling.
Abbreviations
- CPP
conditioned place preference
- DAT
dopamine transporter
- DEGs
differentially expressed genes
- FDR
false discovery rate
- IEGs
immediate early genes
- KO
knockout
- KRH
Krebs–Ringer's HEPES
- MDMA
3,4‐methylenedioxymethamphetamine
- mPFC
medial prefrontal cortex
- NAc
nucleus accumbens
- NET
noradrenaline transporter
- NPS
new psychoactive substances
- PBS‐T
PBS with 0.3% Triton‐X100
- PrL
prelimbic cortex
- qRT‐PCR
quantitative reverse‐transcriptase polymerase chain reaction
- RNA‐Seq
RNA sequencing
- SERT
5‐HT (serotonin) transporter
- SSRI
selective 5‐HT reuptake inhibitor
- WT
wild‐type
Introduction
Cocaine abuse and dependence is a world‐wide health problem. Despite various approaches and many years of preclinical studies, we still lack effective, mechanism‐based therapies to support recovery from dependence and addiction. The development of effective treatment strategies requires a wholistic understanding of in vivo cocaine pharmacodynamics. Primary contributions of dopamine transporter (DAT) inhibition to the reinforcing and addictive properties of cocaine are well‐documented (Luscher and Ungless, 2006), but the psychostimulant also potently blocks the serotonin (5‐HT) and noradrenaline (NE) transporters (SERT and NET, respectively) in vitro and in vivo, at levels of the drug abused by humans (Han and Gu, 2006; Simmler et al., 2013). SERT inhibition has been suggested to be protective with respect to the abuse liability of cocaine (Filip et al., 2005).
SERT knockout (KO) mice have been used to investigate the contributions of 5‐HT to cocaine action (Sora et al., 1998; Sora et al., 2001), including demonstration of increased cocaine conditioned place preference (CPP). SERT KO mice, however, display significant molecular, cellular and behavioural changes that reflect both a developmental and ongoing loss of reuptake inhibition (Murphy and Lesch, 2008). Additionally, cocaine retains the ability to elevate extracellular 5‐HT levels in the striatum of SERT KO animals, most likely due to compensatory 5‐HT uptake by DAT or NET in these animals (Shen et al., 2004). Genetic manipulations of 5‐HT receptors alter cocaine self‐administration (Nair et al., 2013; Brodsky et al., 2016; You et al., 2016), though these studies also may be questioned as to compensatory alterations. As an alternative to genetic manipulation, pharmacological modulation of 5‐HT receptor subtypes has been widely used to study 5‐HT‐mediated cocaine action. Such pharmacological interaction studies also support a role of SERT blockade in different facets of cocaine action (Filip et al., 2005). For example, signalling by prefrontal 5‐HT2A and 5‐HT2C receptors has been implicated in cue reactivity in cocaine self‐administering rats (Anastasio et al., 2014; Cunningham and Anastasio, 2014). However, as the actions of many other 5‐HT receptors may also modulate the actions of cocaine, other approaches are needed to assess the collective actions of serotonergic signalling in both acute and chronic cocaine action.
To ascertain SERT‐specific components of cocaine action, we took advantage of a knock‐in mouse that expresses an Ile172Met substitution (SERT Met172). Because the SERT Met172 variant reduces SERT inhibitory potency for multiple selective 5‐HT reuptake inhibitors (SSRIs), the SERT Met172 model has proved useful in demonstrating requirements (or lack thereof) for SERT antagonism in antidepressant action (Henry et al., 2006; Thompson et al., 2011; Bonnin et al., 2012; Nackenoff et al., 2016). With respect to the current study, the Met172 variant also significantly reduces cocaine potency when tested using radiolabeled 5‐HT uptake in transfected cells (Henry et al., 2006). Importantly, the Met172 mutation does not affect SERT‐mediated 5‐HT recognition or transport kinetics or SERT protein expression levels (Thompson et al., 2011; Nackenoff et al., 2016), providing an opportunity to identify SERT‐dependent actions of cocaine without concerns about compensation arising from loss of in vivo SERT activity. In an initial exploration of the utility of the model to study cocaine action (Prosser et al., 2014), we demonstrated a loss in SERT Met172 mice of cocaine‐induced changes in suprachiasmatic neuron (SCN) firing and circadian free‐running behaviour. Here, we have confirmed a loss of high‐affinity cocaine recognition at SERT in the SERT Met172 mouse model using ex vivo and in vivo paradigms, followed by an assessment of cocaine‐modulated behaviours. To identify 5‐HT‐dependent cellular networks contributing to cocaine action, we examined the effects of the loss of SERT antagonism on cocaine‐induced neuronal activation, as reported by c‐Fos expression, and through analysis of differential patterns of immediate early gene (IEG) expression, targeting molecules thought to trigger addiction‐associated plasticities (Nestler, 2012). Finally, to capture signalling networks linked to 5‐HT‐dependent cocaine actions more broadly, we pursue transcriptome analyses, targeting the prelimbic cortex (PrL) and nucleus accumbens (NAc). Our results elucidate several dimensions of 5‐HT contributions to cocaine action, findings we discuss in relation to earlier genetic and pharmacological studies.
Methods
Construction of homology models
To illustrate the potential for the Met172 substitution to limit cocaine interactions, Phyre2‐based one‐to‐one threading of the amino acid sequences of wild‐type (WT) mouse SERT (Ile172) and SERT Met127 was performed with PDB template structure 4XP4 [Drosophila DAT‐cocaine (Penmatsa et al., 2013)] using PSI‐BLAST alignment and an HMM (hidden Markov model) secondary structure prediction (Kelley et al., 2015). Side chain placements were modelled to minimize backbone and steric hindrance. The resulting structures were aligned with chain A of the 4XP4 structure using PyMOL (PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC).
Animals
All animal care and experimental procedures were conducted under a protocol approved by the Vanderbilt Institutional Animal Care and Use Committee or the University of Texas Medical Branch Institutional Animal Care and Use Committee, compliant with the US National Research Council's Guide for the Care and Use of Laboratory Animals, the US Public Health Service's Policy on Humane Care and Use of Laboratory Animals and Guide for the Care and Use of Laboratory Animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
SERT Ile172 (referred to here as WT) and SERT Met172 mice were maintained on a C57BL/6J background following backcrossing for >10 generations from the original 129S6/S4 background. As previously described, we maintained the 129S6 SERT ER haplotype in WT animals to ensure only the Met172 variant accounted for differences observed (Nackenoff et al., 2016). SERT WT and Met172 genotypes were established by PCR using oligonucleotide primers 5′‐CCTGGCCCTCTTAGCAGGTT‐3′ and 5′‐AGGAGGAGAAGCCAGCAAGG‐3′, assessing the presence of 700 bp or 750 bp bands that are diagnostic for Ile172 or Met172 encoding alleles respectively (Thompson et al., 2011). All experiments were performed with adult, 10–15 week old mice. Unless otherwise stated, experiments and analyses were not conducted blinded for genotypes. Mice were group housed under a 12 h light/dark cycle with water and food ad libitum. Experiments were all performed during the light period. With the exception of the RNA sequencing (RNA‐Seq) studies, which were performed using male mice only, we performed all experiments on both male and female mice in approximately equal numbers. Data from males and females were combined unless a gender effect was evident. Animals were killed by rapid decapitation without prior anaesthesia for synaptosomes and mRNA experiments, with pentobarbital (100 mg·kg−1 i.p.) prior to 4% p‐formaldehyde perfusion for immunohistochemistry, or with euthanasia by CO2 inhalation after behavioural experiments where tissue was not collected for further analysis.
Synaptosomal [3H]5‐HT uptake inhibition
To verify mutation‐induced loss of cocaine sensitivity, we prepared synaptosomes from midbrain (−4 to −5.3 mm posterior from bregma) of WT and SERT Met172 mice. Samples were homogenized in 5 mM HEPES with 0.32 mM sucrose (pH 7.4), and crude synaptosomes were prepared by differential centrifugation as previously described (Ansah et al., 2003). P2 pellets were resuspended in 1 mL of Krebs–Ringer's HEPES (KRH) assay buffer (130 mM NaCl, 1.3 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.8 g·L−1 glucose, 10 mM HEPES, pH 7.4) containing 100 μM ascorbic acid and 100 μM pargyline. Protein content of synaptosomal suspensions was determined using BCA Protein Assay Reagent (Thermo Scientific, Rockford, IL, USA), and the protein concentration of ice cold synaptosomal suspensions was adjusted to 0.1–0.15 mg·mL−1. Synaptosomes were pre‐incubated in triplicate at 37°C for 10 min with agitation prior to the addition of 50 nM [3H]5‐HT (Specific activity 28 Ci·mmol−1; PerkinElmer, NET498, Waltham, MA, USA) and various concentrations of cocaine, D‐amphetamine, RTI‐55, or MDMA (10−11–10−3 M) followed by a further 10 min incubation. Maximal capacity for [3H]5‐HT uptake was assessed by incubation with KRH buffer only, and non‐specific uptake determined by parallel incubations performed in the presence of 10 μM paroxetine, which shows equivalent SERT inhibition potency in WT and SERT Met172 animals (Thompson et al., 2011). Radiolabelled synaptosomes were captured over Whatman GF/B glass fibre filters (Brandel, Gaithersburg, MD, USA) that were presoaked with 0.3% polyethyleneimine using a Brandel Cell Harvester (Brandel, Gaithersburg, MD, USA). Radioactivity was eluted in Ecoscint scintillation fluid (National Diagnostics, Atlanta, GA, USA) for 12–16 h and subsequently quantified by scintillation spectrometry.
Microdialysis assessment of 5‐HT release in vivo
To complement ex vivo studies, we validated loss of high‐affinity cocaine recognition in vivo by assessing the effect of the psychostimulant to elevate extracellular 5‐HT using microdialysis methods. We performed these experiments in the ventral hippocampus as a projection area with a prominent 5‐HT innervation that affords use of a 3 mm microdialysis probe (to maximize recovery) as well as to afford comparison with our earlier studies demonstrating loss of SSRI sensitivity (Thompson et al., 2011). For stereotactic surgery, mice were anaesthetized with 2.5% isoflurane, and betadine solution was used to promote aseptic incisions. A 0.62‐mm‐diameter guide cannula (Synaptech, Marquette, MI, USA) was implanted in the ventral hippocampus of WT and SERT Met172 mice [guide cannula tip coordinates from bregma and dura mater were −3.18 mm AP, 2.5 mm ML and −1.0 mm DV (Paxinos and Franklin, 2004)]. Ketoprofen (5 mg·kg−1 once per 24 h) was administered for analgesia, and the animals were observed after surgery until they resumed normal activity including ambulation. After 24 h of recovery from surgery, a microdialysis probe (3 mm membrane length, active site −1.0 to −4.0 DV from dura mater, 20 000 Da cut‐off; Synaptech, Marquette, MI, USA) was inserted and perfused for 4 h at a rate of 1.0 μL·min−1 with artificial cerebrospinal fluid (149 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, pH 7.2) before initiation of sample collections. Dialysate was collected every 20 min before and after cocaine injections (20 mg·kg−1, i.p.). Experiments were performed in non‐anaesthetized, freely moving animals. 5‐HT levels in the dialysate were quantified with HPLC‐EC in the Vanderbilt Brain Institute Neurochemistry Core Facility. Correct probe locations were confirmed by visual inspection following cresyl violet staining of coronal brain sections, fixed with 4% p‐formaldehyde.
Open field locomotor activity and sensitization
Mice were handled once daily for 5 days before the first day of testing. To record locomotor activity, we used open field chambers (27 × 27 × 20.5 cm) with infrared photobeam tracking (MedAssociates, St. Albans, VT, USA), placed in sound‐attenuating boxes. The boxes were moderately illuminated (33 lux at floor of boxes) and had white flooring. Prior to all open field testing, mice were allowed to acclimatize to the testing room in their home cage for 30 min and for 20 min in the open field chambers before injection. On the first day of testing, mice were injected with saline (20 mL·kg−1, i.p.) and locomotor activity was recorded 20 min prior and 50 min post injection. On the second day, mice were injected with 15 mg·kg−1 cocaine i.p. and locomotor activity was recorded. In addition to horizontal locomotor activity, zone analysis of ambulation in centre and surround zones (with borders defined as 2.5 cm from wall edge), as well as time spent in vertical movements or stereotypical activities (defined by repeated beam breaks), was collected. Then, for sensitization, cocaine (15 mg·kg−1, i.p.) was injected in the home cages for 3 days, followed by one cocaine administration in the open field chambers with recording. The mice were then subjected to 14 days of forced abstinence in their home cages without any injections. During the last 5 days of the abstinent period, mice were handled daily. After the 14 days of abstinence, mice were injected i.p. with 15 mg·kg−1 cocaine and locomotor activity was recorded.
Conditioned place preference
For the conditioned place preference (CPP) paradigm, we used two compartment chambers with distinctive tactile cues (mesh vs. grid flooring) located in sound‐attenuating and moderately illuminated boxes. The locomotion of the mice was tracked by infrared photobeam detectors (MedAssociates, St. Albans, VT). Mice were acclimatized for at least 30 min in the testing room prior to each session. The paradigm is illustrated in Figure 4C. On the habituation day, mice were allowed to explore both sides of the chambers for 20 min. The next day, their preference bias for the chambers was quantified by assessing the time spent on each side of the chamber during 20 min. Typically, the mice had a preference for the mesh flooring, and we therefore assigned the grid flooring to all subjects as CS+ where cocaine was paired with the context, and the mesh flooring as CS− for the saline pairing. On the subsequent 8 days, either cocaine (15 mg·kg−1, i.p.) was injected and mice were placed immediately after the injection into the CS+ side of the chamber or saline was injected (20 mL·kg−1, i.p.) and mice were placed into the CS− side. During the 30 min post injections, mice were conditioned to the stimulus by giving them selective access to only one side of the chamber. Only one CS+ or CS− session took place per day. On day 11, after four CS+ and four CS− sessions, the preference was re‐assessed by giving the mice access to both sides during 20 min without prior injection. Then, 8 days of extinction conditioning followed, on which mice were given saline in the CS+ and in the CS− on alternating days. Place preference post‐extinction was assessed on day 20 without prior injections. The following day, mice were injected with 15 mg·kg−1 cocaine i.p. to assess cocaine‐induced reinstatement of place preference. The time spent in each side of the chamber was recorded during 20 min immediately after the cocaine injection.
Two‐bottle choice test
Mice were singly housed and allowed 1 week of habituation to unlimited access to two bottles of pure drinking water. Following this week of pre‐exposure, a bottle with pure drinking water and a bottle with cocaine in drinking water (0.1 mg·mL−1) were provided in the home cages with 24 h access. The location of the bottles was switched daily to prevent the development of a side preference. The liquid consumption was measured once weekly for 3 weeks, and the relative intake of the cocaine‐containing water was calculated by normalization to total liquid intake (cocaine‐containing water + pure water).
c‐Fos immunohistochemistry
Brains from WT and SERT Met172 mice were collected 120 min after a single dose of cocaine (20 mg·kg−1, i.p.) or saline. Subjects were anaesthetized with 100 mg·kg−1 pentobarbital i.p. and were perfused with 4% p‐formaldehyde (pH 7.5) at 8 mL·min−1 over 5 min. From the perfusion‐fixed brains, 40‐μm‐thick coronal frozen sections were cut on a sledge microtome (Leica, Buffalo Grove, IL, USA) for free‐floating immunohistochemistry. Sections were washed four times (5 min each) in PBS with 0.3% Triton‐X100 (PBS‐T) and then incubated in PBS with 0.3% H2O2 for 30 min. After three additional washes in PBS‐T, sections were incubated for 1 h in PBS‐T with 3% normal goat serum and then incubated with primary rabbit anti mouse c‐Fos antibody (PC38, Calbiochem, Millipore, San Diego, CA, USA), diluted 1:15 000 PBS‐T/normal goat serum, for 3 days at 4°C. Sections were then washed four times in PBS‐T and incubated for 90 min with secondary antibody (diluted 1:500, biotinylated goat anti rabbit, Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Sections were then washed four times in PBS‐T, followed by a 1 h incubation with an avidin‐biotin complex (VECTASTAIN Elite ABC Kit, Vector Laboratories, Burlingame, CA, USA) at room temperature, followed by four washes in PBS‐T. Sections were then incubated with diaminobenzidine (Vector Laboratories, Burlingame, CA) for 4.5 min and then washed in PBS three times. c‐Fos positive nuclei in developed brain sections were quantified from brain sections using ImageJ software (NIH, Bethesda, MD, http://imagej.nih.gov/ij/). Selected brain regions identified based on a mouse brain atlas (Paxinos and Franklin, 2004) were analysed by an experimenter blind to genotype and drug treatment.
Analysis of mRNA levels by qRT‐PCR
Prior to treatment for tissue collection, mice were handled for 5 min·day−1 for 7 days. We singly housed mice (in cages with fresh bedding) 24 h before killing to avoid the confound of gene expression alterations arising from interactions between cocaine‐ and saline‐treated cage mates. To assess acute, single‐dose cocaine effects, mice were injected in their home cage with either saline (20 mL·kg−1, i.p.) or cocaine (20 mg·kg−1, i.p.) by an experimenter blind to genotype and then killed 30 or 120 min later. For chronic cocaine administration, mice were treated with saline or cocaine (15 mg·kg−1, i.p., to parallel sensitization experiments) for 5 days in their home cage and killed 120 min after the last injection. For experiments examining a drug challenge after a period of abstinence, saline or cocaine (15 mg·kg−1, i.p.) administration took place in their home cage for 5 days, then an injection‐free period of 14 days followed, and on day 20, mice were challenged with saline or cocaine (15 mg·kg−1, i.p.). Mice were killed 120 min after the challenge injection, by rapid decapitation and brains were quickly removed, 1 mm coronal sections were obtained on ice with a slice matrix (Ted Pella, Redding, CA USA) and then regional dissections were performed using 1.5‐ or 2‐mm‐diameter punches (Harris Uni‐Core™; Ted Pella, Redding, CA, USA). Total RNA from the PrL and the NAc punches were isolated using the Trizol method (Life Technologies, Grand Island, NY, USA) according to the manufacturer's protocol. Reactions were performed with a one‐step quantitative RT‐PCR (qRT‐PCR) kit (Kapa Biosystems, Woburn, MA, USA) and Sybr‐Green‐based detection, applying a standard three‐step thermal profile. Oligonucleotide primers were developed to be cDNA‐specific and were validated for efficiency. The forward and reverse primers were 5′‐GGGTGAGCTGAAGCCACAAA‐3′; 5′‐CTGGTATGAATCACTGGGGGC‐3′ for Arc, 5′‐ACCTGACCACAGAGTCCTTTT‐3′; 5′‐TGGCTGGGATAACTCGTCTC‐3′ for Egr1, 5′‐CTCGTCGGTGACCATCTTCC‐3′; 5′‐TTGATCATGCCATCTCCCGC‐3′ for Egr2, 5′‐CATCGGCAGAAGGGGCAAAG‐3′; 5′‐TCTGTCTCCGCTTGGAGTGT‐3′ for Fos, 5′‐GAAAAGCGAAGGGTTCGCAG‐3′; 5′‐GCTGATCAGTTTCCGCCTGA‐3′ for Fosb; 5′‐CCTATCGGGGTCTCAAGGGT‐3′; 5′‐TGACCCGAAAAGTAGCTGCC‐3′ for Junb. Tbp mRNA levels, obtained using primers 5′‐GGCGGTTTGGCTAGGTTT‐3′ and 5′‐GGGTTATCTTCACACACCATGA‐3′ were used to normalize candidate gene mRNA levels across samples. Reactions were conducted in duplicates on an Eco Thermal Cycler (Illumina, San Diego, CA, USA).
RNA‐Seq analysis
Paralleling the qRT‐PCR studies described above, mice were pre‐handled for 7 days prior to treatment and singly housed for 24 h prior to sacrifice. Male mice were used only. For acute cocaine action, SERT Met172 and WT mice were injected with cocaine (20 mg·kg−1, i.p.) in their home cages and killed 120 min after injections. For chronic cocaine action, mice were treated with cocaine (15 mg·kg−1, i.p.) once daily for 5 days and killed 120 min after the fifth cocaine injection. Sacrifice took place by rapid decapitation, and tissue punches of the PrL and NAc tissue punches were collected immediately as described above. Total RNA was isolated using RNeasy (Qiagen, Valencia, CA, USA). To reduce RNA‐Seq variability, RNA was pooled from three mice, and three pooled samples per condition were sequenced to allow for statistical analysis. RNA sequencing was performed in the Vanderbilt Technologies for Advanced Genomics (VANTAGE) core facility. Sample preparation was performed with the Illumina True‐seq stranded mRNA sample preparation kit (Illumina, San Diego, CA, USA), and sequencing was performed at Single Read 50 bp on a HiSeq 2500 (acute cocaine action) or Single Read 75 bp on an Illumina NextSeq500 (chronic cocaine action; Illumina, San Diego, CA, USA). Sequence reads were mapped to a reference genome using Tophat2. The rates for mapped reads were >97% for all samples but one (77%; sample ‘WT 2946‐RB‐17’, Supporting Information Table S3). For validation of the data set, we generated correlation blots of the top 1000 genes for all samples within one condition. Strong correlations for all sample combinations within a group were evident by Spearman correlation coefficients of >0.87. Differentially expressed genes (DEGs) between genotypes after cocaine exposure were identified using the EdgeR package. Assessment of genotype‐dependent gene networks regulated in response to drug was performed via Ingenuity Pathway Analysis (QIAGEN Redwood City, CA, www.qiagen.com/ingenuity) for each brain region. Statistically significant DEGs [threshold for DEGs significance set to false discovery rate (FDR) < 5% for NAc with chronic cocaine treatment and P < 0.05 without FDR correction for all other data sets] were analysed to detect which pathways and networks in the Ingenuity Knowledge Base were significantly enriched (P < 0.05) with DEGs. The significant canonical pathways and top five networks are presented in the Supplementary Information. Raw data files showing DEGs are available online.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Statistical analyses and graphical evaluations were performed with Prism 6.0 (GraphPad, Inc., La Jolla, CA, USA) or SAS 9.4 (Cary, NC, USA). All illustrations of coronal brain sections to indicate brain areas used in the experiments were adapted from the mouse brain atlas (Paxinos and Franklin, 2004). Figure legends provide n values and specific statistical tests performed. Unequal group size resulted from efforts to match age of experimental cohorts arising from in‐house breeding, or from exclusion of outliers. Data derive from at least three independent experiments with statistical analyses performed on groups of five or more subjects. In all analyses, P < 0.05 was taken as indicative of statistical significance. For two‐way ANOVAs, we report significant interaction and/or main treatment/genotype effects. F‐values are summarized in Supporting Information Table S9. Sidak post hoc tests for treatment and/or genotype were performed only for significant main effects (F‐values with P < 0.05) and if there was no significant variance inhomogeneity (Barlett's test) or if group sizes were similar (N‐value from largest group/N‐value from smallest group <1.5). Outliers were identified and excluded based on the ROUT outlier test. All data were analysed for sex effects by ANOVA and if no effects were detected, data from male and female animals were combined, unless otherwise noted.
Materials
Cocaine‐HCl, ±3,4‐methylenedioxymethamphetamine (MDMA)‐HCl, D‐amphetamine sulfate, paroxetine‐HCl‐0.5H2O and miscellaneous laboratory reagents were obtained from Sigma‐Aldrich (St. Louis, MO, USA) unless otherwise noted and were of the highest grade available. RTI‐55 ((‐)‐2‐β‐carbomethoxy‐3‐β‐(4‐iodophenyl)tropane) D‐tartrate was obtained from the NIMH Chemical Synthesis and Drug Supply Program (https://nimh‐repository.rti.org). Solutions of cocaine for injections were prepared fresh for each experiment from frozen 20 mg·mL−1 solutions, with sterile 0.9% NaCl as solvent. Cocaine doses for in vivo treatments were calculated as the cocaine salt. Treatment (saline or cocaine) was randomly assigned before each experiment and cohort by an experimenter, matching group sizes of all conditions within every cohort. Unless otherwise stated, experiments and analysis were not conducted blinded for treatment condition.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c,d).
Results
SERT Met172 mice exhibit a loss of cocaine‐sensitivity ex vivo and in vivo
To illustrate the potential for obstruction of the presumptive cocaine binding site by a Met substitution at Ile172 in mouse SERT, we used a homology model based on a cocaine‐bound X‐ray structure of DAT (Penmatsa et al., 2013). In Figure 1A, B, the Met172 residue can be seen to introduce greater bulk into the cocaine binding site that, without other structural accommodations, could diminish cocaine affinity. With ex vivo synaptosomal experiments and in vivo microdialysis experiments, we sought to provide empirical evidence for this loss. As shown in Figure 1C, dose–response 5‐HT uptake inhibition studies of cocaine and the cocaine analogue RTI‐55 demonstrated significantly lower potencies compared to WT synaptosome preparations, as demonstrated by 80‐ and 550‐fold increases in IC50 respectively. In contrast, potencies for SERT inhibition by the substrates MDMA and D‐amphetamine were unaffected. Although a larger potency shift was evident for RTI‐55, we pursued the use of cocaine in subsequent investigations because it is the more clinically relevant substance of abuse. To demonstrate the loss of cocaine potency in vivo, we examined the ability of systemic cocaine injections (20 mg·kg−1, i.p.) to elevate extracellular 5‐HT using a microdialysis approach. As shown in Figure 2, whereas cocaine induced the expected rise in extracellular hippocampal 5‐HT in WT animals, SERT Met172 animals demonstrated a significantly blunted response (Figure 2), whether profiling the temporal course of cocaine effects or by calculating a cumulative response (AUC, inset) from 0 to 100 min after drug injection. Mean baseline 5‐HT values ± SEM prior to drug treatment were 0.23 ± 0.08 nM for WT (N = 7) and 0.17 ± 0.08 nM for Met172 (n = 5), values that did not differ significantly between the two genotypes (P > 0.05, Student's unpaired t‐test).
Figure 1.
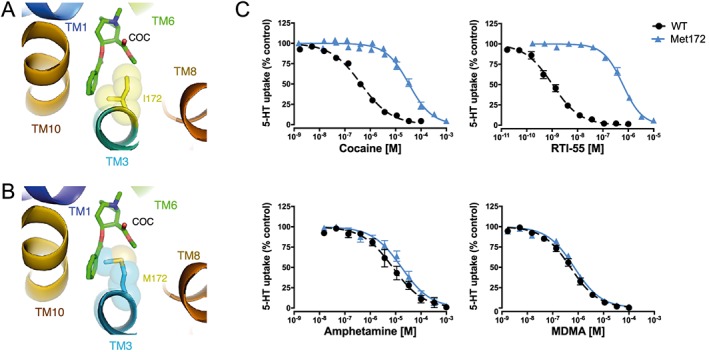
Homology models of mouse SERT Ile172 (A) and Met172 (B) superimposed on the Drosophila DAT‐cocaine co‐crystal structure suggest steric overlap of the Met172 side chain with cocaine. The Ile/Met172 side chain is shown as semi‐transparent spheres and cocaine (COC) from the 4XP4 structure is shown in stick mode. Transmembrane domains (TMs) illustrated are part of the binding pocket. (C) The steric overlap of Met172 with cocaine depicts one potential basis for the loss of high‐affinity binding and a subsequent shift in uptake inhibition potency of cocaine for 5‐HT transport, as shown with ex vivo preparation of synaptosomes from WT and SERT Met172 mice. The potency shift is also evident for the cocaine‐analogue RTI‐55, whereas amphetamine and MDMA potencies are not affected by the mutation. Data are mean ± SEM of independent experiments (n = 5 for all, except n = 4 for MDMA WT and n = 3 for MDMA Met172) performed in triplicate. IC50 values ± SEM (n = 5 for all, except n = 4 for MDMA WT and n = 3 for MDMA Met172) are IC50; cocaine, WT = 0.45 ± 0.03 μM, IC50; cocaine, Met172 = 36 ± 7 μM, IC50; RTI‐55, WT = 1.1 ± 0.2 nM, IC50; RTI‐55, Met172 = 0.62 ± 0.10 μM, IC50; amphetamine, WT = 9.4 ± 2.2 μM, IC50; amphetamine, Met172 = 19 ± 4.8 μM, IC50; MDMA, WT = 0.53 ± 0.11 μM and IC50; MDMA, Met172 = 0.71 ± 0.11 μM. IC50 values for cocaine and RTI‐55 were significantly different between WT and SERT Met172 (P < 0.05; cocaine and RTI‐55 analysed separately with Student's t‐test comparing respective WT IC50 vs. SERT Met172 IC50), but not for amphetamine (P > 0.05). Statistical comparison for MDMA was not conducted since n was <5.
Figure 2.
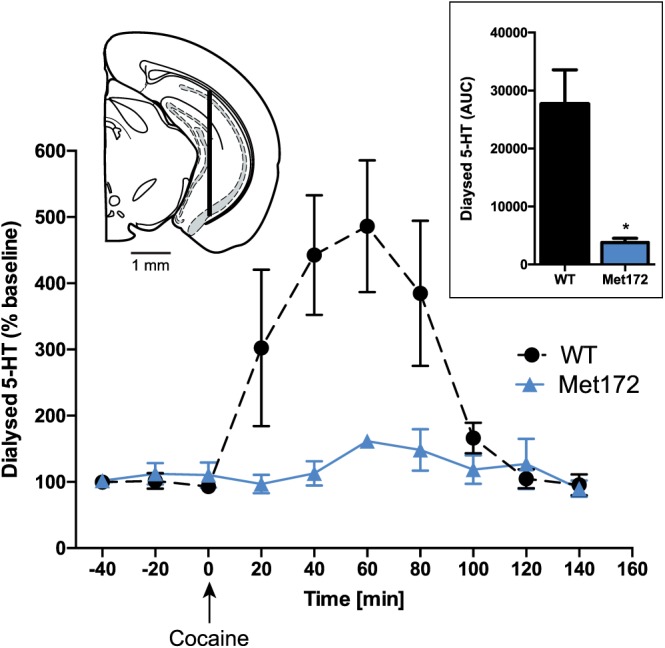
In vivo microdialysis in the ventral hippocampus of SERT Met172 mice reveals a loss of 5‐HT elevations following 20 mg·kg−1 cocaine i.p. Significant main time and genotype effects in two‐way RM‐ANOVA. Inset left: probe location in hippocampus −3.18 anterior from bregma. Inset right: AUC from in vivo microdialysis studies, calculated for 100 min post injection, *P < 0.05, signficantly different from WT; unpaired Student's t‐test. Data plotted are means ± SEM; n = 5 (WT), 7 (Met172).
SERT Met172 mice demonstrate an altered pattern of cocaine‐induced locomotion
In open field tests of horizontal locomotor responses to cocaine, we found no change in basal levels of activity in SERT Met172 mice, when compared with WT animals prior to or after saline injection (Figure 3A, C). SERT Met172 mice also did not differ from WT animals in horizontal locomotion levels (Figure 3B, D) or in total vertical activity or stereotypic behaviour (Supporting Information Figure S1) following systemic cocaine administration (15 mg·kg−1, i.p.). Interestingly, we detected a change in the pattern of horizontal locomotion. When individual centre and surround locomotion levels were normalized to pre‐injection values, we detected a reduced ambulation of SERT Met172 animals in the centre of the open field chamber (thigmotaxis) as compared to WT animals (Figure 3D). Next, we examined cocaine‐induced locomotor sensitization in WT and SERT Met172 animals, treated with cocaine (15 mg·kg−1, i.p.) for five consecutive days and challenged with cocaine (15 mg·kg−1, i.p.) after an abstinence period of 14 days. Compared to hyperlocomotion on day 1 of cocaine treatment, both WT and SERT Met172 mice showed significant locomotor sensitization after 5 days of treatment and challenged after abstinence (Figure 4A). The levels of sensitization were equivalent for both genotypes.
Figure 3.
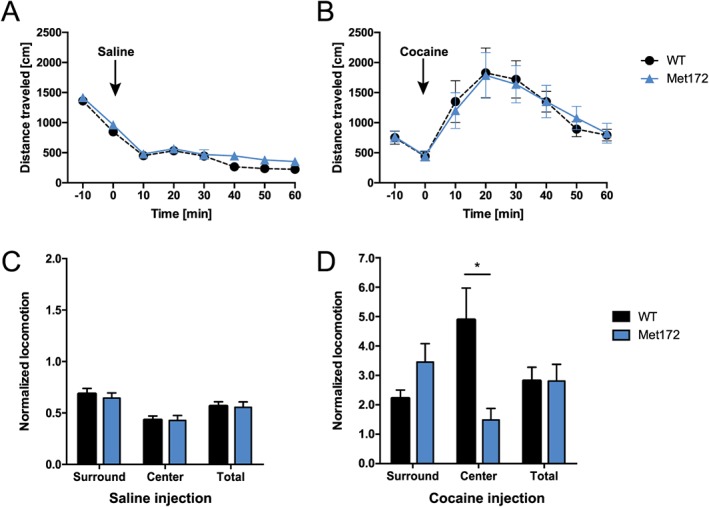
Locomotor response to cocaine in WT and SERT Met172 mice. Acute locomotor activity after 20 mL·kg−1 saline i.p. (A) and 15 mg·kg−1 cocaine i.p. (B) was not different between WT and SERT Met172 mice (two‐way RM‐ANOVA). I.p. injections were administered at time point 0. (C, D) Locomotor activity presented as average distance travelled in 10 min blocks during 50 min post injection normalized to individual baseline activities recorded 10 min prior to the injection. Surround and centre zone in the open field test were differentiated. No genotype differences were observed after saline treatment (C). After cocaine treatment (D), SERT Met 172 mice showed less ambulation in the centre zone than WT mice. Significant interaction effect but main drug and zone effect were not significant. Normalized locomotion is average distance travelled during 30 min post injection divided by average distance during 10 min prior to injection. Data plotted are means ± SEM, n = 27 (WT), 20 (Met172), *P < 0.05, signficantly different as indicated; two‐way ANOVA with Sidak's post hoc test.
Figure 4.
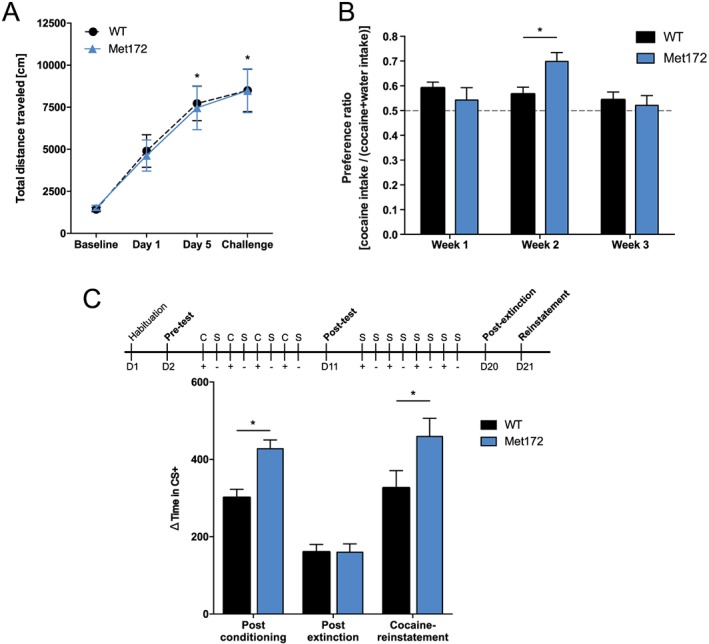
(A) Locomotor sensitization to cocaine (15 mg·kg−1, i.p.) did not differ between WT and SERT Met172 mice. Significant main time effect in two‐way RM‐ANOVA. *P < 0.05, signficantly differences between day 5 and day 1 and between challenge and day 1; Sidak post hoc test, n = 26 (WT), 28 (Met172). (B) Two‐bottle choice test to assess consumption of cocaine (0.1 mg·mL−1) administered in the drinking water. WT mice did not change their preference for cocaine‐supplemented water relative to supplemented drinking water over time. Met172 mice showed a greater cocaine preference compared to WT in the second week (significant interaction and main time effect. Data are means ± SEM, n = 19 (WT), 8 (Met172). *P < 0.05, signficantly different from WT; two‐way ANOVA with Sidak post hoc test,. (C; top) Schematic outline of the paradigm. C, cocaine treatment (15 mg·kg−1, i.p.); S, saline treatment; +, in cocaine‐paired cue (CS+); −, in saline‐paired cue (CS−). Place preference testing occurred on days 1 (D1), 2 (D2), 11 (D11), 20 (D20) and 21 (D21). (C; bottom) Conditioned place preference Δtime in CS+ represents individual time difference spent in the cocaine‐paired chamber on test day versus pre‐conditioning day. Values are seconds from total 1200 s session time. SERT Met172 mice spent more time in CS+ than WT post conditioning and after cocaine (15 mg·kg−1) reinstatement. Significant main time and genotype effect in two‐way ANOVA, All data represent mean ± SEM; n = 76 for post conditioning and post extinction, n = 32 (WT), 28 (Met172) for reinstatement. * P < 0.05, signficantly different from WT; Sidak post hoc test.
Loss of SERT inhibition increases consumption and cocaine place preference
Although the locomotor activating properties of cocaine were intact in the SERT Met172 mice, these assays do not report whether changes in cocaine hedonic value have been altered. To explore this dimension, we implemented the two‐bottle choice test where animals are given free choice to consume cocaine (0.1 mg·mL−1 in water) versus supplemented water in the home cage. For this concentration of cocaine, average daily cocaine intake was as follows: week 1 (WT 15.6 ± 0.86 mg·kg−1·day−1; SERT Met172 14.1 ± 1.6 mg·kg−1·day−1); week 2 (WT 13.4 ± 0.98 mg·kg−1·day−1; SERT Met172 16.4 ± 1.2 mg·kg−1·day−1); week 3 (WT 12.3 ± 0.88 mg·kg−1·day−1; SERT Met172 11.8 ± 1.1 mg·kg−1·day−1); WT mice demonstrated reduced average daily intake of cocaine in week 3 versus week 1 (significant main time effect and genotype × time interaction in two‐way ANOVA, P < 0.05 in Sidak post hoc test). We found time‐dependent genotype differences in preference of cocaine‐containing water relative to non‐supplemented water. WT animals did not exhibit significant changes in the preference for cocaine over the course of 3 weeks of testing (week 1 ~60% vs. water; week 2 ~57% vs. water; week 3 ~54% vs. water). In contrast, SERT Met172 mice showed a significantly increased preference for cocaine in week two (~70% vs. water) compared to week 1 (~54% vs. water) or week 3 (~52% vs. water) of testing (significant main time effect and genotype × time interaction in two‐way ANOVA, P < 0.05 in Sidak post hoc test). Consequently, the preference for cocaine at week 2 was significantly higher for SERT Met172, compared to WT mice (Figure 4B).
As a second test of cocaine ‘liking’, we implemented the CPP paradigm, where animals are able to associate cocaine with cues linked to one side of a two‐sided chamber. Here, we found that after 8 days of conditioning (see Methods section), both WT and SERT Met172 spent more time in the side of the chamber (CS+) associated with cocaine injections (Figure 4C). Notably, SERT Met172 mice exhibited a significantly greater increase in time on the cocaine‐paired side than WT mice did. After 8 days of extinction conditioning, animals of both genotypes displayed a reduced preference for the formerly cocaine‐paired side of the chamber and the genotype differences in side preference had disappeared. However, when animals were administered a single cocaine injection (15 mg·kg−1, i.p.) following extinction training, the increased preference for CS+ in the SERT Met172 mice re‐emerged.
Cocaine‐induced c‐Fos is enhanced in the prelimbic cortex, piriform cortex and the hippocampal CA3 region in SERT Met172 mice
Our behavioural studies above indicated that SERT Met172 genotype affects the actions of cocaine following both acute and chronic administration. To identify brain regions where neuronal activation following acute cocaine administration is supported by enhanced 5‐HT signalling, we quantified c‐Fos expression via immunohistochemistry. We selected specific brain regions for analysis due to their published involvement in cocaine action and/or prominent serotonergic innervation (Zombeck et al., 2010). Compared to saline, cocaine (20 mg·kg−1, i.p.) induced a significant increase in the number of c‐Fos+ cells in all brain areas analysed for both WT and SERT Met172 mice (Figure 5). Whereas we detected no difference in the extent of Fos induction following cocaine in WT and SERT Met172 animals for the NAc shell, basolateral amygdala, infralimbic cortex or dorsal striatum, we detected elevated expression in SERT Met172 versus WT mice in the prelimbic cortex (PrL), the piriform cortex and the dorsal CA3 region of the hippocampus.
Figure 5.
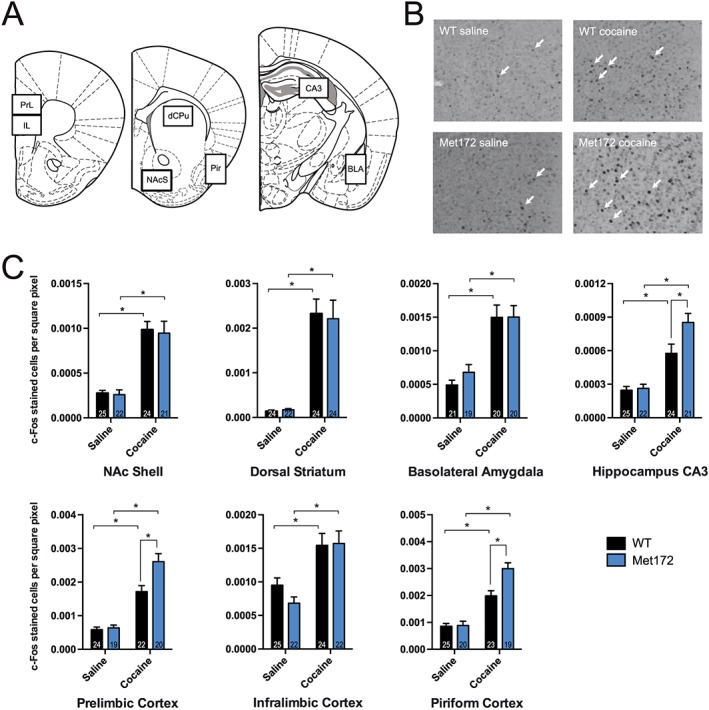
Identification of brain regions where SERT‐inhibition had a significant contribution to c‐Fos expression. Brains from WT and SERT Met172 mice were collected 120 min after a single dose of cocaine (20 mg·kg−1, i.p.) or saline. (A) Areas in which stained nuclei were quantified (BLA, basolateral amygdala; CA3, hippocampal CA3; dCPu, dorsal caudate putamen; IL, infralimbic cortex; NAcS, nucleus accumbens shell; pir, piriform cortex; PrL, prelimbic cortex). (B) Representative pictures of c‐Fos positive stained nuclei in the prelimbic cortex. (C) Quantified c‐Fos expression as marker for neuronal activity in selected brain areas for saline and cocaine‐treated mice. Using two‐way ANOVA, we found significant main treatment effects in all brain areas displayed here, and additionally a significant main genotype effect in the CA3, piriform cortex and PrL. Data plotted are means ± SEM; n = 19–25; exact group sizes are indicated in the graphs. *P < 0.05, signficantly different as indicated; Sidak post hoc test.
Contribution of SERT inhibition to cocaine‐induced IEG expression in the PrL and NAc
To extend our studies of c‐Fos activation to other IEGs known to be responsive to acute cocaine administration (Bhat et al., 1992; Fosnaugh et al., 1995; Nestler, 2012), we quantified changes in mRNA levels by qRT‐PCR, focusing on PrL and NAc. We chose the PrL because it demonstrated a genotype effect in our studies of c‐Fos‐based neuronal activation and has been consistently implicated in addiction. Although the NAc, in contrast to the PrL, did not show differential neuronal activation in our previous experiment, we chose to assess it for mRNA studies because it receives top‐down control from the PrL (Dalley et al., 2011) and is a brain area where cocaine‐induced plasticity of intrinsic neurons (e.g. medium spiny neurons) are thought to drive cocaine addiction (Koob and Volkow, 2010). We assessed mRNA changes at 30 and 120 min following acute cocaine injection to capture the varying dynamics of IEG activation. We observed that, compared to saline, single‐dose cocaine treatment significantly increased mRNA levels in both PrL and NAc for all genes evaluated, at one or both of the time points, except for Egr1 (also known as Zif268), which displayed no cocaine‐induced change at either 30 or 120 min in PrL (Figure 6B). Significant genotype differences in cocaine‐induced IEG up‐regulation were evident in the NAc (Figure 6A) for Egr1 and Egr2 (males only), driven by diminished cocaine‐induced mRNA elevations relative to saline in SERT Met172 animals. In the PrL (Figure 6B), significant genotype differences were evident for Junb, driven by greater mRNA elevations induced by cocaine in SERT Met172 compared to WT animals.
Figure 6.
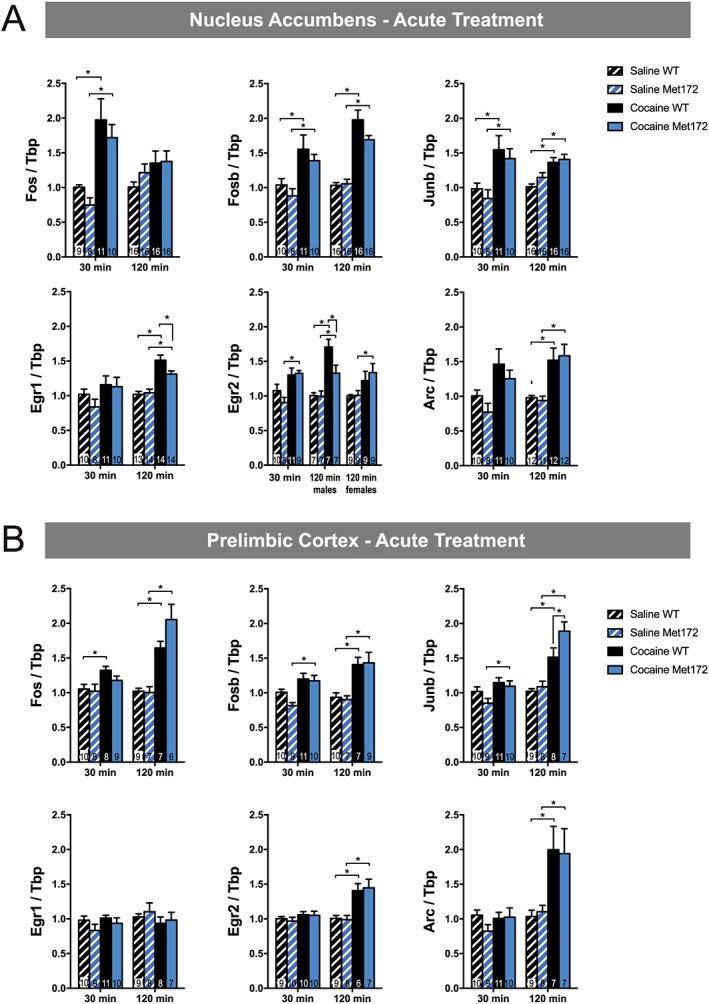
(A) Selected IEG mRNA expression in the nucleus accumbens (NAc) from WT and SERT Met172, 30 and 120 min after a single dose of cocaine (20 mg·kg−1, i.p.) or saline. Gene expression was calculated relative to levels of the housekeeping gene Tbp and normalized to the WT saline control of the respective time point. All genes were analysed separately using two‐way ANOVA with Sidak post hoc test at the 30 and 120 min time points. We found significant main effects of drug in all groups except for Egr1 at 30 min and Fos at 120 min and a significant interaction between the effects of drug and genotype for Egr1 and Egr2 at 120 min. Males and females were analysed separately for Egr2 at 120 min since a significant main sex effect was evident. All other data are males and females combined as no sex differences were observed. n = 7–16; exact group sizes are indicated in the graphs. (B) IEG mRNA expression in the PrL from WT and SERT Met172 30 and 120 min after a single dose of cocaine (20 mg·kg−1, i.p.) or saline. We found significant main treatment effects for Fos, Fosb and Junb at 30 and 120 min and for Arc and Egr2 at 120 min. A significant main effect of genotype was evident for Junb at 120 min. All data are males and females combined as no sex differences were observed. Data plotted represent mean ± SEM; n = 7–9; exact group sizes are indicated in the graphs. *P < 0.05, signficantly different as indicated, two‐way ANOVA with Sidak post hoc test. cocaine versus respective saline or WT versus SERT Met172.
To examine whether the SERT Met172 genotype confers differences in gene expression following chronic cocaine administration (15 mg·kg−1·day−1 for 5 days), we assessed IEGs induced 120 min following the last injection, as well as following a challenge dose administered 14 days following cocaine abstinence. After the fifth daily cocaine injection, all IEGs demonstrated cocaine‐dependent increases in mRNA levels in both the NAc and PrL, as evident by significant main drug effects; however, post hoc tests were not significant for all conditions (see Figure 7A, B). We did not observe any significant main effects of genotype in the chronic treatment. Following a cocaine challenge administered after cocaine abstinence, Fosb, Junb and Arc in the NAc and the PrL were significantly up‐regulated in both genotypes (Figure 8A, B). SERT Met172 mice did not show increased Egr1 levels in the NAc after a cocaine challenge, and post hoc testing confirmed that, in cocaine‐treated animals, SERT Met172 Egr1 mRNA levels are significantly lower compared to WT. Together, these findings document region‐dependent, 5‐HT‐driven effects on IEG induction.
Figure 7.
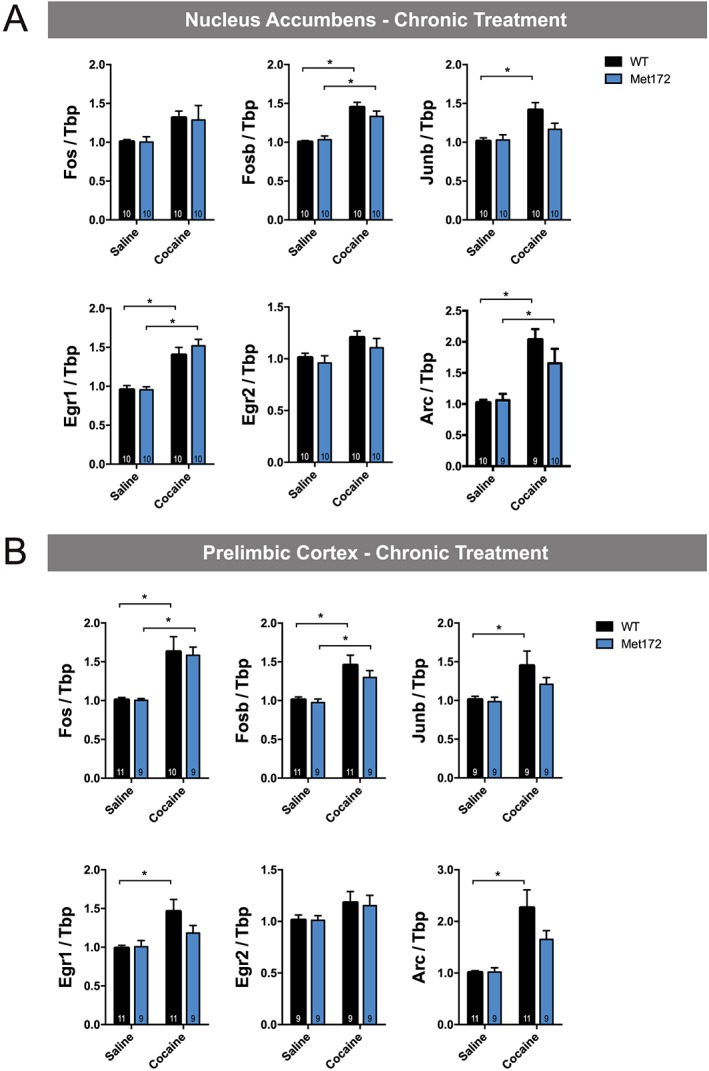
(A) IEGs mRNA expression in the NAc from WT and SERT Met172 chronically treated with cocaine (15 mg·kg−1, i.p.) or saline (20 mL·kg−1) and killed 120 min after the last injection. Main treatment effect was significant for all genes, two‐way ANOVA, * indicates P < 0.05, Sidak post hoc saline versus cocaine, n = 9–10; exact group sizes are indicated in the graphs. (B) IEGs expression in the prelimbic cortex from chronically treated mice. Main treatment effect was significant for all genes except for Egr2, two‐way ANOVA, Data represent mean ± SEM; n = 9–11; exact group sizes are indicated in the graphs. *P < 0.05, signficantly different as indicated; Sidak post hoc test.
Figure 8.
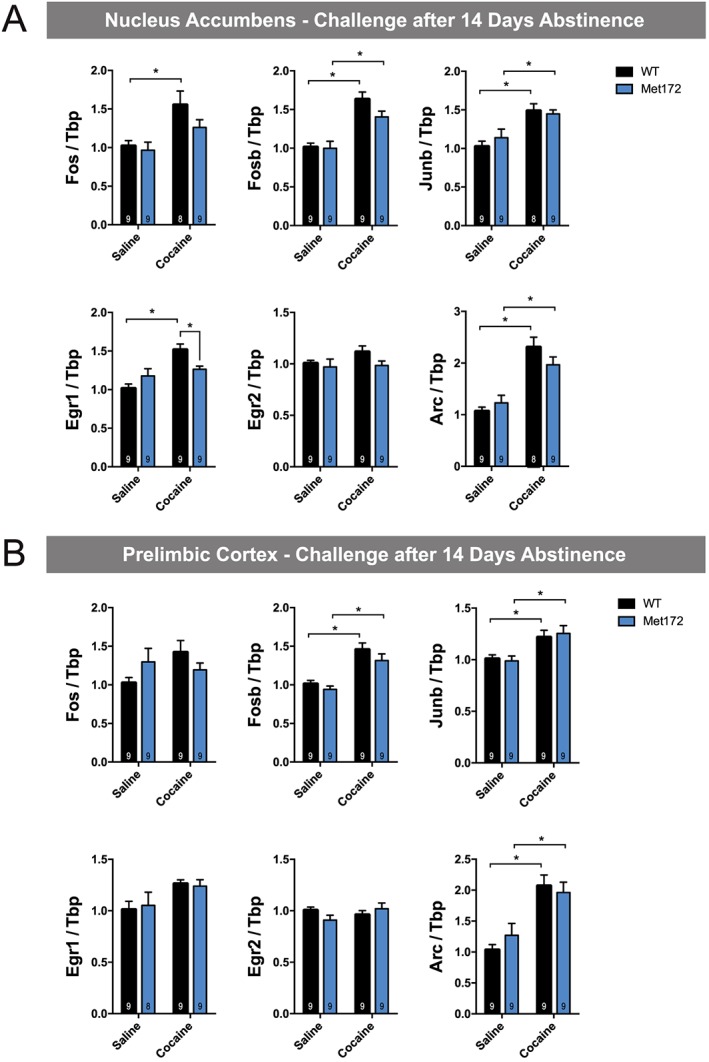
(A) IEGs mRNA expression in the nucleus accumbens from WT and SERT Met172 chronically treated with cocaine (15 mg·kg−1, i.p.) or saline (20 mL·kg−1) followed by 14 days of abstinence and a challenge dose (cocaine 15 mg·kg−1 or saline). Mice were killed 120 min after the challenge dose. Main interaction for Egr1 and main treatment effect for all genes except for Egr2; two‐way ANOVA Data are mean ± SEM.; n = 8–9; exact group sizes are indicated in the graphs. * P < 0.05, signficantly different as indicated; Sidak post hoc test. (B) IEGs expression in the prelimbic cortex from mice challenged with a cocaine or saline dose after 14 days abstinence as described in A. Main treatment effect for Fosb, Junb, Egr1 and Arc, two‐way ANOVA. Data are mean ± SEM; n = 8–9; exact group sizes are indicated in the graphs. *P < 0.05, signficantly different as indicated; Sidak post hoc test.
Gene networks in the PrL related to SERT‐inhibition in acute and chronic cocaine action
To gain insight into a broader network of gene activation arising from cocaine‐induced 5‐HT elevations, we pursued transcriptome‐wide analysis of mRNA levels using RNA sequencing (see Table S1–S8 for normalized expression levels and statistics). Using a nominal cut‐off of P < 0.05 for differentially expressed genes (DEGs; cocaine‐treated WT vs. cocaine‐treated SERT Met172 mice), the PrL showed more 5‐HT sensitive genes than the NAc (370 DEGs in the PrL vs. 55 DEGs in the NAc) following acute treatment, consistent with the greater degree of neuronal activation in the former region observed in c‐Fos studies. In contrast, chronic treatment resulted in a greater genotype‐dependent effect on gene expression in the NAc than in the PrL (319 DEGs in the PrL vs. 1517 DEGs in the NAc). Of the acute DEGs, only one DEG in the PrL, Ano3, survived a 5% FDR, whereas only Arc reached that threshold in the PrL following chronic treatment. In the NAc, no genotype‐dependent changes in gene expression following acute cocaine administration survived FDR correction, whereas 292 genes, including Arc, Junb, Fosb, Egr1 and Egr2, passed this threshold following chronic cocaine treatment.
As false discovery rate corrections impose stringent limitations on the detection of potentially important functional differences in individual genes, we also asked whether specific signalling pathways were overly enriched compared to chance with genes that satisfied the nominal P value < 0.05 to examine pathways enriched for SERT‐dependent DEGs. For the NAc data set obtained after chronic treatment, we used < 5% FDR as in this instance a larger number of genes survived FDR correction. Interestingly, this analysis identified networks associated with NFκB, ERK1/2 and Jnk signalling in both the NAc and PrL after acute and chronic treatment (Table 1, Figure 9 and Supporting Information Figures S2–S20). In the acute treatment, gene networks in the PrL included more DEGs than the gene networks in the NAc, whereas such regional differences were not observed in the chronic treatment. Several networks were observed in the same brain area in both the acute and chronic treatment, for example, the ERK1/2‐related network in the PrL. Although these networks had the same genes as network hubs, the DEGs forming the networks were different between acute and chronic paradigms. Other networks, for example, that linked to PrL Akt signalling (acute cocaine treatment) or NAc ERK1/2 (chronic cocaine treatment) (Figure 9), were specific to the duration of cocaine treatment and brain region of analysis.
Table 1.
Top networks for the NAc and PrL from Ingenuity Pathway Analysis with genes differentially expressed in WT and SERT Met172 mice treated with acute and chronic cocaine
| Brain area; treatment paradigm | Network hub | N of protein‐coding genes in the network that were differentially expressed | Up‐regulated protein‐coding genes in network that reached threshold for differential expression | Down‐regulated protein‐coding genes in network that reached threshold for differential expression |
|---|---|---|---|---|
| NAc; acute | Mapk8 (JNK) | 22 | Cd24a, Cdca7, Col5A1, Dcx, Igfbpl1, Kif23, Marcksl1, Nfe2l3, Nusap1, Pbk, Prc1, Prr11, Rnasel, Rrm2, Thbs4, Top2a | Acvr1c, Cd4, Cep126, Icam1, Mme, Myoc |
| NAc; acute | Ubc (UBC) | 15 | Aspm, Fam83d, Hes6, 2810417H13Rik, Lmbr1l, Mki67, Pole, Tac2, Wdr6 | Dach1, Dgkb, Mpp7, Pde7b, S100pbp, Spock3 |
| NAc; acute | Ubc (UBC) | 13 | Ckap2l, Cpne7, Fkbp10, Grhl1, Mex3a, Olfml2b, Pbk, Rnasel, Slc26a11 | Impg1, Pld5, S100pbp, Slc35d3 |
| NAc; acute | Nfkb1; Mapk1 (NFκB; ERK) | 8 | Calb2, Sp8 | Ano3, Erdr1, Jph1, Musk, Slc22a3, Uhrf1 |
| PrL; acute | Nfkb1 (NFκB) | 29 | Cep164, Dzip1, Frem1, Luc7l3, Mcc, Mtbp, Pdcd5, Slc15a2, Tagln2 | Ano3, Bcl11b, Camk4, Dffa, Inf2, Map3k13, Nnpnt, Nvl, Pcp4l1, Pim3, Ppm1l, Slc16a3, Slc25a4, Slc25a5, Slc25a13, Smpd3, Strn, Syt2, Tmem14c, Trpc3 |
| PrL; acute | Akt1 (Akt) | 27 | Dffb, Dpysl2, Fam107a, Gng13, Hook2, Nek1, Pfdn5, Rgs11, Rpl17, Rpl37, Rpl39, Rpl22l1, Tnfrsf22/Tnfrsf23 | Adcy5, Cdc42ep3, Gnb5, Ina, Kcna1, Kcna5, Kcnab1, Lrrk2, Nefl, Nefm, Parm1, Rgs9, Ric8b, Ttll7 |
| PrL; acute | Mapk8 (JNK) | 23 | Akap8l, Arhgap31, Cbs/LOC102724560, Col5a1, Col5a3, Flnb, Psrc1, Rassf2, Tfr2, Tgfbr1 | Acvr1b, Adamts2, Add2, Cdk17, Clip4, Dach1, Igsf1, Kif11, Osbpl8, Peg10, Ppp6c, Rasd2, Spats2 |
| PrL; acute | Mapk1; Mapk3 (ERK1/2) | 22 | Akap13, C4a/C4b, Dio2, Fcgr2b, Fxyd55, Mfge8, Nktr, Wsb1, Zfp57 | Akap5, Cebpd, Chn2, Cyld, Dgkb, Dgki, Dlk1, Kcnq3, Ndfip2, Pacsin2, Sema3f, Trpc5, Wnt2 |
| PrL; acute | Mapk1 (ERK) | 22 | Arhgef5, Ccdc80, Fau, Rps25, Rps27a, Sst | Cartpt, Epha7, Got1, Irs2, Nkx6–2, Oasl2, Pcsk1, Pde10a, Pde1b, Pde1c, Pde7b, Pdyn, Penk, Rnf130, Slc5a7, Tac1 |
| NAc; chronic | Mapk1; Mapk3 (ERK1/2) | 23 | B3gnt2, Tmem158 | Abca1, Ank2, B3gnt2, Cntnap1, Irs4, Lrp1, Lrp8, Lrp1B, Ptprb, Ptprd, Ptprg, Ptprt, Ptprz1, Rasd1, Ryr2, Scn1a, Scn8a, Scube1, Slc6a11, Smg1, Tnr, Vwf |
| NAc; chronic | Nfkb1 (NFκB) | 23 | Acvrl1, Brms1, Cyp2s1, Nrgn, Peli1, Spata2l, Trim8, | Akap13, Cacna1d, Camk2d, Cyp2j6, Dscam, Epha10, Flnb, Igsf1, Lrrfip1, Myo16, Nrxn1, Rif1, Slc30a3, Slc8a1, Vsnl1, Wnt10a |
| NAc; chronic | Mapk1 (ERK) | 23 | Dlx5, Egr2, Egr3, Fosb, Homer1, Nab2, Nr4a1, Nr4a3, Pdp1, Pou3f1, Sox8, Sox10, Top2a | Adarb1, Fat1, Kcnc1, Mdn1, Mid1, Nf1, Pde1a, Prkdc, Rasgrf2, Shank1 |
| NAc; chronic | Akt1 (Akt) | 20 | Akt1s1, Arc, Dach1, Egr1, Egr4, Foxg1, Foxo1, Foxo6, Mpped2, Rxrg, Vgf | Bsn, Dzip1, Mrpl55, Nrp2, Pclo, Plxna1, Plxna4, Plxnb1, Slit2 |
| NAc; chronic | Junb (JUNB) | 18 | Junb, Klf5, Lmo2, Mme, Spry2, Tiparp | Apc, Birc6, Exph5, Faah, Hipk2, AI314180, Lpp, Macf1, Nav1, Nr2f1, Ubr4, Usp24 |
| PrL; chronic | Map2k1; Map2k2 (MAP2K1/2) | 21 | Bdnf, Btg2, Cartpt, Cck, Dusp1, Dusp6, Klf10, Nudt11, Pvalb | Adcy9, Galnt10, Gdf11, Ksr1, Mef2d, Pde10a, Plxnb2, Plxnd1, Ptprf, Ptprh, Slit1, Ssh1 |
| PrL; chronic | Mapk1; Mapk3 (ERK1/2) | 20 | Arc, Dpysl2, Il17d, Spry4, Trib1, Vapa | Aebp1, Cntnap1, Dag1, Dpysl3, Gnaz, Klhl3, Mid1, Myoc, Ndst1, Pcdhgc3, Slc12a7, Slc17a6, Trpm2, Wnk2 |
| PrL; chronic | Nfkb1 (NFκB) | 19 | 2700029M09Rik, Camk2n1, Gadd45a, Gadd45b, Gas5, Ier2, Nfil3, Pttg1, Wnt10a | C4a/C4b, Cacna1h, Cacnb1, Cacng4, Dvl3, Efs, Fosb, Peli3, Rusc2, Znf668 |
| PrL; chronic | Mapk8 (JNK) | 19 | Clk1, Cox5a, Cycs, Cyr61, Junb, Lsm1, Lsm5, Nr4a1, Snrpf, Tra2a, Zcrb1 | Arhgef10l, Cabp7, Col6a2, Ints1, Myt1l, Nlgn3, Smpd3, Ttc28 |
| PrL; chronic | Racgap1 (GTPase) | 32 | Alkbh2, C9orf40, Crk, Hjurp, Htatip2, Msrb3, Nprl2, Nrsn1, Nxf1, Ovol2 | Abca2, Adap1, Agap2, Arhgef17, Atad2, Ccnd1, Cybrd1, Evi5l, Gltscr1, Kcnd1, Lnx2, Megf8, Mex3b, Myo18a, Ncl, Nfe2l2, Pcdha2, Pcdha3, Pcdha10, Plekhg5, Rabgap1l, Srgap2 |
WT and SERT Met172 mice were treated once with 20 mg·kg−1 cocaine i.p. (acute) or on five consecutive days with 15 mg·kg−1 cocaine i.p. (chronic). Mice were killed 120 min after the last cocaine dose. Each network consists of 35 genes total. Genes within the network that were differentially expressed are listed. Network hub genes are noted in two common nomenclatures. The graphical illustration for each network can be found in the Supplementary Information. Genes with higher expression in WT than in SERT Met172 mice are up‐regulated, and molecules with lower expression in WT than in SERT Met172 mice are down‐regulated.
Figure 9.
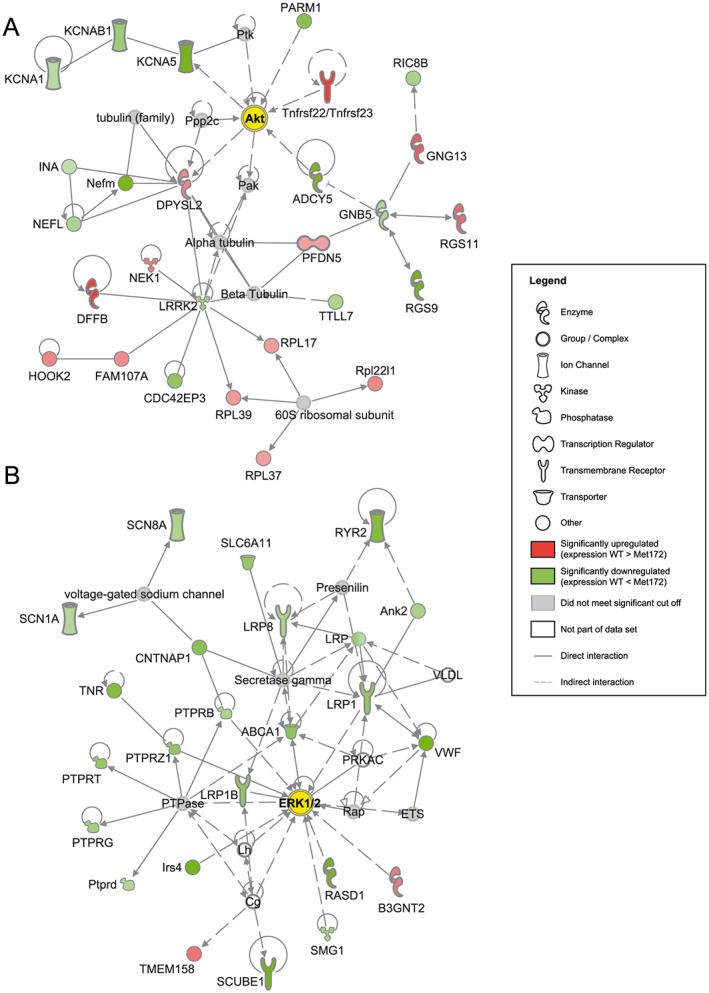
Representative gene networks that were significantly regulated (P < 0.05 with the Fisher's exact test) in response to cocaine in WT compared to SERT Met172 mice. Gene expression was assessed using RNA‐Seq and figures were generated by Ingenuity Pathway Analysis Path Designer (Qiagen). (A) Network centred around the network hub genes Akt (Akt1) in the PrL after acute cocaine treatment (20 mg·kg−1). (B) Network centred around the network hub genes ERK1/2 (Mapk1 and Mapk3) in the NAc after chronic cocaine treatment (5 days, 15 mg·kg−1 daily). Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) or green (down‐regulated in WT), along with other molecules (grey and white) that connect smaller networks to make larger networks. Darker green or red indicates higher statistical significance for differential gene expression. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition). See Supporting Information Figures S7 and S11 for subcellular network versions of panels A and B.
Discussion
The elimination of high‐affinity cocaine recognition in the SERT Met172 mouse in the absence of altered baseline 5‐HT transport provides an ideal opportunity to examine the degree to which SERT inhibition contributes to the molecular and behavioural actions of the psychostimulant, without the confounding aspects of the SERT KO model. In our initial studies (Thompson et al., 2011; Nackenoff et al., 2016), we capitalized on the SERT Met172 model and its loss of high‐affinity SSRI recognition to demonstrate a requirement for 5‐HT signalling in preclinical measures of antidepressant action. The model also proved capable of identifying SERT‐independent actions of the SSRI citalopram, specifically an effect on axon guidance mediated by σ receptor interactions (Bonnin et al., 2012).
In the present study, we first demonstrated the loss of cocaine sensitivity at SERT Met172 ex vivo and in vivo and then assessed the effects of this loss of SERT inhibition to cocaine‐mediated behaviour and gene expression. The slow rise in 5‐HT levels in the hippocampus compared with the faster onset of cocaine locomotor effects suggests a role for signalling by other cocaine‐elevated neurotransmitters, such as neostriatal dopamine signalling, which our genotype comparisons support. The rate of rise of 5‐HT in the hippocampus, compared to the rise of dopamine in the NAc or striatum, will be a complex factor of the density of 5‐HT and dopamine fibres in these regions, the basal and drug‐modulated firing rates of these projections and the differential potencies of cocaine for SERT and DAT. Our initial findings confirmed that acute locomotor response as well as sensitization to the locomotor effects of cocaine are independent of SERT blockade, which was long suspected but not previously demonstrated conclusively. Given the powerful modulation exerted by dopamine on subcortical motor systems and the ability of dopamine receptor antagonists to block acute locomotor effects of cocaine (O'Neill and Shaw, 1999), we expected that acute response to cocaine in the open field would derive predominantly from DAT blockade. Indeed, mice rendered less sensitive to cocaine at DAT via an analogous mutation approach demonstrate a complete loss of locomotor activation following acute cocaine administration (Chen et al., 2006). This model has been used to demonstrate that DAT inhibition is also required for cocaine sensitization (O'Neill et al., 2014). Interestingly, we did detect a significant change in the pattern of locomotion after cocaine treatment in SERT Met172 animals, with reduced ambulation in the centre of the chamber compared with WT animals. To our knowledge, the only previous connection between cocaine‐modulated zone entry and the 5‐HT system are reports by Carey and colleagues who showed that a 5‐HT1A agonist suppressed centre entries after cocaine administration (Carey et al., 2002; Carey et al., 2008). Centre versus surround occupancy and locomotion in open field chambers is a measure of anxiety‐like behaviour and is sensitive to anxiolytic treatments (Prut and Belzung, 2003). Thus, it is tempting to suggest that SERT antagonism by cocaine reduces the potentially anxiogenic effect of being injected with a powerful psychoactivating agent. However, the interpretation of centre/surround measure as an anxiety measure has also been questioned due to the failure of mouse strains (particularly the C57BL/6 background of our SERT Met172 line) to be sensitive to prototypical anxiolytic drugs (Thompson et al., 2015). As the latter agents typically act via GABAergic mechanisms, it is possible that the broader actions of these agents may confound attempts to use pharmacology to attribute predictive validity to this measure. Future studies examining the SERT Met172 model in other tests with predictive validity for anxiolytic agents may prove useful in further analysis of these observations.
The two‐bottle choice and CPP tests provide measures of drug liking, with the CPP test offering opportunities to assess aspects of reward‐driven contextual learning. Both tests provide evidence of a suppressive contribution normally made by augmented 5‐HT signalling following cocaine administration and suggest that SERT inhibition by cocaine attenuates specific cocaine‐related action. Our findings are in accordance with an increased preference for the cocaine‐paired compartment observed in SERT KO mice (Sora et al., 1998). Specific 5‐HT receptors have also been implicated in the modulation of cocaine CPP. For example, pharmacological modulation of the 5‐HT2C receptor with a selective agonist (Craige and Unterwald, 2013) and 5‐HT6 receptor overexpression in the NAc (Ferguson et al., 2008) reduced cocaine CPP. The modulation of the reward system by 5‐HT is a widely accepted hypothesis (Daw et al., 2002) and is supported by a shift to lower abuse liability for amphetamine analogues that display a relative elevation in SERT, compared with DAT inhibition (Wee et al., 2005; Bauer et al., 2013). Our behavioural findings provide further evidence for an opposing role of 5‐HT effects in cocaine abuse liability.
In c‐Fos‐based studies of cocaine‐induced neuronal activation, we found further evidence of a normally suppressive role of SERT inhibition, revealed by increased numbers of c‐Fos immunoreactive neurons in SERT Met172 mice following cocaine injections. This pattern was region‐specific, with significant changes evident in PrL, CA3 hippocampus and piriform cortex. Serotonergic inputs innervate and excite GABAergic interneurons in many cortical regions including these three regions (Freund et al., 1990; Gellman and Aghajanian, 1993; Santana et al., 2004). Loss of 5‐HT‐driven activation of these interneurons due to a lack of SERT inhibition by cocaine and subsequent loss of pyramidal cell inhibition could account for our findings. Together with the infralimbic cortex, the PrL constitutes the medial prefrontal cortex (mPFC), which sends glutamatergic projections to the NAc and mediates cocaine‐evoked plasticity (Pascoli et al., 2014b). The SERT‐dependent modulation of cocaine effects in the PrL could be of significance with respect to an eventual progression to cocaine dependence since this region plays a crucial role in top‐down control over the behaviours driven by networks within the ventral striatum (Dalley et al., 2011), and 5‐HT signalling via 5‐HT2A and 5‐HT2C receptors in the mPFC is implicated in cocaine cue reactivity and self‐administration (Anastasio et al., 2014; Cunningham and Anastasio, 2014).
As IEGs have been demonstrated to be elevated in response to acute cocaine administration, and neuroadaptive changes of relevance to addiction are linked to persistent drug‐induced changes in transcription factors, including IEGs (Nestler, 2012), we evaluated several IEGs to broaden our evaluation of molecular pathways where cocaine‐induced activation is significantly influenced by elevated 5‐HT signalling. Overall, SERT‐mediated IEGs expression changes were subtle, which agrees with our locomotor sensitization data and a prominent role of striatal dopamine D1 receptors in activation of IEGs (Drago et al., 1996). Nevertheless, SERT Met172 and WT comparisons revealed genotype differences for some IEGs in both PrL and the NAc. The reduced activation of IEGs in the NAc suggests that the actions of cocaine on WT SERT provide a stimulatory effect onto principle neurons in this region. As one potential target, antagonists of 5HT2A receptors attenuate c‐Fos activation in the NAc (Szucs et al., 2005). A role of 5‐HT in Egr1 regulation has been shown as lesions of 5‐HT neurons resulted in blunted Egr1 expression after cocaine and amphetamine injections (Bhat et al., 1992). Additionally, cocaethylene, the metabolite that is formed from cocaine in combination with ethanol, acts more potently on DAT than SERT and induces lower Egr1 and Fos expression in the NAc than cocaine (Thiriet et al., 2000).
Further confirming a role of SERT in IEG expression, Arc (activity‐regulated cytoskeletal associated protein) stood out in the RNA‐Seq data set from chronic treatment, with significantly (FDR < 5%) lower expression in SERT Met172 compared to WT, both in the PrL and in the NAc. As an IEG effector protein expressed in dendrites, Arc is regulated via synaptic excitation and has been implicated in cocaine sensitization (Samaha et al., 2004). Arc expression is directly connected to the expression of the IEGs Egr1 and Egr3 (Li et al., 2005), and the lower Egr1 and Egr3 expression levels we found for cocaine‐treated SERT Met172 mice compared to WT in the RNA‐Seq data set are therefore consistent with our finding of relatively lower Arc levels.
Pathway analysis performed with our transcriptome‐wide expression data set offers a strategy to capture 5‐HT‐dependent signalling networks activated by cocaine without the power limitations of individual gene comparisons. This effort revealed networks associated with specific MAP‐kinases (i.e. ERK2, ERK1 and Jnk) and Akt in PrL and NAc in both acute and chronic cocaine treatment. ERK1 and ERK2 (Mapk1 and Mapk3 in mouse gene nomenclature) have been strongly linked to drug‐induced plasticity (Pascoli et al., 2014a), cocaine CPP and locomotor sensitization (Ferguson et al., 2006; Valjent et al., 2006; Pascoli et al., 2011). We also found significant DEG networks associated with the transcription factor NF‐κB, which is activated by ΔFosB (a truncated product of Fosb) and plays a role in mediating cocaine reward and NAc neuronal morphology (Russo et al., 2009; Robison and Nestler, 2011). Genes in networks associated with Akt (Akt1; protein kinase B) were also significantly differentially expressed in our RNA‐Seq experiments, aligning with evidence for the Akt‐GSK3 signalling pathway playing a role in cocaine CPP (Miller et al., 2014). Only a few studies have to date associated these pathways with the serotonergic components of cocaine action. SSRIs induce ERK phosphorylation in the prefrontal cortex (Valjent et al., 2004), and cocaine occludes hippocampal long‐term potentiation via 5‐HT1A receptors and ERK signalling (Grzegorzewska et al., 2010). Importantly, these network hub molecules were not themselves differentially expressed between genotypes, indicating that our network analyses were able to bring their potential roles to light in a way that would not be established by simply examining individual DEGs. Our RNA‐Seq studies thus support a contribution of SERT inhibition to adaptations in specific signalling networks that drive critical plasticity likely to underlie addiction. More targeted studies are now warranted, where, for example, pharmacological or genetic modulation of specific 5‐HT receptors, or more network based interventions, can be employed to limit drug self‐administration or reinstatement.
Our work documents a pattern of neuronal activation and gene expression linked specifically through use of the SERT Met172 model to cocaine inhibition of SERT. To date, most strategies to generate pharmacological approaches to combat cocaine addiction have centred on manipulation of dopamine signalling, for example, the development of non‐reinforcing cocaine analogues that might serve as ‘cocaine antagonists’. Further work is needed to assess whether and/or how the SERT‐dependent actions of cocaine that we reveal relate to cocaine addiction, though evidence indicates that manipulation of specific facets of 5‐HT action may limit substance abuse (Howell and Cunningham, 2015). From this perspective, our data set of specific network influences may be helpful in sharpening pharmacological options that affect the SERT‐dependent networks activated by cocaine. Our work may also provide a better understanding of the actions of new psychoactive substances (NPS), such as bath salts that also act on multiple monoamine transporters. Many NPS resemble in structure or effects the well‐known psychostimulants including cocaine, amphetamine and MDMA (Baumann et al., 2013; Simmler et al., 2013). The variable induction of 5‐HT‐dependent networks by these agents may ultimately dictate the differential characteristics of NPS exposure and the degree to which the generalization of cocaine‐targeted therapies will succeed for these emerging drugs of abuse. Furthermore, if the changes in locomotor patterns we observe (centre vs. surround) following cocaine administration to SERT Met172 as compared with WT animals, are indicative of an anxiogenic response, the SERT‐dependent patterns of neuronal activation and gene networks we have uncovered may also aid in understanding the participation of 5‐HT signalling to anxiety.
Author contributions
L.D.S. and R.D.B. designed the research and L.D.S., M.H.L., N.M.V., S.J.S. and A.S. performed experiments. A.M.J.A. and J.W. processed and analysed RNA‐Seq data. L.K.H. computed and illustrated the drug‐protein interaction model. A.G.N. and P.J.G. provided technical support and input regarding experimental design. L.D.S. and R.D.B. wrote the manuscript with B.Z., J.V.V., N.C.A. and K.A.C. providing research support and revisions of the manuscript. All authors have given their final approval for the manuscript.
Conflict of interest
R.D.B. has consulted with Lundbeck, Pfizer and Prexa and has received research funding from Lundbeck and Prexa. J.V.V. has consulted with Roche, Novartis and Synapdx and has received research funding from Roche, Novartis, SynapDx, Seaside Therapeutics and Forest. K.A.C. is an uncompensated consultant for Arena Pharmaceuticals. All other authors (L.D.S., A.M.J.A., M.H.L., N.M.V., P.J.G., A.G.N., S.J.S., N.C.A., J.W., B.Z., L.K.H. and A.S.) declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Stereotypies and vertical movements in the open field test in response to acute cocaine as a function of genotype. Number of stereotypies (A), time in stereotypy (B), number of vertical counts (C), and time vertical (D) during 50 min post injections. Saline (20 ml kg‐1 i.p.) was administered in the first and cocaine (15 mg kg‐1 cocaine i.p.) in the second session. Data represent mean ± S.E.M., n=28‐31. Main treatment effect was significant in RM‐2‐way ANOVA. *P<0.05, by Sidak's post hoc test.
Figures S2 Jnk‐related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05, with the Fisher's Exact Test) in the NAc in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub gene Jnk. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S3 UBC related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub gene UBC. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S4 UBC related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub gene UBC. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S5 NFκB/ERK related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub genes NFκB and ERK. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S6 NFκB related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub genes NFκB. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S7 Akt related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. Network is centered around the network hub genes Akt. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S8 Jnk related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. Network is centered around the network hub genes Jnk. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S9 ERK1/2 related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. Network is centered around the network hub genes ERK1/2. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S10 ERK related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. Network is centered around the network hub genes ERK. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S11 ERK1/2 related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene ERK1/2. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S12 NFκB related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene NFκB. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S13 ERK related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene ERK. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S14 Akt related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene Akt. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S15 JUNB related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene JUNB, but also actin, caspase, and cyclin A. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S16 MAP2K1/2 related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene MAP2K1/2. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S17 ERK1/2 related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene ERK1/2. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S18 NFκB related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene NFκB. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S19 Jnk related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene Jnk. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S20 GTPase related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene GTPase, but also actin, caspase, and cyclin A. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Table S1 Normalized values from RNA‐Seq in the nucleus accumbens of WT and SERT Met172 mice after acute cocaine treatment.
Table S2 Statistics for RNA‐Seq in the nucleus accumbens of WT and SERT Met172 mice after acute cocaine treatment.
Table S3 Normalized values from RNA‐Seq in the prelimbic cortex of WT and SERT Met172 mice after acute cocaine treamtent.
Table S4 Statistics for RNA‐Seq in the prelimbic cortex of WT and SERT Met172 mice after acute cocaine treatment.
Table S5 Normalized values from RNA‐Seq in the nucleus accumbens of WT and SERT Met172 mice after chronic cocaine treatment.
Table S6 Statistics for RNA‐Seq in the nucleus accumbens of WT and SERT Met172 mice after chronic cocaine treatment.
Table S7 Normalized values from RNA‐Seq in the prelimbic cortex of WT and SERT Met172 mice after chronic cocaine treatment.
Table S8 Statistics of RNA‐Seq in the prelimbic cortex of WT and SERT Met172 mice after chronic cocaine treatment.
Table S9 Summary of F‐values for 2‐way ANOVAs.
Acknowledgements
We gratefully acknowledge the expert laboratory oversight provided by Chris Svitek, Jane Wright, Qiao Han, Angela Steele and Tracy Moore‐Jarrett. The studies were supported by NIH Awards MH094527 (R.D.B.), MH096972 (R.D.B., J.V.V., P.J.G., J.W. and B.Z.), MH094604 (J.V.V.) and T32MH016434 (A.M.J.A.) and an NIH funded COBRE grant P20 GM104360 (L.K.H.). L.D.S. was supported by the Swiss National Science Foundation and a Vanderbilt Conte Center pilot grant P50 MH096972. The Vanderbilt Kennedy Center for Research on Human Development (P30 HD15052) provided infrastructure support for behavioural and neurochemical analyses. The Vanderbilt VANTAGE Core and the University of Texas Medical Branch Center for Addiction Research Rodent In Vivo Assessment Core provided technical assistance for this work. VANTAGE is supported in part by CTSA grant (5UL1 RR024975‐03), the Vanderbilt Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126) and NIH/NCRR (G20 RR030956).
Simmler, L. D. , Anacker, A. M. J. , Levin, M. H. , Vaswani, N. M. , Gresch, P. J. , Nackenoff, A. G. , Anastasio, N. C. , Stutz, S. J. , Cunningham, K. A. , Wang, J. , Zhang, B. , Henry, L. K. , Stewart, A. , Veenstra‐VanderWeele, J. , and Blakely, R. D. (2017) Blockade of the 5‐HT transporter contributes to the behavioural, neuronal and molecular effects of cocaine. British Journal of Pharmacology, 174: 2716–2738. doi: 10.1111/bph.13899.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasio NC, Stutz SJ, Fox RG, Sears RM, Emeson RB, DiLeone RJ et al. (2014). Functional status of the serotonin 5‐HT2C receptor (5‐HT2CR) drives interlocked phenotypes that precipitate relapse‐like behaviors in cocaine dependence. Neuropsychopharmacology 39: 370–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansah TA, Ramamoorthy S, Montanez S, Daws LC, Blakely RD (2003). Calcium‐dependent inhibition of synaptosomal serotonin transport by the alpha 2‐adrenoceptor agonist 5‐bromo‐N‐[4,5‐dihydro‐1H‐imidazol‐2‐yl]‐6‐quinoxalinamine (UK14304). J Pharmacol Exp Ther 305: 956–965. [DOI] [PubMed] [Google Scholar]
- Bauer CT, Banks ML, Blough BE, Negus SS (2013). Use of intracranial self‐stimulation to evaluate abuse‐related and abuse‐limiting effects of monoamine releasers in rats. Br J Pharmacol 168: 850–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Partilla JS, Lehner KR, Thorndike EB, Hoffman AF, Holy M et al. (2013). Powerful cocaine‐like actions of 3,4‐methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive 'bath salts' products. Neuropsychopharmacology 38: 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Cole AJ, Baraban JM (1992). Role of monoamine systems in activation of zif268 by cocaine. J Psychiatry Neurosci 17: 94–102. [PMC free article] [PubMed] [Google Scholar]
- Bonnin A, Zhang L, Blakely RD, Levitt P (2012). The SSRI citalopram affects fetal thalamic axon responsiveness to netrin‐1 in vitro independently of SERT antagonism. Neuropsychopharmacology 37: 1879–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky M, Gibson AW, Smirnov D, Nair SG, Neumaier JF (2016). Striatal 5‐HT6 receptors regulate cocaine reinforcement in a pathway‐selective manner. Neuropsychopharmacology 41: 2377–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey RJ, De Palma G, Damianopoulos E (2002). 5‐HT1A agonist/antagonist modification of cocaine stimulant effects: implications for cocaine mechanisms. Behav Brain Res 132: 37–46. [DOI] [PubMed] [Google Scholar]
- Carey RJ, DePalma G, Shanahan A, Damianopoulos EN, Muller CP, Huston JP (2008). Effects on spontaneous and cocaine‐induced behavior of pharmacological inhibition of noradrenergic and serotonergic systems. Pharmacol Biochem Behav 89: 54–63. [DOI] [PubMed] [Google Scholar]
- Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S et al. (2006). Abolished cocaine reward in mice with a cocaine‐insensitive dopamine transporter. Proc Natl Acad Sci U S A 103: 9333–9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craige CP, Unterwald EM (2013). Serotonin (2C) receptor regulation of cocaine‐induced conditioned place preference and locomotor sensitization. Behav Brain Res 238: 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham KA, Anastasio NC (2014). Serotonin at the nexus of impulsivity and cue reactivity in cocaine addiction. Neuropharmacology 76 (Pt B): 460–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Everitt BJ, Robbins TW (2011). Impulsivity, compulsivity, and top‐down cognitive control. Neuron 69: 680–694. [DOI] [PubMed] [Google Scholar]
- Daw ND, Kakade S, Dayan P (2002). Opponent interactions between serotonin and dopamine. Neural Netw 15: 603–616. [DOI] [PubMed] [Google Scholar]
- Drago J, Gerfen CR, Westphal H, Steiner H (1996). D1 dopamine receptor‐deficient mouse: cocaine‐induced regulation of immediate‐early gene and substance P expression in the striatum. Neuroscience 74: 813–823. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Fasano S, Yang P, Brambilla R, Robinson TE (2006). Knockout of ERK1 enhances cocaine‐evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology 31: 2660–2668. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Mitchell ES, Neumaier JF (2008). Increased expression of 5‐HT6 receptors in the nucleus accumbens blocks the rewarding but not psychomotor activating properties of cocaine. Biol Psychiatry 63: 207–213. [DOI] [PubMed] [Google Scholar]
- Filip M, Frankowska M, Zaniewska M, Golda A, Przegalinski E (2005). The serotonergic system and its role in cocaine addiction. Pharmacol Rep 57: 685–700. [PubMed] [Google Scholar]
- Fosnaugh JS, Bhat RV, Yamagata K, Worley PF, Baraban JM (1995). Activation of arc, a putative “effector” immediate early gene, by cocaine in rat brain. J Neurochem 64: 2377–2380. [DOI] [PubMed] [Google Scholar]
- Freund TF, Gulyas AI, Acsady L, Gorcs T, Toth K (1990). Serotonergic control of the hippocampus via local inhibitory interneurons. Proc Natl Acad Sci U S A 87: 8501–8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellman RL, Aghajanian GK (1993). Pyramidal cells in piriform cortex receive a convergence of inputs from monoamine activated GABAergic interneurons. Brain Res 600: 63–73. [DOI] [PubMed] [Google Scholar]
- Grzegorzewska M, Mackowiak M, Wedzony K, Hess G (2010). 5‐HT1A receptors mediate detrimental effects of cocaine on long‐term potentiation and expression of polysialylated neural cell adhesion molecule protein in rat dentate gyrus. Neuroscience 166: 122–131. [DOI] [PubMed] [Google Scholar]
- Han DD, Gu HH (2006). Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry LK, Field JR, Adkins EM, Parnas ML, Vaughan RA, Zou MF et al. (2006). Tyr‐95 and Ile‐172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J Biol Chem 281: 2012–2023. [DOI] [PubMed] [Google Scholar]
- Howell LL, Cunningham KA (2015). Serotonin 5‐HT2 receptor interactions with dopamine function: implications for therapeutics in cocaine use disorder. Pharmacol Rev 67: 176–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2010). Neurocircuitry of addiction. Neuropsychopharmacology 35: 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Carter J, Gao X, Whitehead J, Tourtellotte WG (2005). The neuroplasticity‐associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Mol Cell Biol 25: 10286–10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Ungless MA (2006). The mechanistic classification of addictive drugs. PLoS Med 3: e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JS, Barr JL, Harper LJ, Poole RL, Gould TJ, Unterwald EM (2014). The GSK3 signaling pathway is activated by cocaine and is critical for cocaine conditioned reward in mice. PLoS One 9: e88026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DL, Lesch KP (2008). Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci 9: 85–96. [DOI] [PubMed] [Google Scholar]
- Nackenoff AG, Moussa‐Tooks AB, McMeekin AM, Veenstra‐VanderWeele J, Blakely RD (2016). Essential contributions of serotonin transporter inhibition to the acute and chronic actions of fluoxetine and citalopram in the SERT Met172 mouse. Neuropsychopharmacology 41: 1733–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SG, Furay AR, Liu Y, Neumaier JF (2013). Differential effect of viral overexpression of nucleus accumbens shell 5‐HT1B receptors on stress‐ and cocaine priming‐induced reinstatement of cocaine seeking. Pharmacol Biochem Behav 112: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ (2012). Transcriptional mechanisms of drug addiction. Clin Psychopharmacol Neurosci 10: 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill B, Tilley MR, Han DD, Thirtamara‐Rajamani K, Hill ER, Bishop GA et al. (2014). Behavior of knock‐in mice with a cocaine‐insensitive dopamine transporter after virogenetic restoration of cocaine sensitivity in the striatum. Neuropharmacology 79: 626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill MF, Shaw G (1999). Comparison of dopamine receptor antagonists on hyperlocomotion induced by cocaine, amphetamine, MK‐801 and the dopamine D1 agonist C‐APB in mice. Psychopharmacology (Berl) 145: 237–250. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Besnard A, Herve D, Pages C, Heck N, Girault JA et al. (2011). Cyclic adenosine monophosphate‐independent tyrosine phosphorylation of NR2B mediates cocaine‐induced extracellular signal‐regulated kinase activation. Biol Psychiatry 69: 218–227. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Cahill E, Bellivier F, Caboche J, Vanhoutte P (2014a). Extracellular signal‐regulated protein kinases 1 and 2 activation by addictive drugs: a signal toward pathological adaptation. Biol Psychiatry 76: 917–926. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O'Connor EC, Luscher C (2014b). Contrasting forms of cocaine‐evoked plasticity control components of relapse. Nature 509: 459–464. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2004). The Mouse Brain in Stereotaxic Coordinates, Compact 2nd edn. Elsevier Academic Press: Amsterdam ; Boston. [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E (2013). X‐ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503: 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser RA, Stowie A, Amicarelli M, Nackenoff AG, Blakely RD, Glass JD (2014). Cocaine modulates mammalian circadian clock timing by decreasing serotonin transport in the SCN. Neuroscience 275: 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prut L, Belzung C (2003). The open field as a paradigm to measure the effects of drugs on anxiety‐like behaviors: a review. Eur J Pharmacol 463: 3–33. [DOI] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ (2011). Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 12: 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Wilkinson MB, Mazei‐Robison MS, Dietz DM, Maze I, Krishnan V et al. (2009). Nuclear factor kappa B signaling regulates neuronal morphology and cocaine reward. J Neurosci 29: 3529–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaha AN, Mallet N, Ferguson SM, Gonon F, Robinson TE (2004). The rate of cocaine administration alters gene regulation and behavioral plasticity: implications for addiction. J Neurosci 24: 6362–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana N, Bortolozzi A, Serrats J, Mengod G, Artigas F (2004). Expression of serotonin1A and serotonin2A receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb Cortex 14: 1100–1109. [DOI] [PubMed] [Google Scholar]
- Shen HW, Hagino Y, Kobayashi H, Shinohara‐Tanaka K, Ikeda K, Yamamoto H et al. (2004). Regional differences in extracellular dopamine and serotonin assessed by in vivo microdialysis in mice lacking dopamine and/or serotonin transporters. Neuropsychopharmacology 29: 1790–1799. [DOI] [PubMed] [Google Scholar]
- Simmler L, Buser T, Donzelli M, Schramm Y, Dieu LH, Huwyler J et al. (2013). Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol 168: 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sora I, Hall FS, Andrews AM, Itokawa M, Li XF, Wei HB et al. (2001). Molecular mechanisms of cocaine reward: combined dopamine and serotonin transporter knockouts eliminate cocaine place preference. Proc Natl Acad Sci U S A 98: 5300–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R et al. (1998). Cocaine reward models: conditioned place preference can be established in dopamine‐ and in serotonin‐transporter knockout mice. Proc Natl Acad Sci U S A 95: 7699–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucs RP, Frankel PS, McMahon LR, Cunningham KA (2005). Relationship of cocaine‐induced c‐Fos expression to behaviors and the role of serotonin 5‐HT2A receptors in cocaine‐induced c‐Fos expression. Behav Neurosci 119: 1173–1183. [DOI] [PubMed] [Google Scholar]
- Thiriet N, Aunis D, Zwiller J (2000). C‐fos and egr‐1 immediate‐early gene induction by cocaine and cocaethylene in rat brain: a comparative study. Ann N Y Acad Sci 914: 46–57. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Jessen T, Henry LK, Field JR, Gamble KL, Gresch PJ et al. (2011). Transgenic elimination of high‐affinity antidepressant and cocaine sensitivity in the presynaptic serotonin transporter. Proc Natl Acad Sci U S A 108: 3785–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson T, Grabowski‐Boase L, Tarantino LM (2015). Prototypical anxiolytics do not reduce anxiety‐like behavior in the open field in C57BL/6J mice. Pharmacol Biochem Behav 133: 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Aubier B, Corbille AG, Brami‐Cherrier K, Caboche J, Topilko P et al. (2006). Plasticity‐associated gene Krox24/Zif268 is required for long‐lasting behavioral effects of cocaine. J Neurosci 26: 4956–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pages C, Herve D, Girault JA, Caboche J (2004). Addictive and non‐addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci 19: 1826–1836. [DOI] [PubMed] [Google Scholar]
- Wee S, Anderson KG, Baumann MH, Rothman RB, Blough BE, Woolverton WL (2005). Relationship between the serotonergic activity and reinforcing effects of a series of amphetamine analogs. J Pharmacol Exp Ther 313: 848–854. [DOI] [PubMed] [Google Scholar]
- You IJ, Wright SR, Garcia‐Garcia AL, Tapper AR, Gardner PD, Koob GF et al. (2016). 5‐HT1A Autoreceptors in the dorsal raphe nucleus convey vulnerability to compulsive cocaine seeking. Neuropsychopharmacology 41: 1210–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zombeck JA, Lewicki AD, Patel K, Gupta T, Rhodes JS (2010). Patterns of neural activity associated with differential acute locomotor stimulation to cocaine and methamphetamine in adolescent versus adult male C57BL/6J mice. Neuroscience 165: 1087–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Stereotypies and vertical movements in the open field test in response to acute cocaine as a function of genotype. Number of stereotypies (A), time in stereotypy (B), number of vertical counts (C), and time vertical (D) during 50 min post injections. Saline (20 ml kg‐1 i.p.) was administered in the first and cocaine (15 mg kg‐1 cocaine i.p.) in the second session. Data represent mean ± S.E.M., n=28‐31. Main treatment effect was significant in RM‐2‐way ANOVA. *P<0.05, by Sidak's post hoc test.
Figures S2 Jnk‐related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05, with the Fisher's Exact Test) in the NAc in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub gene Jnk. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S3 UBC related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub gene UBC. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S4 UBC related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub gene UBC. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S5 NFκB/ERK related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub genes NFκB and ERK. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S6 NFκB related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. The network is centered around the network hub genes NFκB. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S7 Akt related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. Network is centered around the network hub genes Akt. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S8 Jnk related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. Network is centered around the network hub genes Jnk. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S9 ERK1/2 related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. Network is centered around the network hub genes ERK1/2. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figure S10 ERK related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to acute cocaine (20 mg kg‐1, i.p.) in WT compared to SERT Met172 mice. Network is centered around the network hub genes ERK. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S11 ERK1/2 related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene ERK1/2. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S12 NFκB related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene NFκB. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S13 ERK related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene ERK. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S14 Akt related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene Akt. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S15 JUNB related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the NAc in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene JUNB, but also actin, caspase, and cyclin A. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S16 MAP2K1/2 related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene MAP2K1/2. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S17 ERK1/2 related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene ERK1/2. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S18 NFκB related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene NFκB. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S19 Jnk related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene Jnk. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Figures S20 GTPase related network, generated by IPA Path Designer (Qiagen). This gene network was significantly regulated (P<0.05 with the Fisher's Exact Test) in the PrL in response to chronic cocaine (15 mg kg‐1, i.p. for five days) in WT compared to SERT Met172 mice. Network is centered around the network hub gene GTPase, but also actin, caspase, and cyclin A. Seed molecules from the list of differentially regulated genes in WT compared to SERT Met172 mice are shown in the form of protein networks in red (upregulated in WT) and green (downregulated in WT), along with other molecules (gray and white) that connect smaller networks to make larger networks. Interaction or regulation between proteins or complexes is shown with arrows (activation) or blunt‐ended lines (inhibition).
Table S1 Normalized values from RNA‐Seq in the nucleus accumbens of WT and SERT Met172 mice after acute cocaine treatment.
Table S2 Statistics for RNA‐Seq in the nucleus accumbens of WT and SERT Met172 mice after acute cocaine treatment.
Table S3 Normalized values from RNA‐Seq in the prelimbic cortex of WT and SERT Met172 mice after acute cocaine treamtent.
Table S4 Statistics for RNA‐Seq in the prelimbic cortex of WT and SERT Met172 mice after acute cocaine treatment.
Table S5 Normalized values from RNA‐Seq in the nucleus accumbens of WT and SERT Met172 mice after chronic cocaine treatment.
Table S6 Statistics for RNA‐Seq in the nucleus accumbens of WT and SERT Met172 mice after chronic cocaine treatment.
Table S7 Normalized values from RNA‐Seq in the prelimbic cortex of WT and SERT Met172 mice after chronic cocaine treatment.
Table S8 Statistics of RNA‐Seq in the prelimbic cortex of WT and SERT Met172 mice after chronic cocaine treatment.
Table S9 Summary of F‐values for 2‐way ANOVAs.