

Abstract
Background and Purpose
Regulation of μ receptor dynamics such as its trafficking is a possible mechanism underlying opioid tolerance that contributes to inefficient recycling of opioid responses. We aimed to characterize the functional turnover of μ receptors in the noradrenergic nucleus locus coeruleus (LC).
Experimental Approach
We measured opioid effect by single‐unit extracellular recordings of LC neurons from rat brain slices. Immunocytochemical techniques were used to evaluate μ receptor trafficking.
Key Results
After near‐complete, irreversible μ receptor inactivation with β–funaltrexamine (β‐FNA), opioid effect spontaneously recovered in a rapid and efficacious manner. In contrast, α2‐adrenoceptor‐mediated effect hardly recovered after receptor inactivation with the irreversible antagonist EEDQ. When the recovery of opioid effect was tested after various inactivating time schedules, we found that the longer the β‐FNA pre‐exposure, the less efficient and slower the functional μ receptor turnover became. Interestingly, μ receptor turnover was slower when β‐FNA challenge was repeated in the same cell, indicating constitutive μ receptor recycling by trafficking from a depletable pool. Double immunocytochemistry confirmed the constitutive nature of μ receptor trafficking from a cytoplasmic compartment. The μ receptor turnover was slowed down when LC neuron calcium‐ or firing‐dependent processes were prevented or vesicular protein trafficking was blocked by a low temperature or transport inhibitor.
Conclusions and Implications
Constitutive trafficking of μ receptors from a depletable intracellular pool (endosome) may account for its rapid and efficient functional turnover in the LC. A finely‐tuned regulation of μ receptor trafficking and endosomes could explain neuroadaptive plasticity to opioids in the LC.
Abbreviations
- aCSF
artificial CSF
- EEDQ
N‐ethoxycarbonyl‐2‐ethoxy‐1,2‐dihydroquinoline
- Ess
maximal functional recovery at steady‐state
- Et
experimental effect of ME at each post‐β‐FNA time (t)
- β‐FNA
β‐funaltrexamine
- FR
firing rate
- k
rate constant for μ receptor degradation
- k′
rate constant for disappearance of μ receptor function
- Kapp
dissociation constant for ME
- KE
μ receptor occupancy that achieves a half‐maximal effect of ME
- LC
locus coeruleus
- ME
[Met]enkephalin
- n
slope parameter of the concentration‐effect curve for ME
- r
rate constant for μ receptor appearance
- r′
rate constant for reappearance of μ receptor function
- Rss
functionally active μ receptor at steady state
- Rt
functionally active μ receptor at each post‐β‐FNA time
- t1/2′
t 1/2 of recovery of μ receptor function
Introduction
Opioids are efficacious analgesics clinically available for pain relief, but their usefulness is limited by the development of tolerance and dependence (Chu et al., 2012). Most opioids induce their therapeutic effects through activation of Gi/o protein‐coupled μ receptors (Williams et al., 2013). The locus coeruleus (LC), the main noradrenergic nucleus in the brain, has been widely used to explore the cellular mechanisms of opioid tolerance (Nestler and Aghajanian, 1997; Santamarta et al., 2005; Llorente et al., 2012). As such, it mostly expresses postsynaptic μ receptors (Williams and North, 1984; Van Bockstaele et al., 1996), through which opioids inhibit firing activity (Andrade et al., 1983; Williams and North, 1984). Chronic morphine administration induces tolerance to the electrophysiological effects of opioids in the LC (Santamarta et al., 2005). Tolerance evolves from acute opioid desensitization (Dang and Williams, 2004), which is triggered by a strong μ receptor activation and the subsequent receptor phosphorylation, internalization and trafficking (Morgan and Christie, 2011; Williams et al., 2013). Desensitization without endocytosis has been also proposed for certain opioid agonists (Arttamangkul et al., 2006; 2008; Williams et al., 2013). Different signalling pathways are adapted during opioid tolerance and desensitization in the LC (Arttamangkul et al., 2008; Dang and Christie, 2012), including the PKA (Nestler and Aghajanian, 1997), PKC (Bailey et al., 2004) or nitric oxide–ROS/PKG pathways (Santamarta et al., 2005; 2014, Llorente et al., 2012).
Neuronal ionotropic (e.g. AMPA or GABAA) receptors on the cell surface are recycled by a constitutive but finely regulated trafficking of receptor‐containing vesicles from intracellular endosomal compartments upon traditional exocytosis mechanisms (Lüscher, 2002; Stricker and Huganir, 2002). Recycling of GPCRs from a reserve endosome may underlie resensitization of previously internalized receptors (Ferguson, 2001; Koch et al., 2005), although this process has been scarcely examined (Doly and Marullo, 2015). In the LC, μ receptor trafficking from endosomal compartments is required for recycling and resensitization of previously desensitized opioid responses (Dang and Williams, 2004). Impairment of μ receptor recycling that hinders opioid resensitization contributes to the development of cellular morphine tolerance in this nucleus (Dang et al., 2011; Williams et al., 2013). This emerging concept assumes a continuum of μ receptor endocytosis, transport of internalized μ receptors and eventual recycling to the membrane (Law et al., 2000; Tanowitz and von Zastrow, 2003). Alternatively, agonist‐induced μ receptor endocytosis and recycling could occur as two independent processes. The aim of our work was to characterize the recycling mechanisms of functionally active μ receptors. Single‐unit extracellular recordings in vitro and immunocytochemical techniques, combined with μ receptor inactivation with β‐funaltrexamine (β‐FNA) were used to study functional turnover of these receptors. We found that recovery of the opioid effect in LC neurons after μ receptor inactivation was rapid and efficacious, as well as finely regulated by different experimental elements such as the calcium concentration, firing activity, temperature or vesicle trafficking. Immunocytochemistry confirmed these electrophysiological data suggesting a constitutive recycling of μ receptors on the cell surface from a depletable cytoplasmic pool of receptors in LC neurons.
Methods
Animals and ethics statement
Eighty‐nine male adult Sprague Dawley rats (200–300 g) were used to perform the experiments, which are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). This animal species has been long used for functional studies on μ receptor pharmacology (Andrade and Aghajanian, 1984; Mendiguren and Pineda, 2007). For electrophysiological assays, one slice was taken from each animal, and unless stated otherwise, only one neuron was recorded in each slice. The number of experiments in each group was typically five to eight, depending on the level of variability. In order for the use of animals to be minimized, only four complete assays were obtained in those groups in which the efficiency of the technique (i.e. successful recordings) was clearly poorer than the standard, due to the long duration of the assay. Treatments and controls were performed in parallel in a randomized manner. The recording of data could not be blinded to the operator, because electrophysiological outcomes were collected in situ. However, the data analysis performed by the experimenter was confirmed separately by an additional researcher in all cases. Rats were obtained from the animal house of the University of the Basque Country (Leioa, Spain) and housed under standard environmental conditions (22°C, 12:12 h light/dark cycles) with free access to food and water. All the experiments were carried out according to EU Directive 2010/63 on the protection of animals used for scientific purposes and approved by the local Ethical Committee for Research and Teaching of the University of the Basque Country (UPV/EHU, Spain) and the Department of Sustainability and Natural Environment of Provincial Council from Bizkaia (ref. CEEA M20‐2015‐152/170). All the efforts were made to minimize animal suffering and to reduce the number of animals used.
Brain slice preparation
Animals were anaesthetized with chloral hydrate (400 mg·kg−1, i.p.) and decapitated (Mendiguren and Pineda, 2007). The brain was rapidly extracted, and a block of tissue containing the brainstem was immersed in ice‐cold modified artificial CSF (aCSF) where NaCl was equiosmolarly substituted for sucrose to improve neuronal viability. Coronal slices of 500–600 μm thickness containing the LC were cut using a vibratome (FHC Inc., Brunswick, GA, USA). The tissue was allowed to recover from the slicing for 90 min and placed in a modified Haas‐type interface chamber maintained at 33 ± 1°C and continuously perfused with aCSF saturated with 95% O2/5% CO2 (pH = 7.34) at a flow rate of 1.5 mL·min−1. The aCSF contained (in mM) NaCl 126, KCl 3, NaH2PO4 1.25, d‐glucose 10, NaHCO3 25, CaCl2 2 and MgSO4 2. Calcium substitution in low‐calcium aCSF was performed by replacing 90% of the CaCl2 equiosmolarly with MgSO4. The LC was identified visually in the rostral pons as a dark oval area on the lateral borders of the central grey and the fourth ventricle, at or just anterior to the genu of the facial nerve.
Electrophysiological recordings
Single‐unit extracellular recordings of LC cells were made as previously described (Mendiguren and Pineda, 2004; 2007). The recording electrode consisted of an Omegadot glass micropipette pulled (Sutter Instruments, Novato, CA, USA) and filled with 50 mM NaCl, with the tip broken back to a size of 2–5 μm (3–5 MΩ). The extracellular signal from the electrode was passed through a high‐input impedance amplifier (Axoclamp 2B; Molecular devices, Union City, CA, USA) and monitored on an audio amplifier and an oscilloscope (Aumon 14; Cibertec S.A., Madrid, Spain). Individual neuronal spikes were isolated from the background noise with a window discriminator (PDV 225; Cibertec S.A.). The firing rate (FR) was continuously recorded and analysed before, during and after experimental manipulations by a PC‐based custom‐made programme, which generated consecutive 10 s bin histograms of the cumulative number of spikes (HFCP®; Cibertec S.A.). Noradrenergic cells in the LC were identified by their spontaneous and regular discharge activities, their slow FRs and the long‐lasting and positive–negative waveforms (Andrade and Aghajanian, 1984).
Pharmacological procedures and functional μ receptor turnover
Functional turnover of μ receptor‐mediated effects in the LC was evaluated using the method of Mauger et al. (1982), which analysed the recovery of the effect mediated by newly appeared μ receptors after inactivation of the total receptor pool. Thus, the recovery of opioid effect was measured before (control effect), immediately after complete μ receptor inactivation with the irreversible alkylating blocker β‐FNA (300–800 nM, 30 min) (i.e. t 30 = 0) (Williams and North, 1984) and, then, every 15 min over a period of 300 min (i.e. t 30 = 15–300) (Figure 1A). Opioid effect was evaluated by a high concentration of the agonist [Met]enkephalin (ME, 3.2 μM, 1 min), which has been previously shown to induce a maximal inhibitory effect (Williams and North, 1984; Santamarta et al., 2005). In order for the functional turnover of μ receptors to be compared with that of another Gi/o‐coupled receptor (the α2‐adrenoceptor), the inhibitory effect of noradrenaline (NA, 100 μM, 1 min) was tested before (control effect) and, every 15 min, after complete receptor inactivation with the irreversible antagonist EEDQ (10 μM, 30 min) (Adler et al., 1985) (Figure 1B). In some assays, functional μ receptor turnover was further characterized by quantifying the recovery of opioid effect 120 min after 15, 30, 45 or 60 min of β‐FNA administration (i.e. t 15, 30, 45, 60 = 120) (Figure 1C). Moreover, the recovery of opioid effect was also evaluated 120 min after completion of a second 15 min perfusion with β‐FNA following the recovery of the first pre‐application (i.e. t 15(1) = 120 and t 15(2) = 120) (Figure 1D). The possible mechanisms underlying μ receptor turnover were explored by perfusion with a low‐calcium (0.2 mM) aCSF (to block calcium‐dependent mechanisms), administration of the α2‐adrenoceptor agonist NA (100 μM) (to inhibit LC cell firing‐dependent mechanisms), lowering the temperature of the chamber to 22°C (to evaluate vesicle movement mechanisms) or administration of the protein transport inhibitor brefeldin A (10 μM) (to test vesicle trafficking mechanisms; Law et al., 2000) (Figure 1E). In these assays, the aCSF and temperature manipulations were applied during the period of functional recovery after β‐FNA application, but regular conditions were restored 3–5 min before testing the ME effect (45 min).
Figure 1.
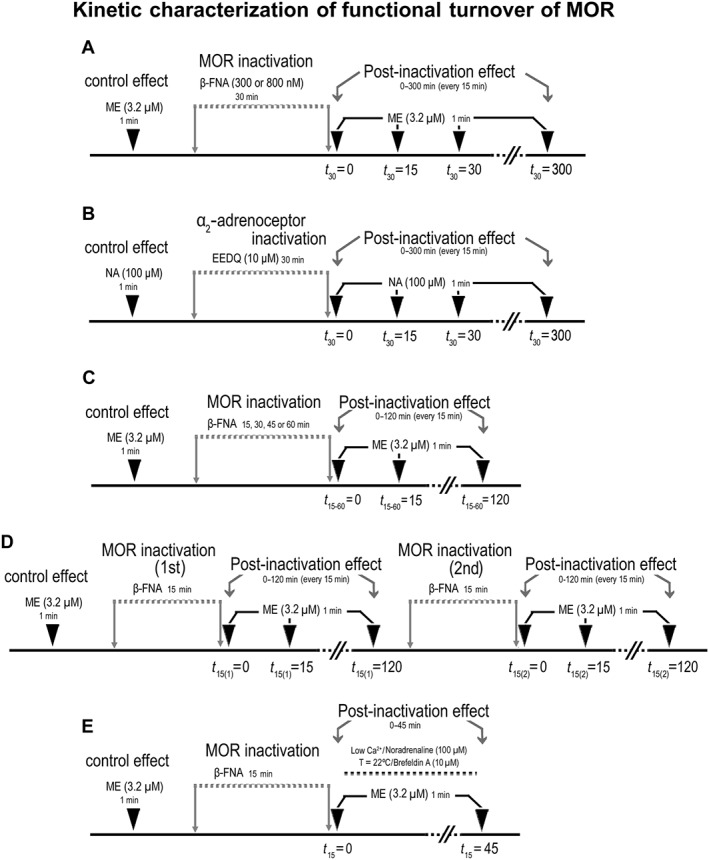
Scheme summarizing the experimental design for functional characterization of μ receptor (MOR) turnover. (A) Functional turnover of μ receptor ‐mediated effect was evaluated by analysing the recovery of ME (3.2 μM, 1 min) effect after complete inactivation of μ receptors with the irreversible alkylating blocker β‐FNA (300–800 nM, 30 min); opioid effect recovery was measured before (control effect), immediately after inactivation (t 30 = 0) and, then, every 15 min over a period of 300 min (t 30 = 15–300). (B) For the μ receptor turnover to be compared with that of α2‐adrenoceptors, NA (100 μM, 1 min) effect was tested before (control effect), immediately after complete receptor inactivation with the irreversible antagonist EEDQ (10 μM, 30 min) (t 30 = 0) and, then, every 15 min over 300 min after inactivation (t 30 = 15–300). (C) Functional μ receptor turnover was further characterized by evaluating the recovery of ME (3.2 μM, 1 min) effect for 120 min after complete μ receptor inactivation with 15, 30, 45 or 60 min of β‐FNA administration (t 15, 30, 45, 60 = 0–120). (D) The recovery of ME (3.2 μM, 1 min) effect was also evaluated for 120 min after completion of a second 15 min perfusion with β‐FNA following the recovery of the first pre‐application (t 15(1) = 0–120 and t 15(2) = 0–120). (E) The possible mechanisms underlying μ receptor turnover were explored by perfusion with a low‐calcium (0.2 mM) aCSF (to block LC cell calcium‐dependent mechanisms), administration of the α2‐adrenoceptor agonist NA (100 μM) (to inhibit LC neuron firing‐dependent mechanisms), lowering the temperature of the chamber to 22°C (to evaluate vesicle movement mechanisms) or administration of the protein transport inhibitor brefeldin A (10 μM) (to test vesicle trafficking mechanisms); these manipulations were applied during the period of functional recovery after β‐FNA application, but regular conditions were restored 3–5 min before testing ME effect (45 min).
Immunocytochemistry and confocal laser scanning microscopy
Midbrain slices containing the LC were taken from the recording chamber at three time points: immediately after β‐FNA perfusion for 15 min (t 15 = 0), 45 min after completion of β‐FNA perfusion for 15 min (t 15 = 45) and immediately after β‐FNA perfusion for 60 min (t 60 = 0). Slices removed from the chamber after perfusion of aCSF without β‐FNA treatment were used as baseline controls. All slices were immersion‐fixed overnight in phosphate‐buffered (0.1 M, pH 7.4) 4% paraformaldehyde at 4°C, cryoprotected for 48 h in 30% sucrose and sectioned on a freezing microtome. Transversal, 40‐μm‐thick sections were pre‐incubated with 1% bovine serum albumin (Sigma, St. Louis, MO, USA) and 1% normal serum (1 h, room temperature) and incubated overnight at 4°C with previously characterized rabbit antiserum against μ receptors (1:2000; Millipore, Bedford, MA, USA) (Kasai et al., 2011) and mouse antibodies to tyrosine hydroxylase (TH; 1:1000; Immunostar, Hudson, WI, USA) (Van Bockstaele and Pickel, 1993). Sections were then washed and further incubated with Cy5 650‐conjugated donkey anti‐rabbit or Dylight 549‐conjugated donkey anti‐mouse fluorescent antibodies (both at 1:400; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and mounted in Mowiol (Vector Labs, Burlingame, CA, USA). Double‐labelled sections were viewed in a Fluoview FV500 Olympus confocal microscope by a 60× Plan APO 1.4 numerical aperture (NA) objective. All images displayed were acquired with identical laser intensity and detector gain parameters. Digital photomicrographs were acquired sequentially to avoid overlapping of fluorescent emission spectra. Micrograph analyses were carried out with ImageJ software (National Institutes of Health, Bethesda, MD, USA). The average fluorescent background subtracted from the total intensity of the entire frame and the green μ receptor‐immunoreactivity channel and the TH/μ receptor overlay were both assigned a white colour value in order to visualize membrane‐associated and cytoplasmic μ receptor collectives.
Analysis and statistics of electrophysiological data
The data and statistical analysis were carried out with the computer programme GraphPad Prism (version 5.0 for Windows; GraphPad Software, Inc., San Diego, CA, USA) and comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The inhibitory effects of the agonists tested (ME and NA) were normalized to the initial FRs in each cell as follows: E = (FR basal − FR post)·100/FR initial, where FR basal is the average FR recorded for 60 s before the agonist application, FR post is the average FR recorded for 60 s after the agonist and FR initial is the FR of each cell at the beginning of the recording. Normalization was employed to obtain comparable measures of the effect across groups. Analysis of a 60 s period after administration of the agonist integrates the whole period of inhibition and its recovery, and so it allows us to obtain a good indication of the maximal effect of the agonist and discriminate between different effects even when a complete cessation of the FR is observed (Llorente et al., 2012).
To calculate the functional turnover of μ receptors, we used the approach proposed by Mauger et al. (1982), which is based on the recovery of the effect after complete blockade of the receptor with an alkylating drug. This approach assumes that the receptor recovery is the difference between the reappearance rate, which is constant, and the disappearance rate, which is proportional to the total receptors at each time (Mauger et al., 1982). It provides two advantages over other methodologies: (i) alkylating drugs are more specific for receptors and less toxic for the neuron (as compared with metabolic inhibitors) and (ii) functional techniques are more suitable for evaluating the relevance of receptor turnover (as opposed to binding studies). Herein, after complete μ receptor blockade with β‐FNA, the recovery of opioid effect was followed for a 300 min period of β‐FNA washout, thereby obtaining the response values to ME (3.2 μM) (E) at each time (t) as stated above. The rate constants of functional reappearance (r′) and functional disappearance (k′) were estimated by nonlinear analysis of the [t, E] pairs to the following exponential function (equation 1): E = r′/k′·(1 − e ‐k′·t) (Mauger et al., 1982; Pineda et al., 1997). The percentage of maximal functional recovery at steady state (E ss), which is the response value to ME (3.2 μM) when time tends to infinity, was calculated as r′/k′. The t 1/2 of functional recovery (t 1/2 ′) was determined from the expression L n2/k′. To evaluate the parameters defining the turnover of the receptor (μ receptor appearance and μ receptor degradation), we first quantified the percentage of functionally active μ receptors at each β‐FNA post‐application time (R t) according to the operational model of Black and Leff (1983), as follows (equation 2): R t = [(K app + A)·K E/A]·[E t/(100 − E t)]1/n (Kenakin, 2006; Kelly, 2013). In the latter function, E t (%) is the experimental effect of ME (3.2 μM; A) at each β‐FNA post‐application time and K E (%) is the occupancy parameter that reaches a half‐maximal effect in the occupancy–effect curve for ME as calculated from the function: K E = 100/{[(K app/EC50) + 1]n − 2}1/n (equation 3); K app is the dissociation constant value for ME obtained from Osborne and Williams (1995) (5.2 μM), and EC50 and n are the parameter values of the concentration–effect curve for ME obtained from Santamarta et al. (2005) (0.15 μM and 1.15 respectively). Finally, the rate constants of receptor appearance (r) and receptor degradation (k) were estimated by nonlinear analysis of functionally active μ receptors (R t) at the different post‐application times (t) with the following function (equation 4): R t = r/k·(1‐e ‐k·t) (Mauger et al., 1982). In this case, the amount of functionally active μ receptor at the steady state (R ss) was calculated as r/k, and the receptor t 1/2 was l n2/k.
Data are expressed as the mean ± SEM of n number of rats. Statistical significances were obtained by a two‐tailed paired Student's t‐test when the FRs or response values were compared before and after test applications within the same cell, and by a two‐tailed two‐sample Student's t‐test when the FRs, response values or turnover parameters were compared between two independent experimental conditions. Response values or basal FRs were statistically compared among more than two experimental conditions by one‐way ANOVA followed by post hoc comparisons in pairs with the Bonferroni's multiple comparison test. Post hoc tests were run only if F achieved the necessary level of statistical significance (i.e. P < 0.05) and there was no significant variance inhomogeneity. The threshold of significance was considered as P = 0.05. Only one level of probability (P < 0.05) is reported.
Materials and drugs
For electrophysiological recordings, the following drugs were used (drug source): brefeldin A (Tocris Bioscience, Bristol, UK), EEDQ (Sigma‐Aldrich Química S.A., Madrid, Spain), β‐FNA hydrochloride (Tocris Bioscience), ME acetate salt (Bachem, Weil am Rhein, Germany) and (−)–NA (+)–bitartrate salt hydrate (Sigma‐Aldrich Química S.A.). Stock solutions of brefeldin A and EEDQ were first prepared in pure DMSO and then diluted in aCSF to obtain a final concentration of 0.1% DMSO. It has been previously shown that 0.1% DMSO does not affect LC cell firing responses (Pineda et al., 1996a). Stock solutions of the rest of the drugs were first prepared in Milli‐Q water and then diluted 1000 to 10 000 fold in aCSF to the desired concentration. Final solutions were prepared freshly just before each experiment, and stock solutions were kept at −20°C. For immunocytochemistry, the following compounds were used (drug source): BSA (Sigma‐Aldrich Química S.A.), Cy5 650‐conjugated donkey anti‐rabbit and Dylight 549‐conjugated donkey anti‐mouse fluorescent antibodies (Jackson ImmunoResearch, Suffolk, UK), mouse anti‐TH (Immunostar), Mowiol (Vector Labs, Peterborough, UK) and rabbit anti‐μ receptor antiserum (Millipore).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Kinetic characterization of functional turnover of μ receptors in the LC
To study the kinetics of μ receptor functional turnover, we followed the recovery of ME effect during a 300 min washout period after complete receptor inactivation with a 30 min administration of β‐FNA (300 and 800 nM) (Figure 1A). Before β‐FNA (300 nM), supramaximal concentrations of ME (3.2–12.8 μM, 1 min) inhibited the FR of LC cells by more than 89% (P < 0.05) (Figure 2A; control in Table 1). The effect of ME (3.2–12.8 μM, 1 min) was almost completely blocked immediately after β‐FNA (300 nM, 30 min) (Figure 2A; see t 30 = 0 in Table 1 for ME 3.2 μM). A higher concentration of β‐FNA (800 nM, 30 min) did not further block ME effect immediately after its administration (Figure 2A; t 30 = 0 in Table 1), confirming the near‐complete and insurmountable μ receptor inactivation achieved by β‐FNA. During the 300 min washout of β‐FNA (300 and 800 nM), the inhibitory effect of ME (3.2 μM) gradually recovered (P < 0.05 vs. t 30 = 0), although it did not reach the control effect found before β‐FNA (t 30 = 300 in Table 1, P < 0.05 vs. control; Figure 2B). The parameters defining functional turnover of opioid response (r′, k′, t ½ ′ and E ss) were obtained by analysing the time course for ME effect recovery with the equation 1 (see the Methods section). Thus, the t 1/2 values for opioid response recovery were shorter than 40 min (t 1/2 ′ in Table 1), whereas the steady‐state recoveries of opioid response during the 300 min period were higher than 60% (E ss in Table 1), suggesting that μ receptor turnover is a fast and efficient process. Finally, turnover parameters were not different when estimated after both β‐FNA (300 or 800 nM) concentrations (Table 1), which argues against the idea that functional recovery is due to β‐FNA dissociation from the receptor during washout.
Figure 2.
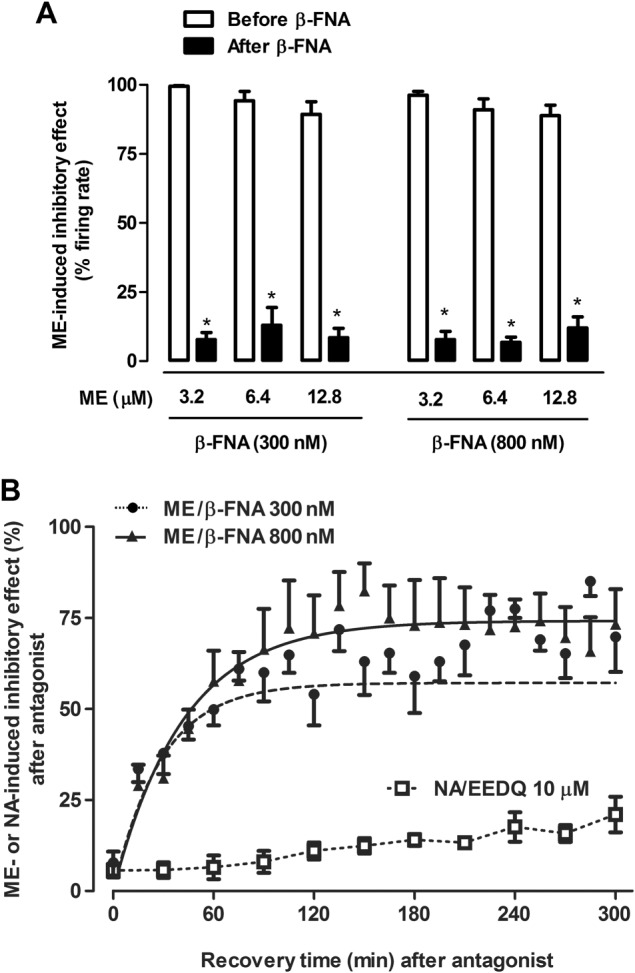
Effect of the opioid agonist ME before and after administration of the irreversible μ receptor antagonist β‐FNA in LC neurons in vitro. (A) Effect of β‐FNA (300 and 800 nM, 30 min) on the inhibition induced by supramaximal concentrations of ME (3.2, 6.4 and 12.8 μM, 1 min). Bars represent the mean ± SEM of ME effect before and after β‐FNA perfusion expressed as a percentage of the initial FR. Note that the inhibitory effect of all supramaximal ME concentrations is almost completely blocked after application of β‐FNA (300 and 800 nM; n = 5). *P < 0.05 compared with the control effect of ME before β‐FNA administration (Student's paired t‐test). (B) Recovery of ME (3.2 μM, 1 min) and NA (100 μM, 1 min)‐induced inhibitory effect following irreversible inactivation of μ receptors or α2‐adrenoceptors by β‐FNA (300 and 800 nM) and EEDQ (10 μM) respectively. Symbols represent the mean ± SEM of the percentage of ME or NA‐induced inhibitory effect after β‐FNA administration (n = 5 in all experimental groups). The horizontal axis represents the time after the end of β‐FNA (300 and 800 nM) or EEDQ (10 μM) application and the vertical axis the inhibitory effect of ME (3.2 μM, 1 min) or NA (100 μM, 1 min) expressed as a percentage of the initial FR. The lines of the figure represent the theoretical curves constructed from the average of the recovery kinetic parameters that were estimated individually by nonlinear regression (Table 1). Since the recovery of NA‐induced inhibitory effect was small, the kinetic parameters could not be calculated. Note that β‐FNA (300 and 800 nM) administration markedly blocks the inhibitory effect of ME (at t = 0), but the effect recovers to the same degree with both concentrations of the antagonist. The recovery of NA‐induced inhibitory effect (100 μM, 1 min) after perfusion with EEDQ (10 μM) was slower and less efficient.
Table 1.
Kinetic parameters of functional turnover for the recovery of ME (3.2 μM, 1 min)‐induced inhibitory effect after β‐FNA (300 and 800 nM, 30 min) administration
| β‐FNA 300 nM (n = 5) | β‐FNA 800 nM (n = 5) | |
|---|---|---|
| ME effect (%) | ||
| Control | 99.5 ± 0.3 | 96.2 ± 1.4 |
| t 30 = 0 min | 7.8 ± 2.6 | 7.8 ± 3.0 |
| t 30 = 300 min | 69.8 ± 9.6 | 73.3 ± 9.6 |
| Functional turnover of ME effect | ||
| r′ (Emax · min−1) | 2.00 ± 0.13 | 1.78 ± 0.32 |
| k′ (min−1) | 0.035 ± 0.007 | 0.024 ± 0.005 |
| t ½′ (min) | 22 ± 3 | 36 ± 8 |
| E ss (%) | 62.0 ± 7.9 | 77.0 ± 7.1 |
| Functional turnover of μ receptor | ||
| r (R t · min−1) | 0.25 ± 0.06* | 0.52 ± 0.20* |
| k (min−1) | 0.022 ± 0.009 | 0.018 ± 0.004 |
| t ½ (min) | 45 ± 9* | 44 ± 9 |
| R ss (%) | 16 ± 3* | 37 ± 15* |
Values are expressed as mean ± SEM obtained by nonlinear regressions of n cells (see the Methods section). Parameters: r′ and k′ are the reappearance and disappearance rate constants of ME effect, t ½ ′ is the t 1/2 of ME effect, E ss is the maximal recovery of ME effect at steady state, r and k are the reappearance and disappearance rate constants of the receptor, t ½ is the t 1/2 of the receptor and R ss is the maximal recovery of the receptor at steady state.
P < 0.05, when compared with the corresponding parameter values for turnover of ME effect. No significant differences were found between the effects of the two concentrations of β‐FNA (300 and 800 nM).
We estimated the percentage of functionally active receptors after β‐FNA (300 and 800 nM) at each washout time point (R t) by substituting in equation 2 the experimental E values for ME (3.2 μM) (see the Methods section) and the parameters n (1.15 from Santamarta et al., 2005), K app (5.2 μM from Osborne and Williams, 1995) and K E (2.89% obtained by equation 3 with the EC50 being 0.15 μM from Santamarta et al., 2005). Next, we calculated the parameters that define the functional turnover of the receptor (r, k, t 1/2 and R ss) by analysing the time course for recovery of functionally active receptors (R t) with equation 4 (see the Methods section). The t 1/2 for receptor recovery obtained after β‐FNA (300 nM) was significantly slower (t 1/2 in Table 1) than the corresponding t 1/2 for opioid response recovery (t 1/2′). The steady‐state recoveries of functionally active receptors (R ss) after β‐FNA (300 and 800 nM) were significantly lower (R ss in Table 1) than the corresponding recoveries of opioid response (E ss). Reappearance rate constants after β‐FNA (300 and 800 nM) were also smaller for the receptor recovery (r in Table 1) than for opioid response recovery (r′). Parameters of receptor turnover were not significantly different when estimated after both β‐FNA (300 or 800 nM) concentrations (Table 1). The differences between response and receptor turnover parameters may reflect the presence of a large fraction of μ receptor reserve for ME effect in the LC.
In contrast to the rapid and efficient functional turnover of μ receptors in the LC, the effect mediated by α2‐adrenoceptors recovered very slowly during the washout of the irreversible receptor blocker EEDQ (10 μM, 30 min) (Figure 1B). Thus, immediately after perfusion with EEDQ (10 μM) (t 30 = 0), the inhibitory effect of NA (10 μM, 1 min) was blocked by 93.6 ± 2.1% (n = 5, P < 0.05), whereas after 300 min of EEDQ (10 μM) washout (t 30 = 300), the inhibitory effect of NA (10 μM) only recovered up to 21.0 ± 4.9% (n = 5, P < 0.05 vs. t 30 = 0) (Figure 2B). Hence, μ receptor turnover in LC neurons is faster and more efficacious than the turnover of the analogous Gi/o‐coupled α2‐adrenoceptor.
Dependence of μ receptor turnover on constitutive receptor trafficking and a depletable receptor pool in LC neurons
Regardless of the duration of β‐FNA pre‐administration, analysis of μ receptor turnover should provide equivalent results on the condition that the magnitude of receptor inactivation is constant at the beginning of the assay (Mauger et al., 1982). To confirm this hypothesis, we compared four duration schedules of β‐FNA (300 nM) pre‐exposure (15, 30, 45 and 60 min) and evaluated the degree of ME (3.2 μM, 1 min) effect recovery 120 min after μ receptor inactivation (i.e. t 15, 30, 45, 60 = 120 respectively) (Figure 1C). This design was chosen to cover perfusion periods shorter and longer than the estimated t ½ ′. As expected, ME effect was almost fully blocked immediately after the four β‐FNA pre‐administrations and no significant difference was found in the amount of this blockade (Figure 3A–C; see t 15–60 = 0 in Table 2). In contrast, the recovery of ME effect during the 120 min of β‐FNA washout was significantly reduced after longer β‐FNA pre‐administrations. Thus, ME effect almost fully recovered after 15 min of β‐FNA pre‐exposure (Figures 3A,B; t 15 = 120 in Table 2), but it recovered to a progressively lesser degree after 30, 45 or 60 min of β‐FNA pre‐administrations (Figure 3A,C; t 30, 45, 60 = 120 in Table 2). The parameters defining functional turnover of opioid response (r′, k′, t ½ ′ and E ss) were obtained in these groups (t 15–60 = 120) by fitting equation 3 to the experimental ME effect values after β‐FNA pre‐administrations (see the Methods section). E ss value in the 15 min pre‐exposure group (t 15 = 120 in Table 2) was close to control (100%), but it was significantly greater than the values in the 30, 45 and 60 min pre‐exposure groups (see t 30,45,60 = 120 in Table 2), suggesting that the longer the pre‐exposure to the blocker, the lesser the recovery of ME effect. Changes in E ss values in these groups were paralleled by reductions in the rate constants of functional reappearance (r′), but not in the rate constants of functional disappearance (k′) (Table 2). Taken together, β‐FNA pretreatment times longer than the t ½ ′ for μ receptor turnover were followed by slower and less complete opioid response recoveries due to reduced reappearance rates of the effect. These results disagree with the predictions of Mauger et al. (1982) in that functional μ receptor turnover depends on the duration of the β‐FNA pretreatment. However, this dependency could be explained by the presence of a constitutive recycling of receptors. To evaluate this hypothesis, we compared the recovery of opioid effect after β‐FNA (300 nM, 15 min) pre‐administration in a neuron that had already been subjected to a previous test with β‐FNA (300 nM, 15 min) and a 120 washout period (i.e. t 15 = 120) (Figure 1D). Thus, during the 120 min washout, the ME (3.2 μM, 1 min) effect recovered to a significantly smaller degree after the second β‐FNA challenge (26.7 ± 2.8%) than after the first one (77.9 ± 3.6%; n = 5, P < 0.05) (Figure 4A, B). The parameters defining functional turnover of opioid response were calculated in these two groups (t 15 = 120) as above. Thus, the E ss obtained after the second pre‐administration was significantly smaller (27.1 ± 3.8%) than that after the first one (85.9 ± 9.3%; n = 5; P < 0.05), which indicates that the recovery of ME response from β‐FNA blockade was partially lost in those cells that had recovered from a previous μ receptor inactivation. Therefore, μ receptor turnover may depend on a depletable pool of receptors that would allow a continuous recycling of μ receptors to the cell membrane under control conditions.
Figure 3.
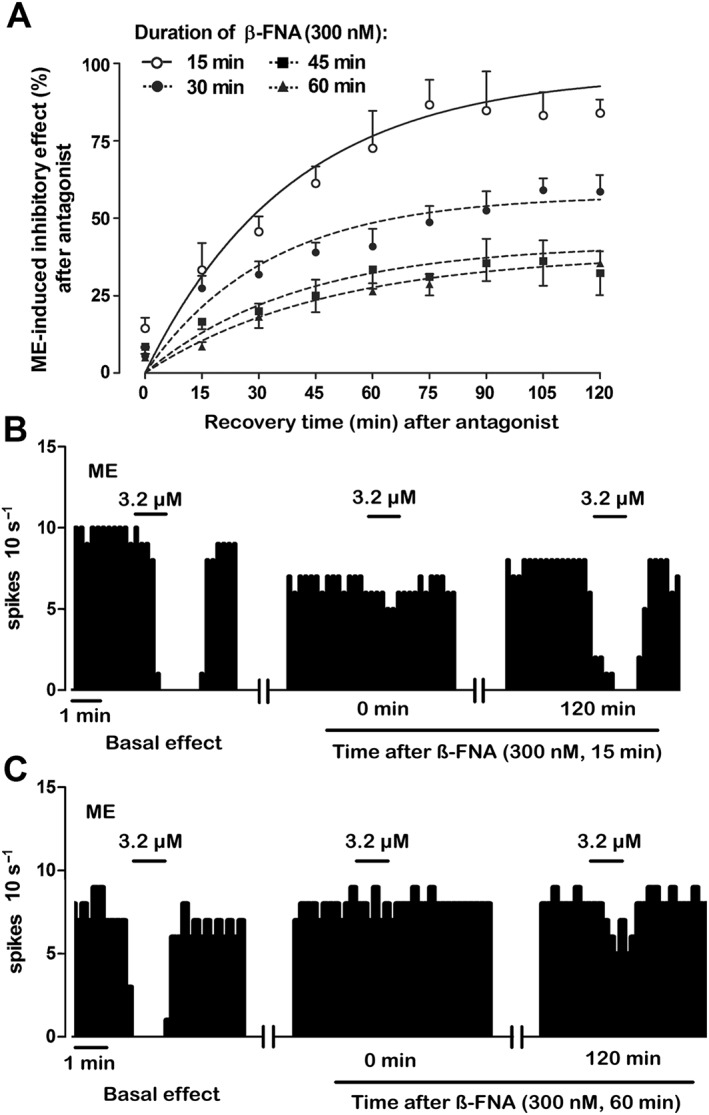
Recovery of the inhibitory effect of ME (3.2 μM, 1 min) following perfusion with the irreversible μ receptor antagonist β‐FNA (300 nM) for different time periods. (A) Symbols represent the mean ± SEM of the percentage of ME‐induced inhibitory effect after β‐FNA perfusion. The horizontal axis represents the time after the end of antagonist application. The vertical axis indicates the inhibitory effect of ME (3.2 μM, 1 min) expressed as a percentage of the initial FR. The lines represent the theoretical curves constructed from the average of the recovery kinetic parameters that define the functional turnover of μ receptors (Table 2). Note that ME effect after 15 min of β‐FNA perfusion (n = 6) at t = 120 min is higher than that obtained after 30 min (n = 8), 45 min (n = 5) or 60 min (n = 5) of β‐FNA administration at t = 120 min. (B, C) Representative examples of FR recordings of LC neurons showing the recovery of ME‐induced inhibitory effect (3.2 μM, 1 min) after complete inactivation of μ receptors with β‐FNA (300 nM, 15 min) (B) and β‐FNA (300 nM, 60 min) (C). Vertical lines represent the number of spikes recorded every 10 s and the horizontal bars the period of drug application. Note that the inhibitory effect of ME is almost blocked 15 min after β‐FNA administration in both recordings in comparison with the control inhibitory effect. The inhibitory effect of ME completely recovers in the neuron receiving β‐FNA (300 nM, 15 min) but not in the neuron perfused with β‐FNA (300 nM, 60 min).
Table 2.
Kinetic parameters of functional turnover for the recovery of ME (3.2 μM, 1 min)‐induced inhibitory effect after different exposure times to β‐FNA (300 nM, 15–60 min)
| β‐FNA 300 nM | ||||
|---|---|---|---|---|
| 15 min (n = 6) | 30 min (n = 8) | 45 min (n = 5) | 60 min (n = 5) | |
| ME effect (%) | ||||
| Control | 100 ± 0.0 | 99.4 ± 0.4 | 100 ± 0.0 | 88.5 ± 9.1 |
| t 15–60 = 0 min | 14.7 ± 3.3 | 5.5 ± 2.6 | 8.5 ± 2.3 | 5.0 ± 3.6 |
| t 15–60 = 120 min | 84.0 ± 4.4 | 58.6 ± 5.4* | 31.0 ± 7.1* | 35.4 ± 3.9* |
| Functional turnover of ME effect | ||||
| r′ (Emax · min−1) | 2.52 ± 0.64 | 1.78 ± 0.29 | 1.04 ± 0.34 | 0.81 ± 0.22* |
| k′ (min−1) | 0.026 ± 0.006 | 0.031 ± 0.006 | 0.025 ± 0.009 | 0.021 ± 0.005 |
| t ½ ′ (min) | 33 ± 6 | 32 ± 8 | 40 ± 10 | 46 ± 13 |
| E ss (%) | 97.3 ± 6.4 | 62.0 ± 5.7* | 45.1 ± 11.6* | 39.5 ± 4.67* |
Values are expressed as mean ± SEM obtained by nonlinear regressions of n cells (see the Methods section). Parameters: r′ and k′ are the reappearance and disappearance rate constants of ME effect, t ½ ′ is the t 1/2 of ME effect and E ss is the maximal recovery of ME effect at steady state.
P < 0.05, when compared with β‐FNA (15 min) by a one‐way ANOVA followed by a post hoc Bonferroni's multiple comparison test.
Figure 4.
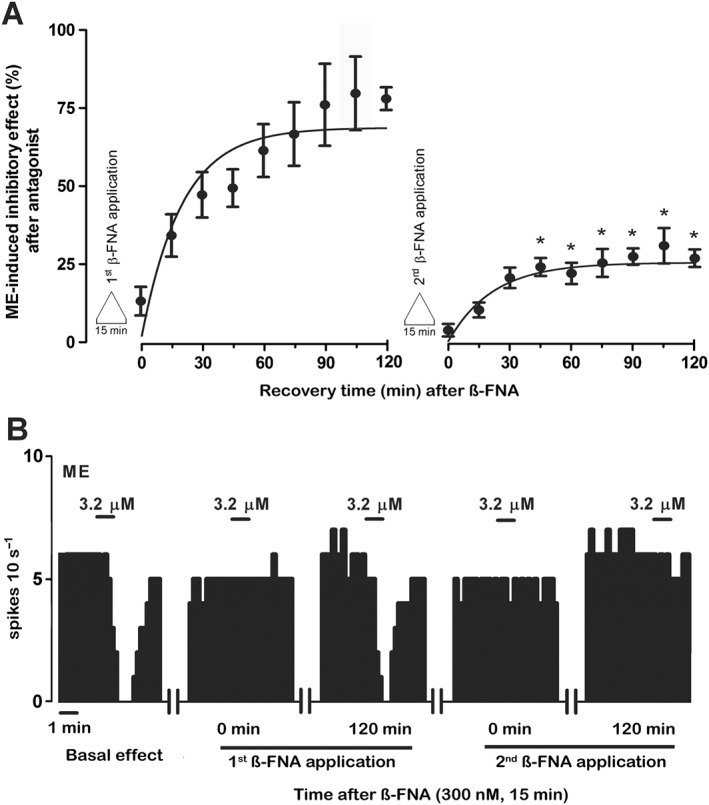
Recovery of the inhibitory effect of ME (3.2 μM, 1 min) following two successive administrations of β‐FNA (300 nM, 15 min). (A) Symbols represent the mean ± SEM of the percentage of ME‐induced inhibitory effect. The horizontal axis represents the time after the end of β‐FNA (300 nM, 15 min) application, whereas the vertical axis indicates the inhibitory effect of ME (3.2 μM, 1 min) expressed as a percentage of its initial FR. The lines of the figure represent the theoretical curve constructed from the average of the recovery kinetic parameters that define the functional turnover of μ receptors. Note that the recovery of the inhibitory effects of ME after the first β‐FNA application is significantly higher than that of the inhibitory effects of ME achieved after the second antagonist perfusion (n = 5; *P < 0.05; Student's paired t‐test). (B) Representative example of FR recording of LC neuron showing the recovery of the inhibitory effect of ME (3.2 μM, 1 min) after two successive administrations of β‐FNA (300 nM, 15 min). Vertical lines represent the number of spikes recorded every 10 s and the horizontal bars the period of drug application. Note that the inhibitory effect of ME is almost blocked 15 min after both β‐FNA administrations. The inhibitory effect of ME completely recovers to the control effect within 120 min after the first β‐FNA administration but not after the second β‐FNA application.
Molecular mechanisms involved in the functional turnover of μ receptors in the LC
To study the molecular mechanisms of μ receptor turnover, we evaluated the recovery of opioid response after μ receptor inactivation under different experimental conditions that are known to block synaptic trafficking: low calcium (2 mM)‐containing aCSF perfusion, neuron activity inhibition (by NA 100 μM), slice temperature lowering (to 22°C) and vesicle trafficking inhibition (by brefeldin A 10 μM). ME effect (3.2 μM, 1 min) was tested before, immediately after μ receptor inactivation with β‐FNA (300 nM, 15 min) (t 15 = 0) and following 45 min of β‐FNA washout (t 15 = 45) (Figure 1E). To remove any interference from acute effects, the slices underwent the experimental manipulations only during the recovery phase from μ receptor inactivation, but not during the testing of ME effect. Immediately after β‐FNA administration (t 15 = 0), the basal FRs of LC cells ranged from 0.42 ± 0.08 to 1.07 ± 0.25 Hz and the degree of inhibition of the ME effect varied from 84.9 ± 4.6 to 94.8 ± 2.2% in the different experimental groups (control, basal FR: 0.55 ± 0.09 Hz, blockade of ME effect: 85.3 ± 3.3%). As expected, these values of basal FR and opioid blockade were not significantly different between groups (one‐way ANOVA: F4,22 = 2.64 and F4,22 = 1.28, respectively; see Figure 5). However, in the presence of low calcium‐containing aCSF, the recovery of ME effect during the 45 min of β‐FNA washout (t 15 = 45) was reduced by 51 ± 17% (n = 6, P < 0.05) with respect to the control (n = 6) (Figure 5A, B). Moreover, when neuron firing activity was experimentally inhibited by NA (100 μM) during the β‐FNA washout (t 15 = 45), the recovery of ME effect was reduced by 85 ± 12% (n = 6, P < 0.05 vs. control) (Figure 5A, B). Finally, when the slice temperature was lowered to 22°C or brefeldin A (10 μM) was perfused during the β‐FNA washout (t 15 = 45), the recovery of ME effect was reduced by 46 ± 11% (n = 5, P < 0.05 vs. control) and 66 ± 8% (n = 5, P < 0.05 vs. control) respectively (Figure 5A, B). These manipulations did not modify acute ME effects in slices that had not been pre‐exposed to β‐FNA (ME effect at t 0 = 45 ranged from 95.5 to 99.7%, n = 5). This indicates that functional turnover of μ receptors, but not ME or β‐FNA effects, is attenuated by restraining calcium‐dependent mechanisms, neuron activity or vesicle trafficking, which suggests that μ receptor turnover in the LC may depend on the presence of a constitutive trafficking of neuronal receptors.
Figure 5.
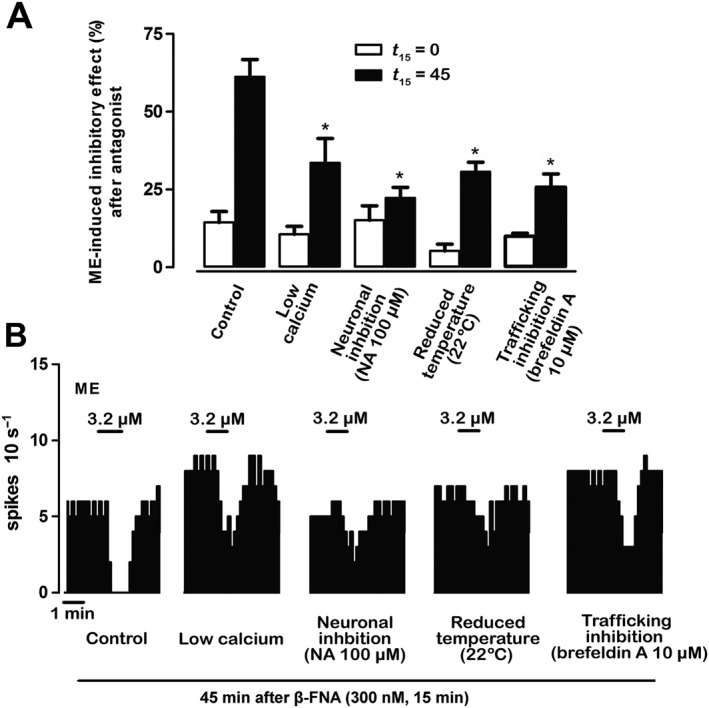
Intracellular mechanisms of the functional turnover of μ receptors. (A) Bar histograms show the mean ± SEM of the percentage of ME‐induced effect immediately (t 15 = 0) and 45 min after 15 min (t 15 = 45) of β‐FNA (300 nM, 15 min) perfusion in different experimental conditions: control (n = 6), in the presence of low‐calcium aCSF (n = 6), during neuronal inhibition by NA (100 μM) (n = 6), at low temperature (n = 5) or during trafficking inhibition by brefeldin A (10 μM) (n = 5). *P < 0.05, compared with the corresponding control by a one‐way ANOVA followed by Bonferroni's post hoc test. (B) Representative examples of FR recordings of five LC neurons showing the recovery of ME (3.2 μM, 1 min)‐induced inhibitory effect 45 min after β‐FNA (300 nM, 15 min) application in different experimental conditions. Vertical lines represent the number of spikes recorded every 10 s and the horizontal bars the period of drug application. Note that the inhibitory effect of ME recovers 45 min after β‐FNA administration only in the control group.
Immunocytochemistry for confocal laser scanning microscopy
To characterize μ receptor turnover morphologically in LC neurons, we examined, by immunofluorescent labelling combined with confocal laser scanning microscopy, the differential expression of μ receptors in neuronal membranes and the cytoplasm of TH‐immunopositive noradrenergic neurons under several experimental conditions: control (before β‐FNA perfusion), immediately after β‐FNA (300 nM, 15 min) (t 15 = 0), 45 min after β‐FNA (300 nM, 15 min) (i.e. after a washout period of 45 min; t 15 = 45) and immediately after β‐FNA (300 nM, 60 min) (t 60 = 0). These settings were chosen because they showed different degrees of functional μ receptor turnover in electrophysiological assays (see above). Under control conditions, abundant μ receptor‐immunopositive structures were found within the neuronal somata of TH‐containing neurons (i.e. the cytoplasm), as well as associated with the external surface of the neuron (i.e. the cell membrane) (control in Figure 6). Immediately after β‐FNA, there was a marked depletion of μ receptor immunoreactivity in the cell membrane of TH‐containing neurons, but μ receptor‐immunoreactive puncta were still observed within the cytoplasm (t 15 = 0 in Figure 6). In contrast, after 45 min of β‐FNA washout, μ receptor immunoreactivity was identified only in close association with the cell membrane of TH‐containing neurons, but not within the cytoplasm (t 15 = 45 in Figure 6). Finally, immediately after β‐FNA (300 nM, 60 min), no immunoreactivity for the μ receptor was detected in any compartment of TH‐containing neurons (t 60 = 0 in Figure 6). Therefore, μ receptor expression on the cell membrane vanishes immediately after a short (15 min) exposure to β‐FNA but reappears after 45 min of β‐FNA washout. The fact that cytoplasmic μ receptor expression follows the opposite pattern to membrane expression (i.e. almost unchanged after a short exposure to β‐FNA, but largely reduced after β‐FNA washout) indicates that recovery of new functional receptors during washout may occur at the expense of μ receptor recycling from a cytoplasmic pool. A β‐FNA perfusion longer than the t ½ ′ (45 min) depletes both cytoplasmic and cell surface μ receptors by removing the trafficking mechanisms from the cytoplasmic compartment.
Figure 6.
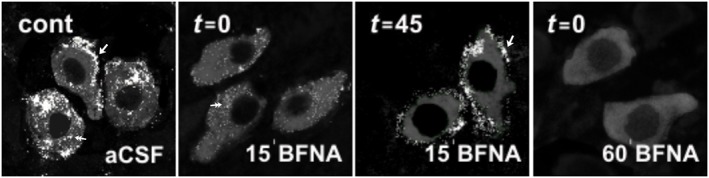
Confocal laser scanning microscopy imaging of μ receptor‐immunoreactivity (ir) in the LC before and after β‐FNA perfusion. In the absence of β‐FNA (control condition, cont), μ receptor‐ir (white) is identified as patches distributed both in the cytoplasm (double arrow) and in association with the plasma membrane (single arrow) in TH‐containing neurons (grey). Immediately after treatment with ß‐FNA (300 nM, 15 min, t 15 = 0), μ receptor‐ir is markedly depleted from the neuronal membrane, but μ receptor‐immunoreactive puncta are still observed within the cytoplasm (double arrow). Forty five min later (t 15 = 45), the μ receptor is present again in the cell membrane (single arrow). Immediately after ß‐FNA (300 nM, 60 min, t 60 = 0) application, μ receptor‐ir is not detected in the cell membrane or the cytoplasmic compartment. All images displayed were acquired with identical laser intensity and detector gain parameters.
Discussion
The present work was undertaken to characterize the functional turnover of μ receptors in noradrenergic neurons of the rat brain in vitro. For this purpose, we evaluated the inhibitory effect of the opioid agonist ME on the FR of LC neurons before and after complete μ receptor inactivation with β‐FNA. ME was used to obtain a functional index of μ receptors in the LC, as its effect is mediated almost exclusively by μ receptors and it washes out very rapidly even at high concentrations (Williams and North, 1984). β‐FNA is an alkylating derivative of naltrexone that has been well characterized as an irreversible and selective μ receptor antagonist in the brain (Portoghese et al., 1980; Ward et al., 1985; Chen et al., 1996). Accordingly, we found a near‐maximal and insurmountable inactivation of ME's effect immediately after β‐FNA (300 or 800 nM). However, a rapid and efficient recovery of ME's effect was observed during the 5 h of β‐FNA washout. The time course of functional recovery was analysed by an exponential function (equation 1; Mauger et al., 1982) to calculate the kinetic parameters defining μ receptor turnover for ME effect. We have previously reported the advantages of this strategy for other receptors in the LC (Pineda et al., 1997).
Analysis of the steady‐state recovery revealed that, 5 h after μ receptor inactivation, LC neurons spontaneously recover a significant fraction of the control response (E ss = 62%). Indeed, μ receptor function recovered very rapidly, as a 50% of ME effect was restored within less than 30 min of β‐FNA washout (t 1/2 ′ = 22 min). As predicted by Mauger et al. (1982), we obtained equivalent estimates of E ss and t 1/2 ′ irrespective of whether they were calculated with 300 or 800 nM of β‐FNA, which indicates that the recovery of μ receptor function is secondary to reappearance of new receptors rather than to dissociation of β‐FNA from μ receptor. In experiments in which the recovery of ME effect was monitored for longer than 5 h, we observed a nearly complete recovery of ME effect during longer β‐FNA washouts (t 30 = 10 and 15 h, recovery = 95.0 ± 2.2 and 97.8 ± 1.2%, respectively; n = 5; data not shown). This suggests that μ receptor turnover follows a double‐phase model: a fast recovery of ME effect during the first h and a late recovery of the remaining ME effect within 15 h. In contrast, functional recovery of α2‐adrenoceptors, as herein measured by the full agonist NA after receptor inactivation with the alkylating agent EEDQ, was much slower and less efficacious than that of μ receptors, despite the fact that both inhibitory receptors are similarly coupled to Gi/o proteins in the LC (Aghajanian and Wang, 1987). EEDQ has been shown to inactive α2‐adrenoceptors and, to a lesser extent, α1‐adrenoceptors (Meller et al., 1988), which are both present in the LC (Nörenberg et al., 1997; Osborne et al., 2002). However, the inhibitory effect of NA on the firing of LC neurons is almost exclusively mediated by somadendritic α2‐adrenoceptors (majorly of the α2A‐subtype), so that α1‐adrenoceptor antagonists fail to affect NA‐induced inhibition (Nörenberg et al., 1997). Therefore, our results on α2‐adrenoceptors opposing those of μ receptor turnover are consistent with a slower turnover rate of α2A‐adrenoceptors (t 1/2 > 5 h). Likewise, slow turnover rates and long half‐lives for recovery of α2A‐adrenoceptors have been found by electrophysiological recordings of LC neurons in vivo (t 1/2 ′ = 14 h; Pineda et al., 1997) and also by binding techniques in other areas of the rat brain (t 1/2 > 50 h; Barturen and Garcia‐Sevilla, 1992). Interestingly, immunocytochemical staining techniques in culture cells have demonstrated that most of the cellular α2A‐adrenoceptors are localized in the plasma membrane after insertion directly from the biosynthetic pathway (Daunt et al., 1997). The observation that only a small amount of α2A‐adrenoceptors undergoes trafficking from an intracellular pool diverges from the relevance of this mechanism proposed herein for μ receptors (see below) and may underlie the differential turnover rates of both inhibitory receptors.
To study the parameters defining the turnover of the receptor itself, we calculated the percentage of functionally active μ receptors at each β‐FNA post‐application time and then fitted the time course for receptor recovery to an exponential function (equation 4). This analysis revealed that turnover of the receptor is less efficient (R ss < E ss) and slower (t 1/2 > t 1/2 ′) than turnover of μ receptor‐mediated effect, which could be explained by the presence of spare receptors for ME's effect in LC neurons (K E = 2.89%). In agreement, substantial fractions of spare receptors for different μ receptor agonists have been shown in the LC (Williams and North, 1984). A dichotomy between receptor and functional turnovers for μ receptors has been also reported for heroin after inactivation with β‐FNA in vivo (Martin et al., 1995, 1998).
Analysis of receptor turnover should yield the same parameter values regardless of the pre‐exposure time of the irreversible antagonist, provided that the degree of receptor inactivation is equivalent at the beginning of the experiment. Unexpectedly, we found that the longer the duration of β‐FNA pre‐administration, the less efficient the recovery of ME effect became (recovery: 84% vs. 31%, after 15 and 45 min of β‐FNA respectively). Longer β‐FNA administrations were accompanied by marked reductions in the reappearance rate (r′) and E ss values, but not in the disappearance rate (k′). This dependency of μ receptor turnover on β‐FNA pre‐exposure cannot be explained by different dissociation rates (k off) of μ receptors, since receptor alkylation by β‐FNA is irreversible (see above). Alternatively, we may consider the presence of a constitutive recycling of μ receptors into the cell membrane by trafficking from a neuronal cytoplasmic pool (functionally named as ‘endosome’; Nagi and Piñeyro, 2011). Thus, the longer the pre‐exposure time to β‐FNA, the greater the choice of endosomal receptors continuously moving into the membrane to be blocked and depleted by β‐FNA and thereby the lesser the efficiency of maximal μ receptor recovery. We supported this hypothesis by evaluating the recovery of opioid effect after two consecutive β‐FNA administrations in the same cell. Thus, the steady‐state recovery of ME effect (E ss) was reduced from 85.9 (after the first administration) to 27.1% (after the second exposure). This indicates that μ receptor turnover is partially lost and, hence, functional recovery is less efficient when the challenge to receptor blockade is repeated in the same cell, as if the reappearance of new functional μ receptor was disturbed by depletion of the endosomal receptors. Double immunocytochemistry experiments showed that control μ receptor staining was expressed on the cell surface and in the cytoplasm of TH‐containing neurons. The immunostaining of μ receptors vanished from the membrane (but not from the cytoplasm) immediately after a short β‐FNA administration (15 min), while it reappeared again in the membrane after 45 min of β‐FNA washout at the expense of a depletion of cytoplasmic μ receptors. Finally, μ receptor immunoreactivity was depleted in both compartments after a longer exposure (60 min) to β‐FNA. Taken together, electrophysiological and immunocytochemical findings suggest that μ receptor turnover may depend on a constitutive trafficking of receptors to the cell membrane from a readily accessible and depletable cytoplasmic pool (Ferguson, 2001). Likewise, a substantial fraction of dendritic AMPA receptors (7%) is maintained in an intracellular pool where they are available for ongoing receptor trafficking or rapid synaptic recruitment (Shi et al., 1999). Similar trafficking processes have been reported for GABAA and GABAB receptors (Lüscher, 2002).
To explore the possible mechanisms underlying μ receptor turnover in the LC, we evaluated the recovery of ME's effect from μ receptor inactivation under different experimental conditions that affect the trafficking of synaptic particles. Thus, the recovery of opioid effect 45 min after μ receptor inactivation was strongly reduced when calcium‐dependent processes were blunted by a low‐calcium aCSF or LC neurons were inhibited by the α2‐adrenoceptor agonist NA. Low‐calcium conditions have been shown to block vesicle trafficking (Hay, 2007). A reduction in discharging activity by α2‐adrenoceptor activation in the LC (Pineda et al., 1996b) should attenuate the spike‐dependent component of receptor trafficking, as proposed for AMPA receptors (Lledo et al., 1998). Moreover, the recovery of opioid effect from μ receptor inactivation was markedly attenuated when the experimental temperature was lowered to 22°C or the protein transport inhibitor brefeldin A (10 μM) was administered. Lowering the experimental temperature (~20°C) has been shown to stop trafficking of lumenal proteins to the membrane by blocking lipid mobilization (Simon et al., 1996; Park et al., 2011). Brefeldin A is a macrocyclic transport inhibitor that has been used to slow down vacuolar protein trafficking in the endomembrane system of eukaryotic cells (Law et al., 2000; Nebenführ et al., 2002). Therefore, these findings suggest that μ receptor turnover in the LC is a fast and highly efficient ongoing process that depends on calcium‐ and spike‐dependent mechanisms and also on the level of protein vesicle trafficking. We hypothesize that functional turnover may share key mechanisms with those of neurotransmitter release in nerve terminals (Neher, 1998; Gaffield and Betz, 2007).
In conclusion, our study shows a constitutive, rapid and efficient turnover of opioid effect after μ receptor inactivation with β‐FNA in the LC. Functional μ receptor turnover appears to be blunted by prolonged or repeated challenges to β‐FNA inactivation in the same cell. Double immunocytochemistry experiments confirmed that μ receptor staining localized in the cytoplasm of TH‐containing cell bodies rapidly moves to the cell membrane during β‐FNA washout. Finally, μ receptor turnover was slowed down by different procedures restraining vesicular protein trafficking, and consequently, it may be explained by constitutive trafficking of μ receptors from an accessible and depletable cytoplasmic pool (endosome) of receptors to the membrane of LC neurons. In the VTA, μ receptor trafficking contributes to the induction of synaptic plasticity during opiate withdrawal (Madhavan et al., 2010; Dacher and Nugent, 2011). In the LC, trafficking of μ receptors is required for resensitization of opioid responses and an impairment of this process contributes to morphine tolerance. A better understanding of how opioids regulate μ receptor turnover and trafficking would facilitate the comprehension of opioid plasticity and tolerance. Moreover, descendant pathways arising from the LC exert a potent supraspinal control of pain transmission (West et al., 1993; Hickey et al., 2014). Since μ receptor activation in the LC contributes to the analgesic effects of opioid agonists (Pertovaara, 2006; Jongeling et al., 2009), we consider that our findings may help to improve the pharmacotherapy of clinical pain.
Author contributions
M.C.M., M.T.S., A.M. and J.P. performed the electrophysiological assays and analysed the data. Z.A., I.B. and J.J.A. performed the immunocytochemistry. M.C.M., P.P., Z.A., I.B., A.M. and J.P. wrote the manuscript. J.P. conceived and designed the study.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by the Ministerio de Ciencia e Innovación [grant SAF2008‐03612] and the UPV/EHU [grant GIU14/29]. Pineda's research group takes part in a network unit supported by the UPV/EHU [UFI 11/35]. M.C.M., M.T.S. and P.P. were supported by predoctoral fellowships from the Basque Government. The experiments comply with the current laws of Spain.
Medrano, M. C. , Santamarta, M. T. , Pablos, P. , Aira, Z. , Buesa, I. , Azkue, J. J. , Mendiguren, A. , and Pineda, J. (2017) Characterization of functional μ opioid receptor turnover in rat locus coeruleus: an electrophysiological and immunocytochemical study. British Journal of Pharmacology, 174: 2758–2772. doi: 10.1111/bph.13901.
References
- Adler CH, Meller E, Goldstein M (1985). Recovery of alpha 2‐adrenoceptor binding and function after irreversible inactivation by N‐ethoxycarbonyl‐2‐ethoxy‐1, 2‐dihydroquinoline (EEDQ). Eur J Pharmacol 116: 175–178. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK, Wang YY (1987). Common alpha 2‐ and opiate effector mechanisms in the locus coeruleus: intracellular studies in brain slices. Neuropharmacology 26: 793–799. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R, Aghajanian GK (1984). Locus coeruleus activity in vitro: intrinsic regulation by a calcium‐dependent potassium conductance but not alpha 2‐adrenoceptors. J Neurosci 4: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R, Vandermaelen CP, Aghajanian GK (1983). Morphine tolerance and dependence in the locus coeruleus: single cell studies in brain slices. Eur J Pharmacol 91: 161–169. [DOI] [PubMed] [Google Scholar]
- Arttamangkul S, Quillinan N, Low MJ, von Zastrow M, Pintar J, Williams JT (2008). Differential activation and trafficking of micro‐opioid receptors in brain slices. Mol Pharmacol 74: 972–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Torrecilla M, Kobayashi K, Okano H, Williams JT (2006). Separation of mu‐opioid receptor desensitization and internalization: endogenous receptors in primary neuronal cultures. J Neurosci 26: 4118–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Kelly E, Henderson G (2004). Protein kinase C activation enhances morphine‐induced rapid desensitization of mu‐opioid receptors in mature rat locus coeruleus neurons. Mol Pharmacol 66: 1592–1598. [DOI] [PubMed] [Google Scholar]
- Barturen F, Garcia‐Sevilla JA (1992). Long term treatment with desipramine increases the turnover of alpha 2‐adrenoceptors in the rat brain. Mol Pharmacol 42: 846–855. [PubMed] [Google Scholar]
- Black JW, Leff P (1983). Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220: 141–162. [DOI] [PubMed] [Google Scholar]
- Chen C, Yin J, Riel JK, Desjarlais RL, Raveglia LF, Zhu J et al. (1996). Determination of the amino acid residue involved in [3H]beta‐funaltrexamine covalent binding in the cloned rat mu‐opioid receptor. J Biol Chem 271: 21422–21429. [DOI] [PubMed] [Google Scholar]
- Chu LF, D'Arcy N, Brady C, Zamora AK, Young CA, Kim JE et al. (2012). Analgesic tolerance without demonstrable opioid‐induced hyperalgesia: a double‐blinded, randomized, placebo‐controlled trial of sustained‐release morphine for treatment of chronic nonradicular low‐back pain. Pain 153: 1583–1592. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacher M, Nugent FS (2011). Opiates and plasticity. Neuropharmacology 61: 1088–1096. [DOI] [PubMed] [Google Scholar]
- Dang VC, Chieng B, Azriel Y, Christie MJ (2011). Cellular morphine tolerance produced by βarrestin‐2‐dependent impairment of μ‐opioid receptor resensitization. J Neurosci 31: 7122–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Christie MJ (2012). Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br J Pharmacol 165: 1704–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Williams JT (2004). Chronic morphine treatment reduces recovery from opioid desensitization. J Neurosci 24: 7699–7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daunt DA, Hurt C, Hein L, Kallio J, Feng F, Kobilka BK (1997). Subtype‐specific intracellular trafficking of alpha2‐adrenergic receptors. Mol Pharmacol 51: 711–720. [DOI] [PubMed] [Google Scholar]
- Doly S, Marullo S (2015). Gatekeepers controlling GPCR export and function. Trends Pharmacol Sci 36: 636–644. [DOI] [PubMed] [Google Scholar]
- Ferguson SS (2001). Evolving concepts in G protein‐coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev 53: 1–24. [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ (2007). Synaptic vesicle mobility in mouse motor nerve terminals with and without synapsin. J Neurosci 27: 13691–13700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay JC (2007). Calcium: a fundamental regulator of intracellular membrane fusion? EMBO Rep 8: 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey L, Li Y, Fyson SJ, Watson TC, Perrins R, Hewinson J et al. (2014). Optoactivation of locus ceruleus neurons evokes bidirectional changes in thermal nociception in rats. J Neurosci 34: 4148–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongeling AC, Johns ME, Murphy AZ, Hammond DL (2009). Persistent inflammatory pain decreases the antinociceptive effects of the mu opioid receptor agonist DAMGO in the locus coeruleus of male rats. Neuropharmacology 56: 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai S, Yamamoto H, Kamegaya E, Uhl GR, Sora I, Watanabe M et al. (2011). Quantitative detection of μ opioid receptor: western blot analyses using μ opioid receptor knockout mice. Curr Neuropharmacol 9: 219–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E (2013). Efficacy and ligand bias at the μ‐opioid receptor. Br J Pharmacol 169: 1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T (2006). Data‐driven analysis in drug discovery. J Recept Signal Transduct Res 26: 299–327. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N et al. (2005). Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol 67: 280–287. [DOI] [PubMed] [Google Scholar]
- Law PY, Erickson LJ, El‐Kouhen R, Dicker L, Solberg J, Wang W et al. (2000). Receptor density and recycling affect the rate of agonist‐induced desensitization of mu‐opioid receptor. Mol Pharmacol 58: 388–398. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, Südhof TC, Malenka RC, Nicoll RA (1998). Postsynaptic membrane fusion and long‐term potentiation. Science 279: 399–403. [DOI] [PubMed] [Google Scholar]
- Llorente J, Santamarta MT, Henderson G, Pineda J (2012). Enhancement of mu‐opioid receptor desensitization by nitric oxide in rat locus coeruleus neurons: involvement of reactive oxygen species. J Pharmacol Exp Ther 342: 552–560. [DOI] [PubMed] [Google Scholar]
- Lüscher B (2002). GABAA and GABAB receptors: regulation of assembly, localization, clustering and turnover In: Moss SJ, Henley J. (eds). Receptor and Ion‐channel Trafficking: Cell Biology of Ligand‐gated and Voltage‐sensitive Ion Channels. Oxford UP: New York, pp. 192–218. [Google Scholar]
- Madhavan A, He L, Stuber GD, Bonci A, Whistler JL (2010). Mu‐opioid receptor endocytosis prevents adaptations in ventral tegmental area GABA transmission induced during naloxone‐precipitated morphine withdrawal. J Neurosci 30: 3276–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TJ, DeMontis MG, Kim SA, Sizemore GM, Dworkin SI, Smith JE (1998). Effects of beta‐funaltrexamine on dose‐effect curves for heroin self‐administration in rats: comparison with alteration of [3H] DAMGO binding to rat brain sections. Drug Alcohol Depend 52: 135–147. [DOI] [PubMed] [Google Scholar]
- Martin TJ, Dworkin SI, Smith JE (1995). Alkylation of mu‐opioid receptors by beta‐funaltrexamine in vivo: comparison of the effects on in situ binding and heroin self‐administration in rats. J Pharmacol Exp Ther 272: 1135–1140. [PubMed] [Google Scholar]
- Mauger JP, Sladeczek F, Bockaert J (1982). Characteristics and metabolism of alpha 1 adrenergic receptors in a nonfusing muscle cell line. J Biol Chem 257: 875–879. [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller E, Goldstein M, Friedhoff AJ, Schweitzer JW (1988). N‐ethoxycarbonyl‐2‐ethoxyl‐1,2‐dihydroquinoline (EEDQ): A new tool to probe CNS receptor function. Adv Exp Med Biol 235: 121–136. [DOI] [PubMed] [Google Scholar]
- Mendiguren A, Pineda J (2004). Cannabinoids enhance N‐methyl‐D‐aspartate‐induced excitation of locus coeruleus neurons by CB1 receptors in rat brain slices. Neurosci Lett 363: 1–5. [DOI] [PubMed] [Google Scholar]
- Mendiguren A, Pineda J (2007). CB(1) cannabinoid receptors inhibit the glutamatergic component of KCl‐evoked excitation of locus coeruleus neurons in rat brain slices. Neuropharmacology 52: 617–625. [DOI] [PubMed] [Google Scholar]
- Morgan MM, Christie MJ (2011). Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br J Pharmacol 164: 1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagi K, Piñeyro G (2011). Regulation of opioid receptor signalling: implications for the development of analgesic tolerance. Mol Brain 4: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebenführ A, Ritzenthaler C, Robinson DG (2002). Brefeldin A: deciphering an enigmatic inhibitor of secretion. Plant Physiol 130: 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (1998). Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20: 389–399. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK (1997). Molecular and cellular basis of addiction. Science 278: 58–63. [DOI] [PubMed] [Google Scholar]
- Nörenberg W, Schöffel E, Szabo B, Starke K (1997). Subtype determination of soma‐dendritic alpha2‐autoreceptors in slices of rat locus coeruleus. Naunyn Schmiedebergs Arch Pharmacol 356: 159–165. [DOI] [PubMed] [Google Scholar]
- Osborne PB, Vidovic M, Chieng B, Hill CE, Christie MJ (2002). Expression of mRNA and functional alpha(1)‐adrenoceptors that suppress the GIRK conductance in adult rat locus coeruleus neurons. Br J Pharmacol 135: 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne PB, Williams JT (1995). Characterization of acute homologous desensitization of mu‐opioid receptor‐induced currents in locus coeruleus neurones. Br J Pharmacol 115: 925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JJ, Gondré‐Lewis MC, Eiden LE, Loh YP (2011). A distinct trans‐Golgi network subcompartment for sorting of synaptic and granule proteins in neurons and neuroendocrine cells. J Cell Sci 124: 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertovaara A (2006). Noradrenergic pain modulation. Prog Neurobiol 80: 53–83. [DOI] [PubMed] [Google Scholar]
- Pineda J, Kogan JH, Aghajanian GK (1996a). Nitric oxide and carbon monoxide activate locus coeruleus neurons through a cGMP‐dependent protein kinase: involvement of a nonselective cationic channel. J Neurosci 16: 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda J, Ruiz‐Ortega JA, Martín‐Ruiz R, Ugedo L (1996b). Agmatine does not have activity at alpha 2‐adrenoceptors which modulate the firing rate of locus coeruleus neurones: an electrophysiological study in rat. Neurosci Lett 219: 103–106. [DOI] [PubMed] [Google Scholar]
- Pineda J, Ruiz‐Ortega JA, Ugedo L (1997). Receptor reserve and turnover of alpha‐2 adrenoceptors that mediate the clonidine‐induced inhibition of rat locus coeruleus neurons in vivo. J Pharmacol Exp Ther 281: 690–698. [PubMed] [Google Scholar]
- Portoghese PS, Larson DL, Sayre LM, Fries DS, Takemori AE (1980). A novel opioid receptor site directed alkylating agent with irreversible narcotic antagonistic and reversible agonistic activities. J Med Chem 23: 233–234. [DOI] [PubMed] [Google Scholar]
- Santamarta MT, Llorente J, Mendiguren A, Pineda J (2014). Involvement of neuronal nitric oxide synthase in desensitization of μ‐opioid receptors in the rat locus coeruleus. J Psychopharmacol 28: 903–914. [DOI] [PubMed] [Google Scholar]
- Santamarta MT, Ulibarri I, Pineda J (2005). Inhibition of neuronal nitric oxide synthase attenuates the development of morphine tolerance in rats. Synapse 57: 38–46. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K et al. (1999). Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 284: 1811–1816. [DOI] [PubMed] [Google Scholar]
- Simon JP, Ivanov IE, Adesnik M, Sabatini DD (1996). The production of post‐Golgi vesicles requires a protein kinase C‐like molecule, but not its phosphorylating activity. J Cell Biol 135: 355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker NL, Huganir RL (2002). AMPA/kainate receptors In: Moss SJ, Henley J. (eds). Receptor and Ion‐channel Trafficking: Cell Biology of Ligand‐gated and Voltage‐sensitive Ion Channels. Oxford UP: New York, pp. 131–155. [Google Scholar]
- Tanowitz M, von Zastrow M (2003). A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J Biol Chem 278: 45978–45986. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Colago EE, Moriwaki A, Uhl GR (1996). Mu‐opioid receptor is located on the plasma membrane of dendrites that receive asymmetric synapses from axon terminals containing leucine‐enkephalin in the rat nucleus locus coeruleus. J Comp Neurol 376: 65–74. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Pickel VM (1993). Ultrastructure of serotonin‐immunoreactive terminals in the core and shell of the rat nucleus accumbens: cellular substrates for interactions with catecholamine afferents. J Comp Neurol 334: 603–617. [DOI] [PubMed] [Google Scholar]
- Ward SJ, Fries DS, Larson DL, Portoghese PS, Takemori AE (1985). Opioid receptor‐binding characteristics of the non‐equilibrium mu‐antagonist, beta‐funaltrexamine (beta‐FNA). Eur J Pharmacol 107: 323–330. [DOI] [PubMed] [Google Scholar]
- West WL, Yeomans DC, Proudfit HK (1993). The function of noradrenergic neurons in mediating antinociception induced by electrical stimulation of the locus coeruleus in two different sources of Sprague–Dawley rats. Brain Res 626: 127–135. [DOI] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S et al. (2013). Regulation of mu‐opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65: 223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, North RA (1984). Opiate–receptor interactions on single locus coeruleus neurones. Mol Pharmacol 26: 489–497. [PubMed] [Google Scholar]